
Abstract
The neurotrophic factor BDNF is an important regulator for the development of brain circuits, for synaptic and neuronal network plasticity, as well as for neuroregeneration and neuroprotection. Up- and downregulations of BDNF levels in human blood and tissue are associated with, e.g., neurodegenerative, neurological, or even cardiovascular diseases. The changes in BDNF concentration are caused by altered dynamics in BDNF expression and release. To understand the relevance of major variations of BDNF levels, detailed knowledge regarding physiological and pathophysiological stimuli affecting intra- and extracellular BDNF concentration is important. Most work addressing the molecular and cellular regulation of BDNF expression and release have been performed in neuronal preparations. Therefore, this review will summarize the stimuli inducing release of BDNF, as well as molecular mechanisms regulating the efficacy of BDNF release, with a focus on cells originating from the brain. Further, we will discuss the current knowledge about the distinct stimuli eliciting regulated release of BDNF under physiological conditions.
Similar content being viewed by others


Introduction
BDNF in health and disease
The neurotrophic factor BDNF plays an important role for the development of brain circuits, the formation and maintenance of neuronal morphology, brain architecture and for synaptic, as well as neuronal network plasticity (Edelmann et al. 2014; Gottmann et al. 2009; Huang and Reichardt 2001; Klein 1994; Lessmann and Brigadski 2009; Park and Poo 2013). Consequently, BDNF crucially regulates learning and memory processes in young and adult mammals (see, e.g., Boschen and Klintsova 2017; Gomez-Pinilla and Vaynman 2005) such that imbalances in BDNF levels and downstream signaling via its cognate TrkB tyrosine kinase receptor are associated with neurodegenerative and psychiatric diseases, like Alzheimer’s disease, major depressive disorder (Castren and Hen 2013), or schizophrenia (Mohammadi et al. 2018). Moreover, BDNF signaling also contributes to physiological functions of the heart and the vasculature and is involved in disorders like coronary artery disease (Ejiri et al. 2005; Jin et al. 2018; Kaess et al. 2015), diabetes mellitus (Eyileten et al. 2017; Suwa et al. 2006), inflammatory diseases such as asthma (Prakash and Martin 2014), different types of cancer (Chopin et al. 2016; Radin and Patel 2017), as well as pain sensation (Deitos et al. 2015; Haas et al. 2010; Laske et al. 2007; Merighi et al. 2008; Sapio et al. 2019). Furthermore, BDNF levels in preterm neonates differ from BDNF levels in full-term neonates (Malamitsi-Puchner et al. 2004), thereby affecting cognitive development in early postnatal life (Chau et al. 2017) and potentially being associated with children’s mental diseases such as autism spectrum disorders (Qin et al. 2016; Zheng et al. 2016).
Measurements of BDNF as a diagnostic marker
In human medical studies, ELISA or Western blot-based measurements of BDNF protein levels in body fluids (e.g., blood or liquor) or tissue samples are considered as a potential proxy of intact brain (or other organ) function and associated diseases. As a result, great efforts are being made to utilize measurements of the BDNF levels as a potential marker for health status, diagnosis and prognosis of diseases and therapeutic outcome (Balietti et al. 2018). Variations in BDNF genotype (compare Intracellular sorting of BDNF in neurons section) are considered to be covariate information in such approaches (Shen et al. 2018). In the blood, the secretory protein BDNF is mainly stored in platelets (Fujimura et al. 2002). However, little is known about the original source of circulating BDNF in blood plasma (Dawood et al. 2007; Klein et al. 2011; Krabbe et al. 2007; Rasmussen et al. 2009; Serra-Millas 2016). BDNF is prominently expressed in the brain, in hippocampus, cortex, amygdala and striatum but also in hypothalamus (reviewed in Edelmann et al. 2014). It is also expressed and released from endothelial cells (Cefis et al. 2020), cells of the immune system (lymphocytes, microglia), megakaryocytes (Chacon-Fernandez et al. 2016; Tamura et al. 2012) and others, like smooth muscle cells (reviewed in Edelmann et al. 2014). Activity-dependent release of BDNF from hypothalamic neurons, which are not shielded from the blood stream by the blood-brain barrier, likely contributes to blood BDNF levels. Transport of BDNF across the blood brain barrier was, although controversially discussed, also described for neurotrophins like BDNF (Pan et al. 1998; Rasmussen et al. 2009; Seifert et al. 2010). Therefore, BDNF released from neurons as a consequence of neural network activity in the brain can act locally by tuning the strength of the neural network but it may also act globally by affecting the function of other organs distant from the brain after diffusion into the bloodstream. Vascular endothelial cells as well as cells of the immune system represent another natural source of BDNF, which likely contribute to the regulation of BDNF levels in the blood (Kerschensteiner et al. 1999; Marie et al. 2018; Nakahashi et al. 2000). However, stimuli-inducing release of BDNF from non-neuronal tissue like platelets and vascular endothelial cells are not well understood.
In the human brain, analysis of post mortem tissue is currently almost the only way to estimate changes in BDNF levels. Decreased levels of BDNF have been detected post mortem in the cortex, hippocampus and nucleus basalis of Meynert in patients suffering from Alzheimer’s disease (Holsinger et al. 2000; Tapia-Arancibia et al. 2008), while region-specific down but also upregulation of BDNF has been shown in major depressive disorder patients (Krishnan et al. 2007; Pandey et al. 2008). However, to understand the relevance of change in BDNF levels in the brain, non-neuronal tissue and blood, a more detailed knowledge of the distinct stimuli-regulating BDNF expression and release in the different cell types is important. Most studies providing information about the molecular and cellular mechanism regulating intra- and extracellular BDNF levels have thus far been performed in cells originating from the brain. The role of distinct subcellular BDNF release sites as well as release kinetics, e.g., for cell survival or synaptic plasticity is best studied in neurons (reviewed, e.g., in Edelmann et al. 2014). Therefore, this review will discuss recent advancements in cell-type specific activity-dependent release of BDNF, with a focus on BDNF release from cells originating from the brain. We will summarize the stimuli inducing or amplifying the release of BDNF as well as the molecular regulation of BDNF secretion and discuss the role of BDNF secretion in the context of synaptic plasticity and specific diseases.
Synthesis, processing and sorting of BDNF
General aspects of BDNF synthesis
The secretory protein BDNF is expressed in neuronal and non-neuronal cells (Katoh-Semba et al. 1997; Maisonpierre et al. 1990; Phillips et al. 1990). In neuronal cells, BDNF-immunoreactivity was found in several regions of the central nervous system as well as in the peripheral and enteric nervous system (Barakat-Walter 1996; Conner et al. 1997; Ernfors et al. 1990; Hoehner et al. 1996; Wetmore and Olson 1995; Yan et al. 1997; Zhang et al. 2007; reviewed, e.g., in Lessmann et al. 2003; Lewin and Barde 1996). In non-neuronal tissue, BDNF is synthesized in cells of the immune system, like T cells, B cell and monocytes (Kerschensteiner et al. 1999; Nakahashi et al. 2000), muscle cells (Matthews et al. 2009; Mousavi and Jasmin 2006), the heart (Donovan et al. 2000; Maisonpierre et al. 1990), thymus, liver and spleen (Katoh-Semba et al. 1997). The tissue-specific expression of BDNF is developmentally regulated (Ernfors et al. 1990; Katoh-Semba et al. 1997). Furthermore, physiological as well as pathophysiological conditions and interventions such as exercise, hypoxia, stress, epileptic seizures and ischemia increase expression of BDNF in a tissue-specific manner (Giannopoulou et al. 2018; Lippi et al. 2020; Thomas et al. 2016; Wetmore et al. 1994).
In humans, the BDNF gene is located on chromosome 11p14.1 consisting of 11 exons (I–XI) and nine promotors that regulate the developmental and regional expression of multiple alternatively spliced mRNA isoforms (Pruunsild et al. 2007; Timmusk et al. 1993). Only exon IX at the 3′ end of the gene locus contains the major coding sequence for the BDNF precursor protein (Pruunsild et al. 2007). This BDNF coding sequence is translated into a pre-pro-protein, with an N-terminal pre-domain that guides the mRNA to the rough endoplasmic reticulum (rER). The pre-sequence is cleaved of co-translationally and the immature uncleaved pro-BDNF protein is synthetized into the ER (compare Lessmann and Brigadski 2009). Importantly, transcription of the BDNF gene into a set of distinctly spliced mRNA variants (Aid et al. 2007; Pruunsild et al. 2007) is tightly regulated by electrical activity-induced Ca2+ elevation in neurons (e.g., (Castrén et al. 1998). The distinct mRNA variants can be transported into dendrites (Baj et al. 2013), where local translation and release of BDNF can be confined to especially active dendritic stretches thereby promoting synaptic plasticity in a BDNF-dependent fashion. This makes BDNF ideally suited to shape developing and fine tune mature synaptic circuits through activity-dependent local synthesis and secretion (compare chapter “Physiological context of regulated release of BDNF section” in this review, see also Johnestone and Mobley 2020).
Posttranslational processing of BDNF
Secretory proteins are posttranslationally processed by different enzymes such as endo- or exopeptidases or glycosyltransferases (reviewed in Thomas 2002). The incidence of the diverse posttranslational modifications depends on the entire set of modifying proteins expressed by a specific cell, as well as on the intracellular ionic composition in cytoplasm, ER and Golgi (Creemers 2002; Gidalevitz et al. 2013). Characteristic features of secreted proteins, such as biological activity, half-life time, or affinity to specific intracellular binding partners are modified by these events. The best-known example for a precursor protein (pro-protein) that undergoes multiple posttranslational modification steps is the hormone pro-opiomelanocortin (POMC). POMC is the precursor of a variety of hormones like ACTH, MSH, or beta-endorphin. These diverse peptides are generated after endoproteolytic cleavage of the precursor protein POMC by subtilisin-like prohormone convertases PC1 or PC2. The thereby generated new N- and C-termini are further processed by exopeptidases, acetyltransferases and/or peptidyl-glycine alpha-amidating monooxygenases (compare Lessmann and Brigadski 2009). However, the corresponding processing of BDNF has not been intensively investigated so far. It is known that the 32-kDa precursor proBDNF can be processed endoproteolytically within the pro-domain by the enzyme subtilisin/kexin-isozyme 1 (SKI-1), generating a 28 kDa protein (Seidah et al. 1999). In addition to the 28-kDa cleavage product of the BDNF precursor, the 14-kDa mature BDNF is generated after cleavage of proBDNF by furin-like protein convertases PACE4, PC1, or PC5 (Mowla et al. 2001; Seidah et al. 1996). Inhibition of furin-like enzymes prevents the synthesis of the 14 kDa mature BDNF without any effect on the generation of the 28 kDa cleavage product (Mowla et al. 2001). Furthermore, extracellular processing of proBDNF to the 14 kDa mature BDNF isoform by the tissue-type plasminogen activator (tPA)/plasmin proteolytic system and by matrix metalloproteinases (MMP) 3, 7 and 9 has been described (Gray and Ellis 2008; Lee et al. 2001; Pang et al. 2004). Further modifications like N-glycosylation and glycosulfation within the prodomain of BDNF increase the half-life time of the protein (Mowla et al. 2001). These posttranslational modifications may also be a prerequisite for interactions of BDNF with chaperones or sorting receptors that guide the secretory protein BDNF predominantly to vesicles of the regulated secretory pathway (reviewed in Leßmann and Brigadski 2009).
Intracellular sorting of BDNF in neurons
Up to now, two sorting proteins have been identified for BDNF. Both, the chaperone sortilin and the exoproteolytic enzyme carboxypeptidase E, bind to the BDNF pro-domain and play an important role in sorting of BDNF to vesicles of the regulated pathway of secretion (Chen et al. 2005; Lou et al. 2005; Kojima et al. 2020). Interestingly, transfection of neurons with a cDNA construct, in which the pre-pro-domain of BDNF is fused to the mature part of the neurotrophin-4 (NT-4) redirects this BDNF-NT-4 chimera efficiently into vesicles of the regulated secretory pathway, whereas wildtype NT-4 is predominantly located in vesicles of the constitutive secretory pathway (Brigadski et al. 2005). This indicates the importance of the BDNF pro-domain for sorting of the protein to the regulated secretory pathway. The significance of the pro-domain for correct sorting of BDNF to secretory granules is also stressed by the commonly observed Val66Met single-nucleotide polymorphism (SNP) located in the BDNF pro-domain. This SNP (carriers in Europe: ~ 60% Val/Val (WT); ~ 35% Val/Met; ~ 5% Met/Met) is associated with reduced sorting of Met-BDNF to vesicles of the regulated secretory pathway, resulting in impaired activity-dependent secretion of BDNF from neuronal cells (Egan et al. 2003). However, this SNP-dependent reduced sorting to secretory granules does not seem to affect all BDNF transcripts to the same extent (Jiang et al. 2009). In addition to the major sorting of BDNF to the regulated secretory pathway (Brigadski et al. 2005), BDNF is also directed to vesicles of the constitutive secretory pathway. However, little is known about the molecular composition of constitutively released vesicles as well as the function of BDNF released in a constitutively manner.
Physiological context of regulated release of BDNF
Just as there exist different cell types expressing and releasing BDNF, diverse stimuli are known to induce regulated release of BDNF. The best characterized stimuli are patterns of neuronal electrical activity like prolonged depolarization, high-frequency stimulation (HFS), or theta-burst stimulation (TBS) that trigger BDNF release in developing and mature neurons (Edelmann et al. 2014) (compare Stimuli-triggering BDNF release from neurons section) (Table 1). Release stimuli in electrically non-excitable cells differ, of course, from the major release stimuli in neuronal cells. Thus, binding of extracellular nucleotides (Coull et al. 2005; Trang et al. 2009; Ulmann et al. 2008; Vignoli and Canossa 2017), pro-inflammatory factors (Jornot et al. 2007), or neuropeptides (Lopez-Benito et al. 2018) to their corresponding receptors has been shown to induce release of BDNF from astrocytes and microglia (compare BDNF release from astrocytes section and BDNF release from microglia section) (Table 2). Next to the different physiological BDNF-releasing stimuli, knowing the subcellular sites where BDNF secretion can take place is important to understand cell-type and release-site specific functions of BDNF, e.g., during neuronal development, synaptic plasticity processes, or in case of disease (e.g., Hartmann et al. 2001; Matsuda et al. 2009; Xia et al. 2009; reviewed in Edelmann et al. 2014; Leßmann and Brigadski 2009; Park and Poo 2013).
Stimuli-triggering BDNF release from neurons
In the central nervous system, BDNF is important for the development of brain circuits and for synaptic as well as for network plasticity processes in the adult brain. These processes require fine-tuning of synaptic activity and structural synaptic rearrangements in an input specific manner. The spatially restricted release of BDNF is ideally suited for this purpose. Since early neuronal networks of immature neurons differ from functionally mature synaptic circuits, it is not surprising that distinct electrical activity patterns are efficient to induce release of BDNF and that the efficacy of releasing BDNF depends on the developmental stage of the neurons as well as on the specific brain area that is investigated.
Stimuli-triggering BDNF release from developing neurons
In developing hippocampus, a sequence of three different electrical activity patterns was described to be important to form synaptic circuitry (Crepel et al. 2007; Egorov and Draguhn 2013; Luhmann and Khazipov 2018). First, intrinsically active neurons generate brief L-type voltage-gated Ca2+-channel (VGCC)-mediated spikes at embryonic stage E16-E19, before an ensemble of neurons coupled by gap-junctions generates synchronous non-synaptic, long lasting calcium-plateaus, called synchronous plateau assemblies (SPA). These SPAs are replaced by giant depolarizing potentials (GDPs), which represent a spontaneous pattern of network activity via chemical synaptic interactions. GDPs consist of large depolarizations associated with a burst of action potentials. These depolarizations last several hundreds of milliseconds, propagate by synaptic interactions as waves through the developing hippocampus and occur at low-frequency (0.003–0.06 Hz) in rodents during the first week of postnatal life (Egorov and Draguhn 2013; Luhmann and Khazipov 2018). GDPs have also been described for other developing brain areas like the neocortex, thalamus, or spinal cord. Development of cortical networks is known to progress in a similar manner as in hippocampal networks (Egorov and Draguhn 2013). However, the time course of onset of cortical development is shifted to postnatal stages (Hanganu-Opatz 2010; Kilb et al. 2011). Interestingly, the onset of BDNF expression in the rodent hippocampus becomes apparent already at E15.5 and differs from the onset of BDNF expression in cortical neurons, which appears at P4 (Baquet et al. 2004; Katoh-Semba et al. 1997). Furthermore, these electrically activity patterns (intrinsically active neurons, SPAs and GDPs) depend on activation of L-type VGCCs. Consistent with this, the release of BDNF has been reported to depend on Ca2+-influx via L-type voltage-gated calcium channels (VGCC) in primary cultures of hippocampal neurons from embryonic or newborn rats and mice (Kolarow et al. 2007; Kuczewski et al. 2008; Matsuda et al. 2009; Kojima et al. 2020). Thus, it could be speculated that BDNF release from intrinsically active neurons, which display uncorrelated calcium transients, or BDNF release from gap-junction-coupled neurons, which generate non-synaptic calcium-plateaus might play a role in shaping neuronal networks during embryonic and early postnatal development. However, this has not been extensively investigated so far.
To date, only GDPs were shown to represent robust activity patterns to induce release of BDNF in this context (Kuczewski et al. 2008; Mohajerani et al. 2007). Kuczewski et al. (2008) combined whole-cell recordings and time-lapse fluorescence imaging of transfected hippocampal neurons and demonstrated BDNF-GFP release after repeated depolarization steps that were strongly reminiscent of GDPs (Kuczewski et al. 2008). Furthermore, Mohajerani et al. reported plasticity of CA3-CA1 synapses after pairing of GDPs with Schaffer collateral stimulation in hippocampal slices during the first postnatal week of development (Mohajerani et al. 2007). They described that the depolarizing action of GABA during GDPs induced calcium influx through L-type VGCC, thereby triggering plasticity of CA3-CA1 synapses by endogenously released BDNF (Gubellini et al. 2005; Mohajerani et al. 2007). The depolarizing effects of GABAergic transmission fade out with ongoing postnatal development and this maturation of hippocampal networks requires a shift in the expression of chloride transporters in neurons from K+-Cl−-cotransporter (KCC2) to Na+-K+-chloride−-cotransporter (NKCC1), which switches GABA transmission to be hyperpolarizing. Since regulation of KCC2 expression is also mediated by BDNF (Rivera et al. 2002, 2005; reviewed in Fiumelli and Woodin 2007), depolarization-induced release of BDNF is important for proper shaping of hippocampal synaptic circuits.
Stimuli-triggering BDNF release from mature neurons
Depolarizing waves propagating through cortical networks represent on the one hand physiological activity patterns during neural circuit development but on the other hand, they have also been implicated in the pathophysiology of stroke, head trauma, or migraine aura (Hartings et al. 2017; Shen et al. 2016). Transient depolarizations of neurons induced by elevated extracellular K+-concentration have been described as a phenomenon called cortical spreading depression (CSD) or spreading depolarizations. CSD is characterized by waves of depolarizations that spread across the cortical surface at a low velocity of 2–5 mm/min. Consistent with activity-dependent regulation of BDNF, increases in BDNF mRNA and protein levels have been observed in response to CSD in rodents (Kawahara et al. 1997; Kokaia et al. 1993). Therefore, a protective role of CSD-induced BDNF expression and release enhancing the tolerance of further injury has been postulated (Kawahara et al. 1997; Shen et al. 2016). Interestingly, a very potent protocol to induce robust neuronal BDNF release is to elevate the extracellular K+-concentration, which in turn leads to depolarization and intracellular Ca2+-elevation. Such an elevated K+-induced release of BDNF was observed, e.g., in embryonic as well as postnatal neuronal cultures, neurons derived from embryonic stem cells and acute and organotypic hippocampal slices (Canossa et al. 1997; Goodman et al. 1996; Griesbeck et al. 1999; Hartmann et al. 2001; Kojima et al. 2001; Leschik et al. 2013; Leschik et al. 2019).
Stimuli-triggering BDNF release in synaptic plasticity
In addition to the development of brain circuits, BDNF is also involved in synaptic and network plasticity processes in mature circuits. Synaptic plasticity (i.e., LTP and LTD) is thought to be a cellular model of learning and memory processes. However, analyzing the activity patterns forming a memory together with the related structural changes of single synapses in a neuronal circuit in vivo is a challenging task. Although it is widely accepted that synaptic changes tune neural circuitry, the contribution of synaptic changes to memory encoding or memory consolidation is not resolved. Furthermore, the physiological correlates of the different activity patterns known to modulate synaptic efficacy, like high or low frequency stimulation, need to be identified. To date, several activity patterns shaping glutamatergic and GABAergic synapses either at pre- and/or postsynaptic sites have been investigated. The efficacy of the different patterns, of course, depends on diverse factors such as the developmental stage of the investigated neuronal circuit. The best-known patterns to induce long-term potentiation (LTP) of synaptic transmission in the hippocampus are high-frequency stimulation (HFS; e.g., repeated 1-s trains at 100 Hz), theta-burst stimulation (TBS; repeated bursts of 4–5 APs at ~ 100 Hz with inter-burst intervals of 0.2 s) or pairing protocols like spike timing-dependent plasticity (STDP; compare, e.g., Gottmann et al. 2009). Protocols that elicit long-term depression (LTD) of synaptic efficacy are characterized by low-frequency synaptic stimulation (LFS at 1 Hz). Both, LTP and LTD, are known to shape synapses at the functional, molecular and structural level. Activity-dependent release of endogenous BDNF or of fluorescently labeled BDNF has been intensively investigated as a cellular mechanism of LTP in brain slices and in neuronal cultures from different brain regions. Release of BDNF induced by HFS was shown, e.g., in PNS-cultured neurons (Balkowiec and Katz 2000), in hippocampal cultures (Balkowiec and Katz 2002; Hartmann et al. 2001; Kojima et al. 2001; Matsuda et al. 2009), amygdala (Meis et al. 2012) and cortico-striatal synapses (Jia et al. 2010), using either BDNF ELISA measurements or live cell imaging of fluorescently labeled BDNF. In contrast, similar patterns of high-frequency stimulation failed to induce efficient release of BDNF in dorsal horn slices from lumbal spinal cord (Lever et al. 2001). However, these authors were among the first to reveal TBS (300 bursts in 75 trains at 100 Hz with an interburst intervals of 0.2 s) as an effective means to induce release of BDNF (compare Balkowiec and Katz 2002). In addition to theta-burst discharges, BDNF release in spinal cord slices could also be elicited by injection of capsaicin, which in turn produces bursting activity reminiscent of theta bursts in the investigated nociceptors (Lever et al. 2001).
Bursting activity, which is repeated at a frequency of the theta rhythm, mimics also electrical activity patterns during hippocampal learning. Accordingly, TBS of Schaffer collateral CA1 synapses in acute brain slices was shown to efficiently release endogenous BDNF, thereby mediating postsynaptically expressed STDP (Edelmann et al. 2015). Correspondingly, TBS was also reported to release fluorescently labeled BDNF from either pre- or from postsynaptic sites in dissociated hippocampal cultures (Bergami et al. 2008; Matsuda et al. 2009) and from cortico-striatal presynaptic terminals (Park 2018). In addition to TBS-induced release of BDNF in hippocampal preparations, many other studies have analyzed the role of endogenously released BDNF in TBS-induced LTP, by either scavenging extracellular BDNF with superfusion of cells with selective BDNF antibodies, or by quantifying released BDNF by ELISA measurements in other brain regions. In this way, BDNF was shown to mediate TBS-LTP, e.g., at glutamatergic inputs to the amygdala (Li et al. 2011) or at retino-optical synapses (Du et al. 2009).
Although the BDNF-dependent LTP-inducing protocols seem to be very similar, the release sites of BDNF as well as the site of BDNF action depend on brain region and context (reviewed in Edelmann et al. 2014; Zagrebelsky et al. 2020). Moreover, even similar patterns of synaptic activity may induce mechanistically distinct types of LTP in the same neuronal circuit and subtle experimental details can determine in which way BDNF is involved in this plasticity. For example, spike timing-dependent long-term potentiation (t-LTP) is characterized by nearly coincident pairing of presynaptically induced excitatory postsynaptic potentials (EPSP) with postsynaptic firing of an action potential (AP) (Bi and Poo 1998). Such activity patterns were shown for the first time in the cortex of tadpoles to elicit BDNF release-dependent t-LTP (Mu and Poo 2006). However, slight changes in t-LTP protocols can decide whether t-LTP is mediated by BDNF or other modulators. In this respect, Edelmann et al. (2015) reported that a burst t-LTP protocol with 4 bAP was mediated by endogenously released BDNF, while t-LTP induced by stimulation protocols including only 1 bAP, occurred independent from BDNF release (Edelmann et al. 2015). These slightly different protocols did not occlude each other, indicating the differential physiological relevance of both protocols at the identical synaptic sites. In another study, regional deletion of BDNF either in the CA1 or the CA3 region revealed a role of presynaptically released BDNF in the induction of HFS-LTP, while postsynaptically released BDNF contributed to the maintenance of HFS-LTP (Lin et al. 2018). Complementary to the above-mentioned in vitro studies, Messaoudi et al. investigated the role of released BDNF in HFS-induced LTP in the hippocampus in vivo. Application of exogenous mature BDNF induced LTP (BDNF-LTP) in perforant path-granule cell synapses of the hippocampus, which occluded gene transcription dependent late-LTP but not early-LTP at the same synapses (Messaoudi et al. 2002). Future studies need to elucidate the role of proBDNF- and BDNF-dependent LTP in vivo in other synaptic circuits of the hippocampus and in additional brain areas (see also Zagrebelsky et al. 2020)
Subsequently to the release of BDNF from BDNF-expressing cells, the protein can also be released after endocytosis of the released BDNF (Fig. 5). Interestingly, the endocytosed BDNF can be further modified by the recipient cell before the re-release event. Such a re-exocytosis or recycling of BDNF has been reported for cultures of mature neurons, astrocytes and platelets (Bergami et al. 2008; Huang et al. 2014; Santi et al. 2006; Vignoli and Canossa 2017). In neurons, recycling of BDNF enables, on the one hand, the replenishment of the BDNF pool of releasing cells and re-use of BDNF after further processing steps. On the other hand, it also enables long distance distribution of synthetized BDNF across synaptically connected neuronal circuits and surrounding astrocytes after transcytosis of BDNF, a mechanism which was previously described or postulated for different neurotrophic factors (Bartheld and Johnson 2001; Butowt and Bartheld 2001; Wirth et al. 2005).
Endocytosis or re-uptake of BDNF is also an important mechanism to fill BDNF stores in platelets (Fujimura et al. 2002; Serra-Milàs 2016). These stores may constitute the major source of BDNF that can be released from platelets in response to blood vessel injury. Of note, the expression of BDNF not only in neurons but also in non-neuronal tissue particularly in the immune system and the cardiovascular system raises the interesting question how sorting and release of BDNF is regulated in these cells. Physiological stimuli, like shear stress in blood vessels but also pathophysiological and oncogenic stimuli, might orchestrate BDNF expression and release in these cell types as well as in cell types in which BDNF expression is under debate or even not proven so far. Future research addressing these important questions is critically needed.
BDNF release from astrocytes
Stimuli-triggering BDNF from astrocytes during plasticity processes
BDNF can be secreted from neuronal cells but also from other cells such as astrocytes. Quantifying BDNF release by ELISA measurements during synaptic plasticity processes or analyzing the contribution of BDNF during LTP by scavenging BDNF from extracellular space (Shelton et al. 1995) do not clarify the original release sites of BDNF. Moreover, a potential permissive function of BDNF for LTP, or acute early or late instructive actions of BDNF during LTP, may depend on BDNF release from different cellular sources. In the central nervous system, astrocytes are closely associated with synapses. One main function of astrocytes is the modulation of synaptic function. It is known that proBDNF is endocytosed by astrocytes in a p75-dependent manner during TBS-LTP in hippocampal or perirhinal cortex slices (Bergami et al. 2008; Vignoli et al. 2016). Extracellular cleavage of proBDNF by plasmin or deletion of glial p75 receptors inhibits endocytosis of proBDNF by astrocytes (Bergami et al. 2008; Vignoli et al. 2016). After recycling of endocytosed proBDNF and intracellular cleavage, the re-released mature BDNF contributes to the maintenance of TBS-LTP in a postsynaptic TrkB-dependent manner (Vignoli et al. 2016). Furthermore, mice that were unable to recycle BDNF failed to recognize familiar from novel objects. Therefore, astrocytic BDNF recycling was shown to be essential for visual recognition memory (Vignoli et al. 2016). Besides the astrocytic recycling of BDNF, neuronal recycling of BDNF has also been shown to contribute to TBS-LTP maintenance in hippocampal slices (Santi et al. 2006). The role of astrocytic recycling has not been addressed in this study. Whether neuronal and astrocytic recyclings of BDNF coexist or whether they are successive processes need to be clarified. In contrast to the endocytosis of pro-BDNF, another study suggests that solely the mature form of BDNF is endocytosed by astrocytes via truncated TrkB receptors in dissociated cultures (Stahlberg et al. 2018). Interestingly, inflammatory stimuli like released interferon-gamma increased astrocytic expression of truncated TrkB, which was associated with increased BDNF recycling capacity of astrocytes (Rubio 1997). Additional studies are needed to clarify whether astrocytic recycling of mature BDNF via truncated TrkB-receptors contributes to astrocyte-dependent synaptic plasticity underlying memory formation or whether it is a part of inflammatory processes.
During plasticity processes, different stimuli might induce release of recycled BDNF from astrocytes. Since glutamate is known to induce the release of astrocytic BDNF (Jean et al. 2008; Santi et al. 2006; Vignoli et al. 2016), the prolonged glutamate release from the presynaptic terminal may be important for the release of recycled BDNF from astrocytes during TBS. These authors showed that the effect of glutamate was mimicked by agonists of AMPA or mGluRI/II receptors (Bergami et al. 2008). Interestingly, TBS activity is likely to induce elevated extracellular potassium levels. Consistent with this, BDNF release from astrocytes was also shown to be induced by elevated extracellular potassium concentration (Stahlberg et al. 2018; Vignoli and Canossa 2017). However, HFS was ineffective in releasing recycled BDNF from pure astrocytic cultures (Bergami et al. 2008).
Another stimulus to induce BDNF release was described by Canossa et al., in hippocampal slices and in neuronal cultures (Canossa et al. 1997). There, BDNF-induced BDNF release was observed (Canossa et al. 1997; Nakajima et al. 2008 but compare Kolarow et al. 2007). This mechanism might also occur in astrocytes during synaptic function to refill stores of BDNF and simultaneously to induce release of a readily releasable pool of BDNF vesicles. Taken together, processing and release of BDNF from astrocytes during synaptic plasticity process and memory formation are factors that should not be neglected.
Stimuli-triggering BDNF from astrocytes in inflammatory or neuroprotective processes
In addition to synaptic functions, astrocytes have several other functions in the CNS. They are important for the regulation of energy supply and for the homeostasis of neurotransmitters and ions. They are involved in function and maintenance of the blood brain barrier and they have immunoregulatory functions. Different stimuli are known to activate one or the other effect of astrocytes, respectively. One player that is associated with inflammation, pain, or immune response in astrocytes is prostaglandin E2 (PGE2). PGE2 has been shown to induce release of BDNF from astrocytes (Hutchinson et al. 2009). In addition to PGE2, the proinflammatory cytokine TNF-alpha was also shown to induce BDNF release from astrocytes (Giralt et al. 2010; Saha et al. 2006).
Furthermore, astrocytes are known to mediate neuroprotective functions, which involve BDNF release (Su et al. 2012). However, neuroprotective functions of astrocyte-derived BDNF were shown to be region-specific (Datta et al. 2018). These authors showed that cultured astrocytes from different brain regions exhibit distinct efficacy of BDNF release in response to oxidative stress induced by 6-hydroxydopamine (6-OHDA).
In line with the neuroprotective function, astrocytes are also important in regulating the glymphatic system. This waste clearance system of the brain has been described as a tunnel system around blood vessels formed by astrocytes. It is important for clearing the brain from toxic compounds like Amyloid-β-, which is itself a stimulus to induce BDNF release from astrocytes (Hou et al. 2011). However, neuronal release of BDNF was unaffected in transgenic Alzheimer models although transport of BDNF was impaired already after acute application of Amyloid-β (Seifert et al. 2016). In conclusion, regulation of the glymphatic system in response to Amyloid-β might be a BDNF-mediated neuroprotective effect of activated astrocytes in the brain.
BDNF release from microglia
Similar to astrocytes, the primary immune cells of the brain, the microglia, also mediate several functions in the CNS, such as neuroprotection and shaping of synaptic spines (Dubbelaar et al. 2018; Stratoulias et al. 2019). Different microglia phenotypes exist, which can be transformed into one another by different chemokines and intercellular interactions, thereby exerting distinct functions in the brain (Orecchioni et al. 2019; Xue et al. 2014). One older simplifying in vitro classification of microglia, the M1/M2 concept, groups the different overlapping activated microglial phenotypes into two extremes: the proinflammatory lipopolysaccharide-(LPS)-induced M1 phenotype and the neuroprotective IL-4-induced M2 phenotype (Werry et al. 2019). BDNF expression and secretion from microglia have been shown for both activated M1 and M2 phenotypes but also for non-activated M0 microglia (Coull et al. 2005; Nakajima et al. 2001; Nakajima et al. 2002; Zhou et al. 2020). For the neuroprotective phenotype, extracellular accumulation of BDNF was shown after polarization of microglial cultures with IL-4 (Zhou et al. 2020). Furthermore, Merlo et al. demonstrated an Amyloid-β 42-induced release of BDNF thereby mediating a neuroprotective effect in cultures of a neuroblastoma cell line (Merlo et al. 2018).
LPS or 8-ceramide-induced polarization of microglia to the proinflammatory phenotype leads to an increase in extracellular mature BDNF that was described to depend on surface expression and activation of adenosine A2a receptor (Gomes et al. 2013; Nakajima et al. 2002). Furthermore, surface expression of P2X4 adenosine receptors (P2X4R) drives microglia polarization towards the pro-inflammatory phenotype. The surface expression of P2X4Rs in microglia was associated with central sensitization like pain hypersensitivity or allodynia (Tsuda et al. 2013). Hypersensitivity in chronic pain was also associated with release of BDNF from microglia (Zhou et al. 2019). The uncovered molecular mechanism underlying BDNF-dependent hypersensitivity in chronic pain involved the small G protein MAPK p38 (Zhou et al. 2019). Activation of this protein is known to be important in P2X4R-mediated release of BDNF from microglia (Coull et al. 2005; Ferrini et al. 2013; Long et al. 2020; Trang et al. 2009). Activation of this purinergic ligand-gated cation channel (P2X4R) by extracellular ATP leads to an increase in intracellular calcium concentration, thereby triggering distinct molecular mechanisms to induce BDNF release from microglia.
Molecular mechanisms underlying release of BDNF
Calcium transients and BDNF secretion
Fusion of transmitter and peptide vesicles depends on calcium ions, which are essential for triggering membrane fusion and on ATP or GTP supply for energy consuming steps of exocytosis such as priming of vesicles and disassembly of the SNARE complex. In recent years, great efforts have been made to uncover the molecular mechanisms regulating exocytosis of BDNF-containing secretory granules. It is well established that the essential calcium transients to trigger membrane fusion may have extracellular and/or intracellular origin. Activation of NMDA receptors (Hartmann et al. 2001), AMPA receptors (Canossa et al. 2001; Hartmann et al. 2001), L-type, N-Type or P/Q-type voltage-gated calcium channels (Buldyrev et al. 2006; Kolarow et al. 2007), transient receptor potential channels (TRPC) (Vohra et al. 2013), or ATP-gated purinergic receptors (P2XR) (Long et al. 2020; Trang et al. 2009) have been shown to contribute to the calcium transients in different BDNF-releasing cell types (see Tables 1, 23 and Fig. 1). Moreover, calcium release from internal calcium stores crucially contributes to BDNF release (Kolarow et al. 2007) and is of particular importance for BDNF release in response to external stimuli acting on G protein-coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs).
Calcium transients and BDNF secretion in neurons
Calcium transients and BDNF secretion in response to electrical stimulation
In neurons, calcium influx via pre- or postsynaptic NMDA receptors contributes to electrically induced BDNF release at the respective release site (Hartmann et al. 2001; Matsuda et al. 2009; Park 2018). At least for presynaptic terminals of cortico-striatal synapses, calcium influx from internal calcium stores was shown to prevent presynaptic BDNF release mediated via presynaptic NMDA receptors containing the GluN1 subunit (Park 2018) (Fig. 3). At postsynaptic release sites, electrically evoked BDNF release was solely dependent on extracellular calcium influx and not on calcium release from internal calcium stores (Kuczewski et al. 2008) (Fig. 2a). However, elevated potassium-induced BDNF release, depolarization-induced BDNF release and electrically evoked BDNF release in young cultures occur independent from AMPAR and NMDAR activation (Balkowiec and Katz 2002; Hartmann et al. 2001; Kuczewski et al. 2008) (Fig. 2b). In these cases, calcium influx was mediated via L-type VGCC (Balkowiec and Katz 2002; Buldyrev et al. 2006; Hartmann et al. 2001; Kolarow et al. 2007; Kuczewski et al. 2008), although the specific type of VGCC contributing to BDNF release might depend on the developmental stage and the brain area of the investigated neuron (Fig. 2b). Furthermore, dendritic elevated potassium-induced release of BDNF in hippocampal cultures was—in contrast to bAP-induced dendritic BDNF release—dependent on calcium efflux from internal calcium stores (Griesbeck et al. 1999; Kolarow et al. 2007; Kuczewski et al. 2008) (Fig. 2). This is in line with the role of BDNF during development of neural networks (compare Stimuli-triggering BDNF release from developing neurons section). The maturation of functional synapses might depend on BDNF release triggered by calcium-influx via VGCC and calcium release from internal calcium stores. In contrast, at more mature synapses, the calcium nanodomains defined by calcium influx via only NMDARs and VGCCs seem to be sufficient for bAP-induced site-specific local BDNF release from dendrites. Since calcium influx from internal stores was shown to be important for presynaptic AP-induced NMDAR-mediated BDNF release (Park 2018), calcium nanodomains might differ between dendrites and axons.

Schematic illustration of different release sites for BDNF. Release of BDNF takes place from somatic and dendritic compartments (green: ① + ②) and from axonal structures (yellow: ③) of glutamatergic neurons. Presynaptic neuron (yellow) and the postsynaptic glutamatergic neuron (green) are connected via glutamatergic synapses. The postsynaptic neuron additionally receives input from GABAergic interneurons (red). Astrocytic ④ and microglial ⑤ BDNF release has also been described. Recycling of BDNF ⑥ has been observed in neurons and in astrocytes. bAP back-propagating action potential, ER endoplasmic reticulum, GABABR gamma-aminobutyric acid receptor B, IP3-R inositol trisphosphate receptor, mGluR metabotropic glutamate receptor, NaV voltage-gated sodium channel, NMDAR N-methyl d-aspartate receptor, P2XR P2X purinergic receptor, PKC protein Kinase C., PLC phospholipase C, TRPC transient receptor potential channel, VGCC voltage gated calcium channels. Adapted from Brigadski and Leßmann, Neuroforum, 2014.

Suggested mechanisms for somatic and dendritic release of BDNF. BDNF release is dependent on extracellular Ca2+-influx (a) and/or intracellular Ca2+-release from internal stores (ER) Ca2+-influx (b). Ca2+-influx from extracellular space is mediated via VGCC and/or NMDAR (a, b). Ca2+ release from ER is mediated via IP3R or RyR (b). Increased burst firing activity, glutamate, or other ligands of GPCR mediate transient intracellular Ca2+-increase important for vesicle exocytosis. AC adenylate cyclase, CAMKII calmodulin-dependent protein kinase II; DAG diacylglycerol, ER endoplasmatic reticulum, IP3 inositol triphosphate, IP3R inositol-3-phosphate receptor, NaV voltage-gated sodium channel, NMDAR N-methyl d-aspartate receptor, PKA protein kinase A, PKC protein kinase C; PLC phospholipase C; RyR ryanodine, VGCC voltage-gated calcium channel. Adapted from Brigadski and Leßmann, Neuroforum, 2014
Calcium transients and BDNF secretion in response to chemical stimulation
Increased bursting activity of neural networks driven by glutamate or ketamine application or disinhibition of GABAergic transmission by GABAAR inhibitors represent potent stimuli for the induction of BDNF release (Canossa et al. 1997; Griesbeck et al. 1999; Lepack et al. 2014; Nakajima et al. 2008; Porcher et al. 2011; Santi et al. 2006). Interestingly, in contrast to bAP-induced BDNF release, glutamate-induced BDNF release from brain slices or primary neuronal cultures has been shown to be independent from extracellular calcium but dependent on calcium release from internal calcium stores (Canossa et al. 2001; Griesbeck et al. 1999; Santi et al. 2006) (Fig. 2b). This glutamate-induced BDNF release was also dependent on AMPA receptor or mGluR activation, respectively (Canossa et al. 1997; Canossa et al. 2001; Canossa et al. 2002; Nakajima et al. 2008; Santi et al. 2006), as well as on IP3-signaling that activates calcium release from internal calcium stores (Canossa et al. 2002; Gartner and Staiger 2002) (Fig. 2b). In most of these studies, BDNF levels in the extracellular medium was quantified by ELISA measurements. Therefore, release of BDNF from non-neuronal cells could not be excluded. Ketamine-induced BDNF release was blocked in the presence of AMPA receptor inhibitors but also in the presence of L-type VGCC inhibitors (Lepack et al. 2014). In this study, increased glutamatergic transmission induced by ketamine application was discussed as a reason for induction of BDNF release (Duman et al. 2019; Lepack et al. 2014). However, the molecular mechanisms underlying action of ketamine are not well understood (Lester et al. 2012; Wei et al. 2020). Therefore, it is possible that the ketamine-induced BDNF release acts via different mechanisms than glutamate-induced release. It is also conceivable that depolarization-induced conformational change of VGCC elicited by glutamate or ketamine-induced increase in bursting activity of the neural network is a prerequisite for BDNF release, which leads to structural reorganization of release, sites (Marom et al. 2010) irrespective of calcium influx from the extracellular medium.

Suggested mechanisms for axonal release of BDNF. BDNF release is dependent on Ca2+-influx from extracellular space via presynaptic NMDAR and intracellular Ca2+-release from internal Ca2+-stores. ER endoplasmic reticulum, IP3R inositol-3-phosphate receptor, NMDAR N-methyl d-aspartate receptor, TBS theta burst stimulation
Calcium transients and BDNF secretion in response to activation of GPCRs and RTKs
In addition to calcium influx through VGCC or NMDA receptors, calcium release from internal calcium stores is mediated by activation of G protein-coupled receptors (GPCRs) or receptor tyrosine kinases (RTKs). Different GPCRs and RTKs are known to be important for BDNF release. Neurotrophin-induced BDNF release was shown to depend on Trk receptor activation (Lopez-Benito et al. 2018; Nakajima et al. 2008; Santi et al. 2006). Furthermore, neurotrophin-induced BDNF release was solely dependent on calcium release from internal calcium stores (Canossa et al. 1997; Canossa et al. 2001; Lopez-Benito et al. 2018; Nakajima et al. 2008; Santi et al. 2006) (Fig. 2b). Similarly, GPCR-mediated BDNF release was also only dependent on calcium release from internal calcium stores (Buldyrev et al. 2006; Canossa et al. 2001; Lao-Peregrin et al. 2017; Santi et al. 2006). In addition, intracellular application of the non-hydrolysable guanosine diphosphate (GDP) analog GDPβ-S inhibits giant depolarization (GDP)-induced BDNF release (Kuczewski et al. 2008). In neuronal preparations, GCPRs activated by caffeine, glutamate via mGluR, GABA via GABAB receptors, or calcitonin-gene-related protein (CGRP) receptor via CGRP have been shown to induce release of BDNF (Balkowiec and Katz 2002; Buldyrev et al. 2006; Canossa et al. 2001; Lao-Peregrin et al. 2017; Santi et al. 2006). Nevertheless, GPCR-mediated release seems to be even more prominent in non-neuronal cells.
Intracellular calcium transients and BDNF secretion from glial cells
Ligands for GPCRs such as prostaglandine E2 or adenosine A2 (AA2R) induce BDNF release via activation of PKA or PLC in astrocytic and microglial cultures (Gomes et al. 2013; Hsieh et al. 2010; Hutchinson et al. 2009; Jean et al. 2008) (Fig. 4a). In addition to neuronal preparations, glutamate-induced release of BDNF was also observed in astrocytic (Jean et al. 2008) but not in microglial cultures (Trang et al. 2009). Glutamate-induced release of BDNF via mGluR-activation was observed within 10 min of stimulation, while kainate or NMDA application failed to induce release of BDNF in astrocytic cultures. The glutamate-induced release was dependent on activation of phospholipase C (PLC) and inositol-3-phosphate receptor (IP3-R) as well as on calcium release from internal calcium stores (Jean et al. 2008) (Fig. 4a). Besides the glutamate-induced BDNF release, release was also elicited after ATP treatment of astrocytic and microglial cultures (Long et al. 2020; Trang et al. 2009) (Fig. 4b). Trang et al. demonstrated a biphasic release of BDNF with peaks at 5 and 60 min after stimulation with ATP (Trang et al. 2009). SiRNA-mediated knockdown or pharmacological inhibition of purinergic P2X4Rs prevented the ATP-induced BDNF release (Long et al. 2020; Trang et al. 2009). Calcium influx from extracellular but not from internal calcium stores was important for BDNF release after ATP treatment (Fig. 4b). Application of TAT-NSF700, which prevents the disassembly activity of NSF, as well as inhibition of p38MAPK phosphorylation, reduced BDNF release from microglia (Long et al. 2020; Trang et al. 2009; Zhou et al. 2019). BDNF release from spinal dorsal horn microglia in response to p38-MAPK activation was associated with chronic pain hypersensitivity (Zhou et al. 2019). In addition to hydrophilic messengers, membrane permeable first messengers like progesterone or C8-ceramide are also effective stimuli to accumulate BDNF in extracellular medium after 16-h incubation time in primary glial cultures (Nakajima et al. 2002; Su et al. 2012).

Suggested mechanisms for astrocytic and microglial release of BDNF. Glial BDNF release is dependent on GPCR activation and Ca2+-release from internal Ca2+-stores (a) and on Ca2+-influx via P2X-R (b). AC adenylate cyclase, DAG diacylglycerol, ER endoplasmatic reticulum, IP3 inositol triphosphate, IP3R inositol-3-phosphate receptor, p38MAPK p38-mitogen-activated protein kinase; P2XR purinergic P2X receptor, PKA protein kinase A, PLC phospholipase C; TRPC transient receptor potential channel
Contribution of small G proteins, SNARE proteins and signaling pathways to BDNF secretion
Different proteins are involved in the organization of the release site, the transition of BDNF-containing vesicles from the reserve pool to a readily releasable pool, fusion pore opening, as well as disassembly of the SNARE complex. The exact contribution of all the proteins and direct interactions between the proteins with all their multiple binding sites are unknown also for synaptic vesicles. For BDNF-containing granules, some proteins and their binding partners have been identified. Among them are important cytosolic kinases (Gimenez-Cassina et al. 2012; Gomes et al. 2013; Hsieh et al. 2010; Hutchinson et al. 2009; Kolarow et al. 2007; Santi et al. 2006; Trang et al. 2009), small G proteins (Hong et al. 2016; Kato et al. 2018; Persoon et al. 2019), SNARE proteins (Dean et al. 2009; Shimojo et al. 2015; Wong et al. 2015) and other priming factors (Eckenstaler et al. 2016; Lee et al. 2019).
Contribution of cytosolic-signaling pathways to BDNF secretion
Important cytosolic kinases, which are also implicated in BDNF release, are the protein kinase A (PKA), calcium calmodulin kinase II (CAMKII), protein kinase C (PKC) via phospholipase C (PLC), as well as protein kinase G (PKG) via the NO/cGMP/PKG-signaling pathway. The NO/cGMP/PKG-signaling pathway was shown to negatively regulate calcium release from internal calcium stores, thereby reducing BDNF secretion (Canossa et al. 2002; Kolarow et al. 2014; Santi et al. 2006). BDNF release was also dependent on activation of the PLC/IP3/IP3R-pathway, thereby mediating calcium release from internal calcium stores (Canossa et al. 2002; Gartner and Staiger 2002; Jean et al. 2008). Activation of PKA was shown to be important for BDNF release from neurons and glial cells (Gomes et al. 2013; Hsieh et al. 2010; Hutchinson et al. 2009; Kolarow et al. 2007) although the exact downstream effectors contributing to cAMP/PKA-dependent BDNF release are unknown. Furthermore, inhibition of CAMKII prevented BDNF release from neurons (Kolarow et al. 2007) while chronic inhibition of GSK-3 reduced accumulation of BDNF in culture media of neuroblastoma cells (Gimenez-Cassina et al. 2012). These serine/threonine kinases are known to regulate open channel probability of VGCCs, IP3Rs, the activity of effector proteins important for actin dynamics, as well as the activity of different other proteins, such as small G-proteins.
Contribution of small G-proteins to BDNF secretion
The Ras superfamily of small G proteins is an important regulator of several cellular processes. Based on structural and functional similarities, the superfamily is divided into 5 main subfamilies: Ras, Ran, Rho, Arf and Rab family. While the Ras family controls cell proliferation, differentiation and survival, the Ran family contributes to nucleocytoplasmic transport of RNA and proteins. The Rho family is important for actin dynamics and Rab and Arf subfamilies have been shown to regulate vesicular transport (Wennerberg et al. 2005). Different small G proteins seem to contribute to the release of BDNF. Activation of p38 MAPK is mediated via Ras. Ras/p38-signaling is important for BDNF release in astrocytes and microglia (Long et al. 2020; Trang et al. 2009). The small G-protein rab3a, which is important for vesicular trafficking, is localized on membranes of BDNF-containing granules, in astrocytes. Interactions between huntingtin (htt) and rab3a prevent binding of rab3a GTPase-activating protein1 (rab3a-GAP1) to rab3a, which leads to an impaired attachment of the granules to the plasma membrane (Hong et al. 2016). In synaptic vesicle release, it could be shown that Rab3a interacting protein Rim1alpha directly interacts with the priming factor munc13 thereby recruiting munc13 to the active zone. Inhibition of this interaction reduces the number of readily releasable vesicles (Andrews-Zwilling et al. 2006; Betz et al. 2001; Dulubova et al. 2005). A similar finding has been reported recently for BDNF-containing vesicles. Recruitment of BDNF-containing granules to release sites and thus fusion depends on the interaction of Rab3a-Rim1 and Munc13 (Persoon et al. 2019).
Contribution of regulatory proteins and SNARE proteins to BDNF secretion
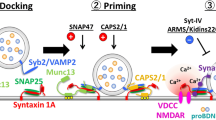
The priming protein Munc13 further catalyzes the transition of the closed inhibitory form of the Munc18/syntaxin complex to the opened active form, thus enabling the formation of the SNARE complex. Therefore, Munc-18 is an essential protein for synaptic vesicle priming and stabilizing the core complex by binding to the SNARE protein syntaxin. Accordingly, in knockout mice heterozygous for munc-18, BDNF release is negatively affected (Lee et al. 2019).
The Ca2+-dependent activator protein for secretion (CAPS1) is another priming factor during exocytosis of synaptic vesicles and granule exocytosis in neurons (Farina et al. 2015; Jockusch et al. 2007). CAPS1, which is expressed in many brain regions, is localized in axons and dendrite of hippocampal neurons (Eckenstaler et al. 2016; Farina et al. 2015; Sadakata et al. 2006; Speidel et al. 2003). For BDNF-containing granules, a function of CAPS1 in regulating intravesicular pH value could be demonstrated (Eckenstaler et al. 2016). A transient knockdown of CAPS1 significantly increased the pH value in BDNF-containing granules while the pH in the cytosolic compartment was unaffected. Furthermore, knockdown of CAPS1 reduced the number of fusion-competent BDNF-containing vesicles as well as the amount of released BDNF per single vesicle by an unknown mechanism (Eckenstaler et al. 2016).
Furthermore, the contribution of different SNARE proteins to the release of BDNF was analyzed (Shimojo et al. 2015). In cortical cultures, the SNARE proteins involved in BDNF release slightly differ for axonal and dendritic release. While Syb2 and SNAP 25 were important for dendritic and axonal release of BDNF, the SNARE protein SNAP 47, which is irrelevant for synaptic vesicle exocytosis, contributed more to axonal than to dendritic BDNF release (Shimojo et al. 2015). Another protein that interacts with SNARE proteins is synaptotagmin (Syt). Synaptotagmins are localized in the vesicular membrane. One member of this protein family, synaptotagmin-4 (Syt-4), is upregulated by activity and seizure and also localized on BDNF vesicles. Syt-4 reduces depolarization-induced release of BDNF by preventing the fusion step (Dean et al. 2009). However, TBS-induced release of previously endocytosed BDNF was shown to be mediated by syt-6 and not syt-4 at postsynaptic sites in hippocampal neurons (Wong et al. 2015). Knockdown of the SNARE protein syt-6 as well as inhibition of direct interaction of syt-6 with the SNARE complex interacting protein complexin inhibited re-release of BDNF quantum dots. The recycling of BDNF was dependent on extracellular calcium as well as AMPAR or NMDAR activation, respectively (Wong et al. 2015). Wong et al. showed that endogenous and endocytosed BDNFs are both targeted to axons as well as to dendrites in distinct non-overlapping vesicles (Wong et al. 2015). While syt-6 is an important protein for recycling of BDNF in neurons, the release of endogenous post-Golgi BDNF was regulated by the SNARE protein syt-4 (Wong et al. 2015).
Altogether, great effort has been done to uncover the cellular and molecular mechanisms of BDNF release in recent years. A meshwork of proteins such as serine/threonine kinases, small G-proteins and SNARE complex (-interacting) proteins have been demonstrated to participate in regulated release of BDNF, while constitutive release of BDNF is mainly neglected in almost all studies. Since regulated release of synaptic vesicles and BDNF-containing vesicles share multiple proteins during fusion, the regulated release of vesicles seems to be a highly conserved evolutionary process.)

Suggested mechanisms for recycling of BDNF. Endocytosed BDNF is recycled for re-release event in neurons and astrocytes. ER endoplasmatic reticulum, IP3R inositol-3-phosphate receptor, NMDAR N-methyl d-aspartate receptor, TBS theta burst stimulation
Conclusion
BDNF and the other neurotrophins were initially identified as target-derived survival factors that are secreted from non-neuronal tissue to secure survival of innervating neurons. In the early 1990s, the prominent function of BDNF in synaptic plasticity emerged, followed by first insights into activity-dependent release of BDNF from neurons. While sustained firing of action potentials or longer lasting depolarization are keys to neuronal BDNF secretion, the subsequent signaling cascades and the molecular machinery triggering and regulating BDNF vesicle exocytosis have just begun to be elucidated. Importantly, BDNF and potentially also the other neurotrophins can be released from many non-neuronal cell types using similar molecular mechanisms. Therefore, future studies will need to establish the following:
-
1.
What are the physiological and pathophysiological correlates of the diverse BDNF-releasing stimuli in neuronal and non-neuronal cells?
-
2.
How are the molecular pathways triggered in non-neuronal cells that can release BDNF (e.g.. microglia, astrocytes, lymphocytes, endothelial cells, muscle cells)?
-
3.
What are the molecular differences between the release machinery for TGN-derived new vesicles vs. recycled vesicles from neuronal and non-neuronal cells, respectively?
-
4.
What are the subcellular release sites of the distinct BDNF vesicles (new or recycled vesicles) in neuronal and non-neuronal cells?
-
5.
What are the exact components of BDNF species (e.g., mBDNF, proBDNF and propeptide) and other cargo proteins (e.g., PCs, other neuropeptides, ATP and pH) in BDNF secretory granules that might help to fine tune the effects of released BDNF in target cells?
-
6.
How do these differences in release sites, release machinery, distinct subsets of BDNF vesicles and components of BDNF species contribute to the distinct physiological and behavioral functions?
-
7.
Last but not least: what is the origin and function of BDNF in the blood stream?
References
Adachi N, Numakawa T, Kumamaru E, Itami C, Chiba S, Iijima Y, Richards M, Katoh-Semba R, Kunugi H (2013) Phencyclidine-induced decrease of synaptic connectivity via inhibition of BDNF secretion in cultured cortical neurons. Cerebral cortex (New York, N.Y. : 1991) 23(4):847–858. https://doi.org/10.1093/cercor/bhs074
Aicardi G, Argilli E, Cappello S, Santi S, Riccio M, Thoenen H, Canossa M (2004) Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 101(44):15788–15792. https://doi.org/10.1073/pnas.0406960101
Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T (2007) Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res 85(3):525–535. https://doi.org/10.1002/jnr.21139
Andrews-Zwilling YS, Kawabe H, Reim K, Varoqueaux F, Brose N (2006) Binding to Rab3A-interacting molecule RIM regulates the presynaptic recruitment of Munc13-1 and ubMunc13-2. J Biol Chem 281(28):19720–19731. https://doi.org/10.1074/jbc.M601421200
Arancibia S, Lecomte A, Silhol M, Aliaga E, Tapia-Arancibia L (2007) In vivo brain-derived neurotrophic factor release and tyrosine kinase B receptor expression in the supraoptic nucleus after osmotic stress stimulus in rats. Neuroscience 146(2):864–873. https://doi.org/10.1016/j.neuroscience.2007.01.057
Atasoy İL, Dursun E, Gezen-Ak D, Metin-Armağan D, Öztürk M, Yılmazer S (2017) Both secreted and the cellular levels of BDNF attenuated due to tau hyperphosphorylation in primary cultures of cortical neurons. J Chem Neuroanat 80:19–26. https://doi.org/10.1016/j.jchemneu.2016.11.007
Ba F, Ren J, Greer JJ (2005) Brain-derived neurotrophic factor release with neuronal activity in fetal rats. Neuroreport 16(2):141–143. https://doi.org/10.1097/00001756-200502080-00014
Babu H, Ramirez-Rodriguez G, Fabel K, Bischofberger J, Kempermann G (2009) Synaptic network activity induces neuronal differentiation of adult hippocampal precursor cells through BDNF signaling. Front Neurosci 3:49. https://doi.org/10.3389/neuro.22.001.2009
Baj G, Carlino D, Gardossi L, Tongiorgi E (2013) Toward a unified biological hypothesis for the BDNF Val66Met-associated memory deficits in humans: a model of impaired dendritic mRNA trafficking. Front Neurosci 7:188. https://doi.org/10.3389/fnins.2013.00188
Baj G, Pinhero V, Vaghi V, Tongiorgi E (2016) Signaling pathways controlling activity-dependent local translation of BDNF and their localization in dendritic arbors. J Cell Sci 129(14):2852–2864. https://doi.org/10.1242/jcs.177626
Balietti M, Giuli C, Conti F (2018) Peripheral blood brain-derived neurotrophic factor as a biomarker of Alzheimer’s disease. Are there methodological biases? Mol Neurobiol 55(8):6661–6672. https://doi.org/10.1007/s12035-017-0866-y
Balkowiec A, Katz DM (2000) Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci 20(19):7417–7423
Balkowiec A, Katz DM (2002) Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci 22(23):10399–10407
Baquet ZC, Gorski JA, Jones KR (2004) Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci 24(17):4250–4258. https://doi.org/10.1523/JNEUROSCI.3920-03.2004
Barakat-Walter I (1996) Brain-derived neurotrophic factor-like immunoreactivity is localized mainly in small sensory neurons of rat dorsal root ganglia. J Neurosci Methods 68(2):281–288. https://doi.org/10.1016/0165-0270(96)00093-3
von Bartheld CS, Johnson JE (2001) Target-derived BDNF (brain-derived neurotrophic factor) is essential for the survival of developing neurons in the isthmo-optic nucleus. J Comp Neurol 433(4):550–564. https://doi.org/10.1002/cne.1159
Baumbauer KM, Huie JR, Hughes AJ, Grau JW (2009) Timing in the absence of supraspinal input II: regularly spaced stimulation induces a lasting alteration in spinal function that depends on the NMDA receptor, BDNF release, and protein synthesis. J Neurosci 29(46):14383–14393. https://doi.org/10.1523/JNEUROSCI.3583-09.2009
Bergami M, Santi S, Formaggio E, Cagnoli C, Verderio C, Blum R, Berninger B, Matteoli M, Canossa M (2008) Uptake and recycling of pro-BDNF for transmitter-induced secretion by cortical astrocytes. J Cell Biol 183(2):213–221. https://doi.org/10.1083/jcb.200806137
Betz A, Thakur P, Junge HJ, Ashery U, Rhee JS, Scheuss V, Rosenmund C, Rettig J, Brose N (2001) Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron 30(1):183–196. https://doi.org/10.1016/s0896-6273(01)00272-0
Bi GQ, Poo MM (1998) Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci 18(24):10464–10472
Boschen KE, Klintsova AY (2017) Neurotrophins in the brain: interaction with alcohol exposure during development Vitamins and hormones 104:197–242. https://doi.org/10.1016/bs.vh.2016.10.008
Brigadski T, Hartmann M, Lessmann V (2005) Differential vesicular targeting and time course of synaptic secretion of the mammalian neurotrophins. J Neurosci 25(33):7601–7614. https://doi.org/10.1523/JNEUROSCI.1776-05.2005
Briz V, Liu Y, Zhu G, Bi X, Baudry M (2015) A novel form of synaptic plasticity in field CA3 of hippocampus requires GPER1 activation and BDNF release. J Cell Biol 210(7):1225–1237. https://doi.org/10.1083/jcb.201504092
Buldyrev I, Tanner NM, Hsieh HY, Dodd EG, Nguyen LT, Balkowiec A (2006) Calcitonin gene-related peptide enhances release of native brain-derived neurotrophic factor from trigeminal ganglion neurons. J Neurochem 99(5):1338–1350. https://doi.org/10.1111/j.1471-4159.2006.04161.x
Butowt R, von Bartheld CS (2001) Sorting of internalized neurotrophins into an endocytic transcytosis pathway via the Golgi system. Ultrastructural analysis in retinal ganglion cells. J Neurosci 21(22):8915–8930
Canossa M, Gartner A, Campana G, Inagaki N, Thoenen H (2001) Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and Trk receptor activation is mediated via phospholipase C signalling pathways. EMBO J 20(7):1640–1650. https://doi.org/10.1093/emboj/20.7.1640
Canossa M, Giordano E, Cappello S, Guarnieri C, Ferri S (2002) Nitric oxide down-regulates brain-derived neurotrophic factor secretion in cultured hippocampal neurons. Proc Natl Acad Sci U S A 99(5):3282–3287. https://doi.org/10.1073/pnas.042504299
Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H (1997) Neurotrophin release by neurotrophin: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci U S A 94(24):13279–13286. https://doi.org/10.1073/pnas.94.24.13279
Castren E, Hen R (2013) Neuronal plasticity and antidepressant actions. Trends Neurosci 36(5):259–267. https://doi.org/10.1016/j.tins.2012.12.010
Castrén E, Berninger B, Leingärtner A, Lindholm D (1998) Chapter 6 Regulation of brain-derived neurotrophic factor mRNA levels in hippocampus by neuronal activity. In: van Leeuwen FW (ed) Neuronal degeneration and regeneration. From basic mechanisms to prospects for therapy ; proceedings of the 20th International Summer School of Brain Research, held at the Royal Netherlands Academy of Sciences, Amsterdam, The Netherlands from 25 to 29 August 1997, vol 117. Elsevier, Amsterdam, New York, pp 57–64
Cefis M, Quirié A, Pernet N, Marie C, Garnier P, Prigent-Tessier A (2020) Brain-derived neurotrophic factor is a full endothelium-derived factor in rats. Vasc Pharmacol:106674. https://doi.org/10.1016/j.vph.2020.106674
Chacon-Fernandez P, Sauberli K, Colzani M, Moreau T, Ghevaert C, Barde YA (2016) Brain-derived neurotrophic factor in megakaryocytes. J Biol Chem 291(19):9872–9881. https://doi.org/10.1074/jbc.M116.720029
Chau CM, Cepeda IL, Devlin AM, Weinberg J, Grunau RE (2017) The Val66Met brain-derived neurotrophic factor gene variant interacts with early pain exposure to predict cortisol dysregulation in 7-year-old children born very preterm. Implications for cognition Neuroscience 342:188–199. https://doi.org/10.1016/j.neuroscience.2015.08.044
Chen G, Kolbeck R, Barde YA, Bonhoeffer T, Kossel A (1999) Relative contribution of endogenous neurotrophins in hippocampal long-term potentiation. J Neurosci 19(18):7983–7990
Chen M, Russo-Neustadt A (2013) Kinetics of norepinephrine- and serotonin-induced BDNF release in cultured embryonic hippocampal neurons. NM 04(04):194–207. https://doi.org/10.4236/nm.2013.44031
Chen Z-Y, Ieraci A, Teng H, Dall H, Meng C-X, Herrera DG, Nykjaer A, Hempstead BL, Lee FS (2005) Sortilin controls intracellular sorting of brain-derived neurotrophic factor to the regulated secretory pathway. J Neurosci 25(26):6156–6166. https://doi.org/10.1523/JNEUROSCI.1017-05.2005
Chopin V, Lagadec C, Toillon RA, Le Bourhis X (2016) Neurotrophin signaling in cancer stem cells. Cellular and molecular life sciences : CMLS 73(9):1859–1870. https://doi.org/10.1007/s00018-016-2156-7
Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S (1997) Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS. evidence for anterograde axonal transport. J Neurosci 17(7):2295–2313
Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, de Koninck Y (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438(7070):1017–1021. https://doi.org/10.1038/nature04223
Creemers J (2002) Proteolytic enzymes as therapeutic targets - keystone symposium. 3–8 February 2002, Keystone, CO, USA. IDrugs 5(3):216–219
Crepel V, Aronov D, Jorquera I, Represa A, Ben-Ari Y, Cossart R (2007) A parturition-associated nonsynaptic coherent activity pattern in the developing hippocampus. Neuron 54(1):105–120. https://doi.org/10.1016/j.neuron.2007.03.007
Datta I, Ganapathy K, Razdan R, Bhonde R (2018) Location and number of astrocytes determine dopaminergic neuron survival and function under 6-OHDA stress mediated through differential BDNF release. Mol Neurobiol 55(7):5505–5525. https://doi.org/10.1007/s12035-017-0767-0
Dawood T, Anderson J, Barton D, Lambert E, Esler M, Hotchkin E, Haikerwal D, Kaye D, Lambert G (2007) Reduced overflow of BDNF from the brain is linked with suicide risk in depressive illness. Mol Psychiatry 12(11):981–983. https://doi.org/10.1038/sj.mp.4002059
Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER (2009) Synaptotagmin-IV modulates synaptic function and long-term potentiation by regulating BDNF release. Nat Neurosci 12(6):767–776. https://doi.org/10.1038/nn.2315
Deitos A, Dussan-Sarria JA, Souza A, Medeiros L, Tarrago Mda G, Sehn F, Chassot M, Zanette S, Schwertner A, Fregni F, Torres IL, Caumo W (2015) Clinical value of serum neuroplasticity mediators in identifying the central sensitivity syndrome in patients with chronic pain with and without structural pathology. Clin J Pain 31(11):959–967. https://doi.org/10.1097/AJP.0000000000000194
Donovan MJ, Lin MI, Wiegn P, Ringstedt T, Kraemer R, Hahn R, Wang S, Ibanez CF, Rafii S, Hempstead BL (2000) Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development 127(21):4531–4540
Du J-l, Wei H-p, Wang Z-r, Wong ST, Poo M-m (2009) Long-range retrograde spread of LTP and LTD from optic tectum to retina. Proc Natl Acad Sci U S A 106(45):18890–18896. https://doi.org/10.1073/pnas.0910659106
Dubbelaar ML, Kracht L, Eggen BJL, Boddeke E (2018) The kaleidoscope of microglial phenotypes. Front Immunol 9:1753. https://doi.org/10.3389/fimmu.2018.01753
Dulubova I, Lou X, Lu J, Huryeva I, Alam A, Schneggenburger R, Sudhof TC, Rizo J (2005) A Munc13/RIM/Rab3 tripartite complex: from priming to plasticity? EMBO J 24(16):2839–2850. https://doi.org/10.1038/sj.emboj.7600753
Duman RS, Deyama S, Fogaça MV (2019) Role of BDNF in the pathophysiology and treatment of depression: activity-dependent effects distinguish rapid-acting antidepressants. Eur J Neurosci. https://doi.org/10.1111/ejn.14630
Eckenstaler R, Lessmann V, Brigadski T (2016) CAPS1 effects on intragranular pH and regulation of BDNF release from secretory granules in hippocampal neurons. J Cell Sci 129(7):1378–1390. https://doi.org/10.1242/jcs.178251
Edelmann E, Cepeda-Prado E, Franck M, Lichtenecker P, Brigadski T, Leßmann V (2015) Theta burst firing recruits BDNF release and signaling in postsynaptic CA1 neurons in spike-timing-dependent LTP. Neuron 86(4):1041–1054. https://doi.org/10.1016/j.neuron.2015.04.007
Edelmann E, Lessmann V, Brigadski T (2014) Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 76 Pt C:610–627. https://doi.org/10.1016/j.neuropharm.2013.05.043
Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, Zaitsev E, Gold B, Goldman D, Dean M, Lu B, Weinberger DR (2003) The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112(2):257–269. https://doi.org/10.1016/s0092-8674(03)00035-7
Egorov AV, Draguhn A (2013) Development of coherent neuronal activity patterns in mammalian cortical networks: common principles and local hetereogeneity. Mech Dev 130(6–8):412–423. https://doi.org/10.1016/j.mod.2012.09.006
Ejiri J, Inoue N, Kobayashi S, Shiraki R, Otsui K, Honjo T, Takahashi M, Ohashi Y, Ichikawa S, Terashima M, Mori T, Awano K, Shinke T, Shite J, Hirata K, Yokozaki H, Kawashima S, Yokoyama M (2005) Possible role of brain-derived neurotrophic factor in the pathogenesis of coronary artery disease. Circulation 112(14):2114–2120. https://doi.org/10.1161/CIRCULATIONAHA.104.476903
Ernfors P, Ibanez CF, Ebendal T, Olson L, Persson H (1990) Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc Natl Acad Sci U S A 87(14):5454–5458. https://doi.org/10.1073/pnas.87.14.5454
Eyileten C, Kaplon-Cieslicka A, Mirowska-Guzel D, Malek L, Postula M (2017) Antidiabetic effect of brain-derived neurotrophic factor and its association with inflammation in type 2 diabetes mellitus. J Diabetes Res 2017:2823671. https://doi.org/10.1155/2017/2823671
Farina M, van de Bospoort R, He E, Persoon CM, van Weering JR, Broeke JH, Verhage M, Toonen RF (2015) Correction. CAPS-1 promotes fusion competence of stationary dense-core vesicles in presynaptic terminals of mammalian neurons. Elife 4:e12968. https://doi.org/10.7554/eLife.12968
Ferrini F, Trang T, Mattioli TA, Laffray S, Del’Guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, de Koninck Y (2013) Morphine hyperalgesia gated through microglia-mediated disruption of neuronal cl(−) homeostasis. Nat Neurosci 16(2):183–192. https://doi.org/10.1038/nn.3295
Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B (1996) Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 381(6584):706–709. https://doi.org/10.1038/381706a0
Fiorentino H, Kuczewski N, Diabira D, Ferrand N, Pangalos MN, Porcher C, Gaiarsa J-L (2009) GABA(B) receptor activation triggers BDNF release and promotes the maturation of GABAergic synapses. J Neurosci 29(37):11650–11661. https://doi.org/10.1523/JNEUROSCI.3587-09.2009
Fiumelli H, Woodin MA (2007) Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr Opin Neurobiol 17(1):81–86. https://doi.org/10.1016/j.conb.2007.01.002
Fogaça MV, Fukumoto K, Franklin T, Liu R-J, Duman CH, Vitolo OV, Duman RS (2019) N-Methyl-D-aspartate receptor antagonist d-methadone produces rapid, mTORC1-dependent antidepressant effects. Neuropsychopharmacology 44(13):2230–2238. https://doi.org/10.1038/s41386-019-0501-x
Fritsch B, Reis J, Martinowich K, Schambra HM, Ji Y, Cohen LG, Lu B (2010) Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron 66(2):198–204. https://doi.org/10.1016/j.neuron.2010.03.035
Fujimura H, Altar CA, Chen R, Nakamura T, Nakahashi T, Kambayashi J, Sun B, Tandon NN (2002) Brain-derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost 87(4):728–734
Fukumoto K, Fogaça MV, Liu R-J, Duman C, Kato T, Li X-Y, Duman RS (2019) Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci U S A 116(1):297–302. https://doi.org/10.1073/pnas.1814709116
Gartner A, Staiger V (2002) Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci U S A 99(9):6386–6391. https://doi.org/10.1073/pnas.092129699
Giannopoulou I, Pagida MA, Briana DD, Panayotacopoulou MT (2018) Perinatal hypoxia as a risk factor for psychopathology later in life: the role of dopamine and neurotrophins. Hormones (Athens, Greece) 17(1):25–32. https://doi.org/10.1007/s42000-018-0007-7
Gidalevitz T, Stevens F, Argon Y (2013) Orchestration of secretory protein folding by ER chaperones. Biochim Biophys Acta 1833(11):2410–2424. https://doi.org/10.1016/j.bbamcr.2013.03.007
Gimenez-Cassina A, Lim F, Diaz-Nido J (2012) Chronic inhibition of glycogen synthase kinase-3 protects against rotenone-induced cell death in human neuron-like cells by increasing BDNF secretion. Neurosci Lett 531(2):182–187. https://doi.org/10.1016/j.neulet.2012.10.046
Giralt A, Friedman HC, Caneda-Ferron B, Urban N, Moreno E, Rubio N, Blanco J, Peterson A, Canals JM, Alberch J (2010) BDNF regulation under GFAP promoter provides engineered astrocytes as a new approach for long-term protection in Huntington’s disease. Gene Ther 17(10):1294–1308. https://doi.org/10.1038/gt.2010.71
Gomes C, Ferreira R, George J, Sanches R, Rodrigues DI, Goncalves N, Cunha RA (2013) Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor-dependent manner. A2A receptor blockade prevents BDNF release and proliferation of microglia. J Neuroinflammation 10:16. https://doi.org/10.1186/1742-2094-10-16
Gomez-Pinilla F, Vaynman S (2005) A “deficient environment” in prenatal life may compromise systems important for cognitive function by affecting BDNF in the hippocampus. Exp Neurol 192(2):235–243. https://doi.org/10.1016/j.expneurol.2004.12.001
Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F (1996) Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci 7(3):222–238. https://doi.org/10.1006/mcne.1996.0017
Gottmann K, Mittmann T, Lessmann V (2009) BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res 199(3–4):203–234. https://doi.org/10.1007/s00221-009-1994-z
Gray K, Ellis V (2008) Activation of pro-BDNF by the pericellular serine protease plasmin. FEBS Lett 582(6):907–910. https://doi.org/10.1016/j.febslet.2008.02.026
Griesbeck O, Canossa M, Campana G, Gartner A, Hoener MC, Nawa H, Kolbeck R, Thoenen H (1999) Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc Res Tech 45(4–5):262–275. https://doi.org/10.1002/(SICI)1097-0029(19990515/01)45:4/5<262:AID-JEMT10>3.0.CO;2-K
Gubellini P, Ben-Ari Y, Gaiarsa JL (2005) Endogenous neurotrophins are required for the induction of GABAergic long-term potentiation in the neonatal rat hippocampus. J Neurosci 25(24):5796–5802. https://doi.org/10.1523/JNEUROSCI.0824-05.2005
Guo W, Robbins MT, Wei F, Zou S, Dubner R, Ren K (2006) Supraspinal brain-derived neurotrophic factor signaling: a novel mechanism for descending pain facilitation. J Neurosci 26(1):126–137. https://doi.org/10.1523/JNEUROSCI.3686-05.2006
Haas L, Portela LV, Bohmer AE, Oses JP, Lara DR (2010) Increased plasma levels of brain derived neurotrophic factor (BDNF) in patients with fibromyalgia. Neurochem Res 35(5):830–834. https://doi.org/10.1007/s11064-010-0129-z
Hanganu-Opatz IL (2010) Between molecules and experience: role of early patterns of coordinated activity for the development of cortical maps and sensory abilities. Brain Res Rev 64(1):160–176. https://doi.org/10.1016/j.brainresrev.2010.03.005
Hartings JA, Shuttleworth CW, Kirov SA, Ayata C, Hinzman JM, Foreman B, Andrew RD, Boutelle MG, Brennan KC, Carlson AP, Dahlem MA, Drenckhahn C, Dohmen C, Fabricius M, Farkas E, Feuerstein D, Graf R, Helbok R, Lauritzen M, Major S, Oliveira-Ferreira AI, Richter F, Rosenthal ES, Sakowitz OW, Sanchez-Porras R, Santos E, Scholl M, Strong AJ, Urbach A, Westover MB, Winkler MK, Witte OW, Woitzik J, Dreier JP (2017) The continuum of spreading depolarizations in acute cortical lesion development. Examining Leao’s legacy. J Cereb Blood Flow Metab 37(5):1571–1594. https://doi.org/10.1177/0271678X16654495
Hartmann M, Heumann R, Lessmann V (2001) Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20(21):5887–5897. https://doi.org/10.1093/emboj/20.21.5887
Harward SC, Hedrick NG, Hall CE, Parra-Bueno P, Milner TA, Pan E, Laviv T, Hempstead BL, Yasuda R, McNamara JO (2016) Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 538(7623):99–103. https://doi.org/10.1038/nature19766
Hedrick NG, Harward SC, Hall CE, Murakoshi H, McNamara JO, Yasuda R (2016) Rho GTPase complementation underlies BDNF-dependent homo- and heterosynaptic plasticity. Nature 538(7623):104–108. https://doi.org/10.1038/nature19784
Henry FE, Wang X, Serrano D, Perez AS, Carruthers CJL, Stuenkel EL, Sutton MA (2018) A unique homeostatic signaling pathway links synaptic inactivity to postsynaptic mTORC1. J Neurosci 38(9):2207–2225. https://doi.org/10.1523/JNEUROSCI.1843-17.2017
Hoehner JC, Wester T, Pahlman S, Olsen L (1996) Localization of neurotrophins and their high-affinity receptors during human enteric nervous system development. Gastroenterology 110(3):756–767. https://doi.org/10.1053/gast.1996.v110.pm8608885
Holsinger RMD, Schnarr J, Henry P, Castelo VT, Fahnestock M (2000) Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer’s disease. Brain research. Mol Brain Res 76(2):347–354. https://doi.org/10.1016/s0169-328x(00)00023-1
Hong Y, Zhao T, Li XJ, Li S (2016) Mutant huntingtin impairs BDNF release from astrocytes by disrupting conversion of Rab3a-GTP into Rab3a-GDP. J Neurosci 36(34):8790–8801. https://doi.org/10.1523/JNEUROSCI.0168-16.2016
Hou L, Liu Y, Wang X, Ma H, He J, Zhang Y, Yu C, Guan W, Ma Y (2011) The effects of amyloid-beta42 oligomer on the proliferation and activation of astrocytes in vitro. In Vitro Cell Dev Biol Anim 47(8):573–580. https://doi.org/10.1007/s11626-011-9439-y
Hsieh CH, Li HY, Chen JC (2010) Nitric oxide and interleukin-1beta mediate noradrenergic induced corticotrophin-releasing hormone release in organotypic cultures of rat paraventricular nucleus. Neuroscience 165(4):1191–1202. https://doi.org/10.1016/j.neuroscience.2009.12.003
Huang EJ, Reichardt LF (2001) Neurotrophins. roles in neuronal development and function. Annu Rev Neurosci 24:677–736. https://doi.org/10.1146/annurev.neuro.24.1.677
Huang S-L, Wang J, He X-J, Li Z-F, Pu J-N, Shi W (2014) Secretion of BDNF and GDNF from free and encapsulated choroid plexus epithelial cells. Neurosci Lett 566:42–45. https://doi.org/10.1016/j.neulet.2014.02.017
Hutchinson AJ, Chou CL, Israel DD, Xu W, Regan JW (2009) Activation of EP2 prostanoid receptors in human glial cell lines stimulates the secretion of BDNF. Neurochem Int 54(7):439–446. https://doi.org/10.1016/j.neuint.2009.01.018
Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJL, Sutton MA (2010) Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 68(6):1143–1158. https://doi.org/10.1016/j.neuron.2010.11.034
Jean YY, Lercher LD, Dreyfus CF (2008) Glutamate elicits release of BDNF from basal forebrain astrocytes in a process dependent on metabotropic receptors and the PLC pathway. Neuron Glia Biol 4(1):35–42. https://doi.org/10.1017/S1740925X09000052
Jia Y, Gall CM, Lynch G (2010) Presynaptic BDNF promotes postsynaptic long-term potentiation in the dorsal striatum. J Neurosci 30(43):14440–14445. https://doi.org/10.1523/JNEUROSCI.3310-10.2010
Jiang X, Zhou J, Mash DC, Marini AM, Lipsky RH (2009) Human BDNF isoforms are differentially expressed in cocaine addicts and are sorted to the regulated secretory pathway independent of the Met66 substitution. NeuroMolecular Med 11(1):1–12. https://doi.org/10.1007/s12017-008-8051-0
Jin H, Chen Y, Wang B, Zhu Y, Chen L, Han X, Ma G, Liu N (2018) Association between brain-derived neurotrophic factor and von Willebrand factor levels in patients with stable coronary artery disease. BMC Cardiovasc Disord 18(1):23. https://doi.org/10.1186/s12872-018-0762-z
Jockusch WJ, Speidel D, Sigler A, Sørensen JB, Varoqueaux F, Rhee J-S, Brose N (2007) CAPS-1 and CAPS-2 are essential synaptic vesicle priming proteins. Cell 131(4):796–808. https://doi.org/10.1016/j.cell.2007.11.002
Johnestone and Mobley (2020). Local TrkB signaling: themes in development and neural plasticity. Cell Tissue Res.
Jornot L, Grouzmann E, Lacroix JS, Rochat T (2007) BDNF and DPP-IV in polyps and middle turbinates epithelial cells. Rhinology 45(2):129–133
Jourdi H, Hamo L, Oka T, Seegan A, Baudry M (2009) BDNF mediates the neuroprotective effects of positive AMPA receptor modulators against MPP+-induced toxicity in cultured hippocampal and mesencephalic slices. Neuropharmacology 56(5):876–885. https://doi.org/10.1016/j.neuropharm.2009.01.015
Kaess BM, Preis SR, Lieb W, Beiser AS, Yang Q, Chen TC, Hengstenberg C, Erdmann J, Schunkert H, Seshadri S, Vasan RS, CardioGram ATL, Deloukas P, Holm H, Kathiresan S, Konig IR, McPherson R, Reilly MP, Roberts R, Samani NJ, Stewart AF (2015) Circulating brain-derived neurotrophic factor concentrations and the risk of cardiovascular disease in the community. J Am Heart Assoc 4(3):e001544. https://doi.org/10.1161/JAHA.114.001544
Kang H, Welcher AA, Shelton D, Schuman EM (1997) Neurotrophins and time: different roles for TrkB signaling in hippocampal long-term potentiation. Neuron 19(3):653–664. https://doi.org/10.1016/S0896-6273(00)80378-5
Kato T, Fogaca MV, Deyama S, Li XY, Fukumoto K, Duman RS (2018) BDNF release and signaling are required for the antidepressant actions of GLYX-13. Mol Psychiatry 23(10):2007–2017. https://doi.org/10.1038/mp.2017.220
Katoh-Semba R, Takeuchi IK, Semba R, Kato K (1997) Distribution of brain-derived neurotrophic factor in rats and its changes with development in the brain. J Neurochem 69(1):34–42. https://doi.org/10.1046/j.1471-4159.1997.69010034.x
Kawahara N, Croll SD, Wiegand SJ, Klatzo I (1997) Cortical spreading depression induces long-term alterations of BDNF levels in cortex and hippocampus distinct from lesion effects: implications for ischemic tolerance. Neurosci Res 29(1):37–47. https://doi.org/10.1016/s0168-0102(97)00069-2
Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, Kolbeck R, Hoppe E, Oropeza-Wekerle RL, Bartke I, Stadelmann C, Lassmann H, Wekerle H, Hohlfeld R (1999) Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med 189(5):865–870. https://doi.org/10.1084/jem.189.5.865
Kilb W, Kirischuk S, Luhmann HJ (2011) Electrical activity patterns and the functional maturation of the neocortex. Eur J Neurosci 34(10):1677–1686. https://doi.org/10.1111/j.1460-9568.2011.07878.x
Klein AB, Williamson R, Santini MA, Clemmensen C, Ettrup A, Rios M, Knudsen GM, Aznar S (2011) Blood BDNF concentrations reflect brain-tissue BDNF levels across species. Int J Neuropsychopharmacol 14(3):347–353. https://doi.org/10.1017/S1461145710000738
Klein R (1994) Role of neurotrophins in mouse neuronal development. FASEB J 8(10):738–744. https://doi.org/10.1096/fasebj.8.10.8050673
Kohara K, Kitamura A, Morishima M, Tsumoto T (2001) Activity-dependent transfer of brain-derived neurotrophic factor to postsynaptic neurons. Science (New York, N.Y.) 291(5512):2419–2423. https://doi.org/10.1126/science.1057415
Kojima M, Ishii C, Sano Y, Mizui T, Furuichi T (2020) Journey of brain-derived neurotrophic factor: from intracellular tracfficking to secretion. Cell Tissue Res.
Kojima M, Takei N, Numakawa T, Ishikawa Y, Suzuki S, Matsumoto T, Katoh-Semba R, Nawa H, Hatanaka H (2001) Biological characterization and optical imaging of brain-derived neurotrophic factor-green fluorescent protein suggest an activity-dependent local release of brain-derived neurotrophic factor in neurites of cultured hippocampal neurons. J Neurosci Res 64(1):1–10. https://doi.org/10.1002/jnr.1080
Kokaia Z, Gidö G, Ringstedt T, Bengzon J, Kokaia M, Siesjö BK, Persson H, Lindvall O (1993) Rapid increase of BDNF mRNA levels in cortical neurons following spreading depression: regulation by glutamatergic mechanisms independent of seizure activity. Brain Res Mol Brain Res 19(4):277–286. https://doi.org/10.1016/0169-328x(93)90126-a
Kolarow R, Brigadski T, Lessmann V (2007) Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. J Neurosci 27(39):10350–10364. https://doi.org/10.1523/JNEUROSCI.0692-07.2007
Kolarow R, Kuhlmann CRW, Munsch T, Zehendner C, Brigadski T, Luhmann HJ, Lessmann V (2014) BDNF-induced nitric oxide signals in cultured rat hippocampal neurons: time course, mechanism of generation, and effect on neurotrophin secretion. Front Cell Neurosci 8:323. https://doi.org/10.3389/fncel.2014.00323
Krabbe KS, Nielsen AR, Krogh-Madsen R, Plomgaard P, Rasmussen P, Erikstrup C, Fischer CP, Lindegaard B, Petersen AMW, Taudorf S, Secher NH, Pilegaard H, Bruunsgaard H, Pedersen BK (2007) Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 50(2):431–438. https://doi.org/10.1007/s00125-006-0537-4
Krishnan V, Han M-H, Graham DL, Berton O, Renthal W, Russo SJ, Laplant Q, Graham A, Lutter M, Lagace DC, Ghose S, Reister R, Tannous P, Green TA, Neve RL, Chakravarty S, Kumar A, Eisch AJ, Self DW, Lee FS, Tamminga CA, Cooper DC, Gershenfeld HK, Nestler EJ (2007) Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell 131(2):391–404. https://doi.org/10.1016/j.cell.2007.09.018
Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R, Lessmann V, Medina I, Gaiarsa JL (2008) Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci 28(27):7013–7023. https://doi.org/10.1523/JNEUROSCI.1673-08.2008
Lao-Peregrin C, Ballesteros JJ, Fernandez M, Zamora-Moratalla A, Saavedra A, Gomez Lazaro M, Perez-Navarro E, Burks D, Martin ED (2017) Caffeine-mediated BDNF release regulates long-term synaptic plasticity through activation of IRS2 signaling. Addict Biol 22(6):1706–1718. https://doi.org/10.1111/adb.12433
Laske C, Stransky E, Eschweiler GW, Klein R, Wittorf A, Leyhe T, Richartz E, Kohler N, Bartels M, Buchkremer G, Schott K (2007) Increased BDNF serum concentration in fibromyalgia with or without depression or antidepressants. J Psychiatr Res 41(7):600–605. https://doi.org/10.1016/j.jpsychires.2006.02.007
Lee R, Kermani P, Teng KK, Hempstead BL (2001) Regulation of cell survival by secreted proneurotrophins. Science 294(5548):1945–1948. https://doi.org/10.1126/science.1065057
Lee YI, Kim YG, Pyeon HJ, Ahn JC, Logan S, Orock A, Joo KM, Lőrincz A, Deák F (2019) Dysregulation of the SNARE-binding protein Munc18-1 impairs BDNF secretion and synaptic neurotransmission: a novel interventional target to protect the aging brain. GeroScience 41(2):109–123. https://doi.org/10.1007/s11357-019-00067-1
Lepack AE, Fuchikami M, Dwyer JM, Banasr M, Duman RS (2014) BDNF release is required for the behavioral actions of ketamine. Int J Neuropsychopharmacol 18(1). https://doi.org/10.1093/ijnp/pyu033
Leschik J, Eckenstaler R, Endres T, Munsch T, Edelmann E, Richter K, Kobler O, Fischer K-D, Zuschratter W, Brigadski T, Lutz B, Lessmann V (2019) Prominent postsynaptic and dendritic exocytosis of endogenous BDNF vesicles in BDNF-GFP Knock-in mice. Mol Neurobiol 56(10):6833–6855. https://doi.org/10.1007/s12035-019-1551-0
Leschik J, Eckenstaler R, Nieweg K, Lichtenecker P, Brigadski T, Gottmann K, Lessmann V, Lutz B (2013) Embryonic stem cells stably expressing BDNF-GFP exhibit a BDNF-release-dependent enhancement of neuronal differentiation. J Cell Sci 126(Pt 21):5062–5073. https://doi.org/10.1242/jcs.135384
Lessmann V, Brigadski T (2009) Mechanisms, locations, and kinetics of synaptic BDNF secretion: an update. Neurosci Res 65(1):11–22. https://doi.org/10.1016/j.neures.2009.06.004
Leßmann V, Brigadski T (2009) Mechanisms, locations, and kinetics of synaptic BDNF secretion: an update. Neurosci Res 65(1):11–22
Lessmann V, Gottmann K, Malcangio M (2003) Neurotrophin secretion: current facts and future prospects. Prog Neurobiol 69(5):341–374. https://doi.org/10.1016/s0301-0082(03)00019-4
Lester HA, Miwa JM, Srinivasan R (2012) Psychiatric drugs bind to classical targets within early exocytotic pathways: therapeutic effects. Biol Psychiatry 72(11):907–915. https://doi.org/10.1016/j.biopsych.2012.05.020
Lever IJ, Bradbury EJ, Cunningham JR, Adelson DW, Jones MG, McMahon SB, Marvizon JC, Malcangio M (2001) Brain-derived neurotrophic factor is released in the dorsal horn by distinctive patterns of afferent fiber stimulation. J Neurosci 21(12):4469–4477
Lewin GR, Barde YA (1996) Physiology of the neurotrophins. Annu Rev Neurosci 19:289–317. https://doi.org/10.1146/annurev.ne.19.030196.001445
Li C, Dabrowska J, Hazra R, Rainnie DG (2011) Synergistic activation of dopamine D1 and TrkB receptors mediate gain control of synaptic plasticity in the basolateral amygdala. PLoS One 6(10):e26065. https://doi.org/10.1371/journal.pone.0026065
Li Y, Calfa G, Inoue T, Amaral MD, Pozzo-Miller L (2010) Activity-dependent release of endogenous BDNF from mossy fibers evokes a TRPC3 current and Ca2+ elevations in CA3 pyramidal neurons. J Neurophysiol 103(5):2846–2856. https://doi.org/10.1152/jn.01140.2009
Lin PY, Kavalali ET, Monteggia LM (2018) Genetic dissection of presynaptic and postsynaptic BDNF-TrkB signaling in synaptic efficacy of CA3-CA1 synapses. Cell Rep 24(6):1550–1561. https://doi.org/10.1016/j.celrep.2018.07.020
Lippi G, Mattiuzzi C, Sanchis-Gomar F (2020) Updated overview on interplay between physical exercise, neurotrophins, and cognitive function in humans. J Sport Health Sci 9(1):74–81. https://doi.org/10.1016/j.jshs.2019.07.012
Liu D-W, Ma L, Zhang X-H, Wang Y-Y (2019) Conditioned taste aversion memory extinction temporally induces insular cortical BDNF release and inhibits neuronal apoptosis. Neuropsychiatr Dis Treat 15:2403–2414. https://doi.org/10.2147/NDT.S215289
Long T, He W, Pan Q, Zhang S, Zhang D, Qin G, Chen L, Zhou J (2020) Microglia P2X4R-BDNF signalling contributes to central sensitization in a recurrent nitroglycerin-induced chronic migraine model. J Headache Pain 21(1):4. https://doi.org/10.1186/s10194-019-1070-4
Lopez-Benito S, Sanchez-Sanchez J, Brito V, Calvo L, Lisa S, Torres-Valle M, Palko ME, Vicente-Garcia C, Fernandez-Fernandez S, Bolanos JP, Gines S, Tessarollo L, Arevalo JC (2018) Regulation of BDNF release by ARMS/Kidins220 through modulation of synaptotagmin-IV levels. J Neurosci 38(23):5415–5428. https://doi.org/10.1523/JNEUROSCI.1653-17.2018
Lou H, Kim S-K, Zaitsev E, Snell CR, Lu B, Loh YP (2005) Sorting and activity-dependent secretion of BDNF require interaction of a specific motif with the sorting receptor carboxypeptidase e. Neuron 45(2):245–255. https://doi.org/10.1016/j.neuron.2004.12.037
Lu H, Park H, Poo MM (2014) spike-timing-dependent BDNF secretion and synaptic plasticity. Philos Trans R Soc Lond Ser B Biol Sci 369(1633):20130132. https://doi.org/10.1098/rstb.2013.0132
Luhmann HJ, Khazipov R (2018) Neuronal activity patterns in the developing barrel cortex. Neuroscience 368:256–267. https://doi.org/10.1016/j.neuroscience.2017.05.025
Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD (1990) NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron 5(4):501–509. https://doi.org/10.1016/0896-6273(90)90089-x
Malamitsi-Puchner A, Economou E, Rigopoulou O, Boutsikou T (2004) Perinatal changes of brain-derived neurotrophic factor in pre- and fullterm neonates. Early Hum Dev 76(1):17–22. https://doi.org/10.1016/j.earlhumdev.2003.10.002
Marie C, Pedard M, Quirie A, Tessier A, Garnier P, Totoson P, Demougeot C (2018) Brain-derived neurotrophic factor secreted by the cerebral endothelium. A new actor of brain function? J Cereb Blood Flow Metab 38(6):935–949. https://doi.org/10.1177/0271678X18766772
Marom M, Hagalili Y, Sebag A, Tzvier L, Atlas D (2010) Conformational changes induced in voltage-gated calcium channel Cav1.2 by BayK 8644 or FPL64176 modify the kinetics of secretion independently of Ca2+ influx. J Biol Chem 285(10):6996–7005. https://doi.org/10.1074/jbc.M109.059865
Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, Poo MM (2009) Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci 29(45):14185–14198. https://doi.org/10.1523/JNEUROSCI.1863-09.2009
Matthews VB, Astrom MB, Chan MH, Bruce CR, Krabbe KS, Prelovsek O, Akerstrom T, Yfanti C, Broholm C, Mortensen OH, Penkowa M, Hojman P, Zankari A, Watt MJ, Bruunsgaard H, Pedersen BK, Febbraio MA (2009) Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52(7):1409–1418. https://doi.org/10.1007/s00125-009-1364-1
Meis S, Endres T, Lessmann V (2012) Postsynaptic BDNF signalling regulates long-term potentiation at thalamo-amygdala afferents. J Physiol 590(1):193–208. https://doi.org/10.1113/jphysiol.2011.220434
Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, Bardoni R (2008) BDNF as a pain modulator. Prog Neurobiol 85(3):297–317. https://doi.org/10.1016/j.pneurobio.2008.04.004
Merlo S, Spampinato SF, Beneventano M, Sortino MA (2018) The contribution of microglia to early synaptic compensatory responses that precede beta-amyloid-induced neuronal death. Sci Rep 8(1):7297. https://doi.org/10.1038/s41598-018-25453-1
Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR (2002) Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci 22(17):7453–7461
Mohajerani MH, Sivakumaran S, Zacchi P, Aguilera P, Cherubini E (2007) Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3 CA1 connections in the hippocampus. Proc Natl Acad Sci U S A 104(32):13176–13181. https://doi.org/10.1073/pnas.0704533104
Mohammadi A, Amooeian VG, Rashidi E (2018) Dysfunction in brain-derived neurotrophic factor signaling pathway and susceptibility to schizophrenia, Parkinson’s and Alzheimer’s diseases. Curr Gene Ther 18(1):45–63. https://doi.org/10.2174/1566523218666180302163029
Mousavi K, Jasmin BJ (2006) BDNF is expressed in skeletal muscle satellite cells and inhibits myogenic differentiation. J Neurosci 26(21):5739–5749. https://doi.org/10.1523/JNEUROSCI.5398-05.2006
Mowla SJ, Farhadi HF, Pareek S, Atwal JK, Morris SJ, Seidah NG, Murphy RA (2001) Biosynthesis and post-translational processing of the precursor to brain-derived neurotrophic factor. J Biol Chem 276(16):12660–12666. https://doi.org/10.1074/jbc.M008104200
Mu Y, Poo M-m (2006) Spike timing-dependent LTP/LTD mediates visual experience-dependent plasticity in a developing retinotectal system. Neuron 50(1):115–125. https://doi.org/10.1016/j.neuron.2006.03.009
Nakahashi T, Fujimura H, Altar CA, Li J, Kambayashi J, Tandon NN, Sun B (2000) Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett 470(2):113–117. https://doi.org/10.1016/s0014-5793(00)01302-8
Nakajima K, Honda S, Tohyama Y, Imai Y, Kohsaka S, Kurihara T (2001) Neurotrophin secretion from cultured microglia. J Neurosci Res 65(4):322–331. https://doi.org/10.1002/jnr.1157
Nakajima K, Tohyama Y, Kohsaka S, Kurihara T (2002) Ceramide activates microglia to enhance the production/secretion of brain-derived neurotrophic factor (BDNF) without induction of deleterious factors in vitro. J Neurochem 80(4):697–705. https://doi.org/10.1046/j.0022-3042.2001.00752.x
Nakajima T, Sato M, Akaza N, Umezawa Y (2008) Cell-based fluorescent indicator to visualize brain-derived neurotrophic factor secreted from living neurons. ACS Chem Biol 3(6):352–358. https://doi.org/10.1021/cb800052v
Orecchioni M, Ghosheh Y, Pramod AB, Ley K (2019) Macrophage polarization. Different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front Immunol 10:1084. https://doi.org/10.3389/fimmu.2019.01084
Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ (1998) Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology 37(12):1553–1561. https://doi.org/10.1016/s0028-3908(98)00141-5
Pandey GN, Ren X, Rizavi HS, Conley RR, Roberts RC, Dwivedi Y (2008) Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int J Neuropsychopharmacol 11(8):1047–1061. https://doi.org/10.1017/S1461145708009000
Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B (2004) Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science 306(5695):487–491. https://doi.org/10.1126/science.1100135
Park H (2018) Cortical axonal secretion of BDNF in the striatum is disrupted in the mutant-huntingtin Knock-in mouse model of Huntington’s disease. Exp Neurobiol 27(3):217–225. https://doi.org/10.5607/en.2018.27.3.217
Park H, Poo MM (2013) Neurotrophin regulation of neural circuit development and function. Nature reviews. Neuroscience 14(1):7–23. https://doi.org/10.1038/nrn3379
Patterson SL, Pittenger C, Morozov A, Martin KC, Scanlin H, Drake C, Kandel ER (2001) Some forms of cAMP-mediated long-lasting potentiation are associated with release of BDNF and nuclear translocation of phospho-MAP kinase. Neuron 32(1):123–140. https://doi.org/10.1016/s0896-6273(01)00443-3
Persoon CM, Hoogstraaten RI, Nassal JP, van Weering JRT, Kaeser PS, Toonen RF, Verhage M (2019) The RAB3-RIM pathway is essential for the release of neuromodulators. Neuron 104(6):1065–1080.e12. https://doi.org/10.1016/j.neuron.2019.09.015
Petoukhov E, Fernando S, Mills F, Shivji F, Hunter D, Krieger C, Silverman MA, Bamji SX (2013) Activity-dependent secretion of progranulin from synapses. J Cell Sci 126(Pt 23):5412–5421. https://doi.org/10.1242/jcs.132076
Phillips HS, Hains JM, Laramee GR, Rosenthal A, Winslow JW (1990) Widespread expression of BDNF but not NT3 by target areas of basal forebrain cholinergic neurons. Science 250(4978):290–294. https://doi.org/10.1126/science.1688328
Porcher C, Hatchett C, Longbottom RE, McAinch K, Sihra TS, Moss SJ, Thomson AM, Jovanovic JN (2011) Positive feedback regulation between gamma-aminobutyric acid type a (GABA(a)) receptor signaling and brain-derived neurotrophic factor (BDNF) release in developing neurons. J Biol Chem 286(24):21667–21677. https://doi.org/10.1074/jbc.M110.201582
Prakash YS, Martin RJ (2014) Brain-derived neurotrophic factor in the airways. Pharmacol Ther 143(1):74–86. https://doi.org/10.1016/j.pharmthera.2014.02.006
Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T (2007) Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics 90(3):397–406. https://doi.org/10.1016/j.ygeno.2007.05.004
Qin XY, Feng JC, Cao C, Wu HT, Loh YP, Cheng Y (2016) Association of peripheral blood levels of brain-derived neurotrophic factor with autism spectrum disorder in children. A systematic review and meta-analysis. JAMA Pediatr 170(11):1079–1086. https://doi.org/10.1001/jamapediatrics.2016.1626
Radin DP, Patel P (2017) BDNF. An oncogene or tumor suppressor? Anticancer Res 37(8):3983–3990. https://doi.org/10.21873/anticanres.11783
Rasmussen P, Brassard P, Adser H, Pedersen MV, Leick L, Hart E, Secher NH, Pedersen BK, Pilegaard H (2009) Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol 94(10):1062–1069. https://doi.org/10.1113/expphysiol.2009.048512
Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M (2002) BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol 159(5):747–752. https://doi.org/10.1083/jcb.200209011
Rivera C, Voipio J, Kaila K (2005) Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. J Physiol 562(Pt 1):27–36. https://doi.org/10.1113/jphysiol.2004.077495
Rubio N (1997) Mouse astrocytes store and deliver brain-derived neurotrophic factor using the non-catalytic gp95trkB receptor. Eur J Neurosci 9(9):1847–1853. https://doi.org/10.1111/j.1460-9568.1997.tb00751.x
Sadakata T, Itakura M, Kozaki S, Sekine Y, Takahashi M, Furuichi T (2006) Differential distributions of the Ca2+ -dependent activator protein for secretion family proteins (CAPS2 and CAPS1) in the mouse brain. J Comp Neurol 495(6):735–753. https://doi.org/10.1002/cne.20947
Sadakata T, Shinoda Y, Oka M, Sekine Y, Sato Y, Saruta C, Miwa H, Tanaka M, Itohara S, Furuichi T (2012) Reduced axonal localization of a Caps2 splice variant impairs axonal release of BDNF and causes autistic-like behavior in mice. Proc Natl Acad Sci U S A 109(51):21104–21109. https://doi.org/10.1073/pnas.1210055109
Saha RN, Liu X, Pahan K (2006) Up-regulation of BDNF in astrocytes by TNF-alpha: a case for the neuroprotective role of cytokine. J NeuroImmune Pharmacol 1(3):212–222. https://doi.org/10.1007/s11481-006-9020-8
Santi S, Cappello S, Riccio M, Bergami M, Aicardi G, Schenk U, Matteoli M, Canossa M (2006) Hippocampal neurons recycle BDNF for activity-dependent secretion and LTP maintenance. EMBO J 25(18):4372–4380. https://doi.org/10.1038/sj.emboj.7601303
Sapio MR, Iadarola MJ, LaPaglia DM, Lehky T, Thurm AE, Danley KM, Fuhr SR, Lee MD, Huey AE, Sharp SJ, Tsao JW, Yanovski JA, Mannes AJ, Han JC (2019) Haploinsufficiency of the brain-derived neurotrophic factor gene is associated with reduced pain sensitivity. Pain 160(5):1070–1081. https://doi.org/10.1097/j.pain.0000000000001485
Schildt S, Endres T, Lessmann V, Edelmann E (2013) Acute and chronic interference with BDNF/TrkB-signaling impair LTP selectively at mossy fiber synapses in the CA3 region of mouse hippocampus. Neuropharmacology 71:247–254. https://doi.org/10.1016/j.neuropharm.2013.03.041
Seidah NG, Benjannet S, Pareek S, Chrétien M, Murphy RA (1996) Cellular processing of the neurotrophin precursors of NT3 and BDNF by the mammalian proprotein convertases. FEBS Lett 379(3):247–250. https://doi.org/10.1016/0014-5793(95)01520-5
Seidah NG, Mowla SJ, Hamelin J, Mamarbachi AM, Benjannet S, Toure BB, Basak A, Munzer JS, Marcinkiewicz J, Zhong M, Barale JC, Lazure C, Murphy RA, Chretien M, Marcinkiewicz M (1999) Mammalian subtilisin/kexin isozyme SKI-1. A widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proc Natl Acad Sci U S A 96(4):1321–1326. https://doi.org/10.1073/pnas.96.4.1321
Seifert B, Eckenstaler R, Ronicke R, Leschik J, Lutz B, Reymann K, Lessmann V, Brigadski T (2016) Amyloid-beta induced changes in vesicular transport of BDNF in hippocampal neurons. Neural Plast 2016:4145708. https://doi.org/10.1155/2016/4145708
Seifert T, Brassard P, Wissenberg M, Rasmussen P, Nordby P, Stallknecht B, Adser H, Jakobsen AH, Pilegaard H, Nielsen HB, Secher NH (2010) Endurance training enhances BDNF release from the human brain. Am J Phys Regul Integr Comp Phys 298(2):R372–R377. https://doi.org/10.1152/ajpregu.00525.2009
Sen A, Nelson TJ, Alkon DL (2017) ApoE isoforms differentially regulates cleavage and secretion of BDNF. Molecular brain 10(1):19. https://doi.org/10.1186/s13041-017-0301-3
Serra-Millas M (2016) Are the changes in the peripheral brain-derived neurotrophic factor levels due to platelet activation? World J Psychiatry 6(1):84–101. https://doi.org/10.5498/wjp.v6.i1.84
Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD (1995) Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci 15(1):477–491. https://doi.org/10.1523/JNEUROSCI.15-01-00477.1995
Shen P-P, Hou S, Di M, Zhao M-M, Zhu M-Q, Zhang J-D, Feng L-S, Cui L, Feng J-C (2016) Cortical spreading depression-induced preconditioning in the brain. Neural Regen Res 11(11):1857–1864. https://doi.org/10.4103/1673-5374.194759
Shen T, You Y, Joseph C, Mirzaei M, Klistorner A, Graham SL, Gupta V (2018) BDNF polymorphism. A review of its diagnostic and clinical relevance in neurodegenerative disorders. Aging Dis 9(3):523–536. https://doi.org/10.14336/AD.2017.0717
Shimojo M, Courchet J, Pieraut S, Torabi-Rander N, Sando, R., 3rd, Polleux F, Maximov A (2015) SNAREs controlling vesicular release of BDNF and development of callosal axons. Cell Rep 11(7):1054–1066. https://doi.org/10.1016/j.celrep.2015.04.032
Shinoda Y, Sadakata T, Nakao K, Katoh-Semba R, Kinameri E, Furuya A, Yanagawa Y, Hirase H, Furuichi T (2011) Calcium-dependent activator protein for secretion 2 (CAPS2) promotes BDNF secretion and is critical for the development of GABAergic interneuron network. Proc Natl Acad Sci U S A 108(1):373–378. https://doi.org/10.1073/pnas.1012220108
Speidel D, Varoqueaux F, Enk C, Nojiri M, Grishanin RN, Martin TF, Hofmann K, Brose N, Reim K (2003) A family of Ca2+-dependent activator proteins for secretion: comparative analysis of structure, expression, localization, and function. J Biol Chem 278(52):52802–52809. https://doi.org/10.1074/jbc.M304727200
Stahlberg MA, Kügler S, Dean C (2018) Visualizing BDNF cell-to-cell transfer reveals astrocytes are the primary recipient of neuronal BDNF bioRxiv 255935; https://doi.org/10.1101/255935
Stratoulias V, Venero JL, Tremblay ME, Joseph B (2019) Microglial subtypes: diversity within the microglial community. EMBO J 38(17):e101997. https://doi.org/10.15252/embj.2019101997
Su C, Cunningham RL, Rybalchenko N, Singh M (2012) Progesterone increases the release of brain-derived neurotrophic factor from glia via progesterone receptor membrane component 1 (Pgrmc1)-dependent ERK5 signaling. Endocrinology 153(9):4389–4400. https://doi.org/10.1210/en.2011-2177
Su W-F, Wu F, Jin Z-H, Gu Y, Chen Y-T, Fei Y, Chen H, Wang Y-X, Xing L-Y, Zhao Y-Y, Yuan Y, Tang X, Chen G (2019) Overexpression of P2X4 receptor in Schwann cells promotes motor and sensory functional recovery and remyelination via BDNF secretion after nerve injury. Glia 67(1):78–90. https://doi.org/10.1002/glia.23527
Sun F, Nguyen T, Jin X, Huang R, Chen Z, Cunningham RL, Singh M, Su C (2016) Pgrmc1/BDNF signaling plays a critical role in mediating glia-neuron cross talk. Endocrinology 157(5):2067–2079. https://doi.org/10.1210/en.2015-1610
Suwa M, Kishimoto H, Nofuji Y, Nakano H, Sasaki H, Radak Z, Kumagai S (2006) Serum brain-derived neurotrophic factor level is increased and associated with obesity in newly diagnosed female patients with type 2 diabetes mellitus. Metab Clin Exp 55(7):852–857. https://doi.org/10.1016/j.metabol.2006.02.012
Tamura S, Nagasawa A, Masuda Y, Tsunematsu T, Hayasaka K, Matsuno K, Shimizu C, Ozaki Y, Moriyama T (2012) BDNF, produced by a TPO-stimulated megakaryocytic cell line, regulates autocrine proliferation. Biochem Biophys Res Commun 427(3):542–546. https://doi.org/10.1016/j.bbrc.2012.09.093
Tanaka J-I, Horiike Y, Matsuzaki M, Miyazaki T, Ellis-Davies GCR, Kasai H (2008) Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science (New York, N.Y.) 319(5870):1683–1687. https://doi.org/10.1126/science.1152864
Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S (2008) New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Res Rev 59(1):201–220. https://doi.org/10.1016/j.brainresrev.2008.07.007
Thomas AX, Cruz Del Angel Y, Gonzalez MI, Carrel AJ, Carlsen J, Lam PM, Hempstead BL, Russek SJ, Brooks-Kayal AR (2016) Rapid increases in proBDNF after pilocarpine-induced status epilepticus in mice are associated with reduced proBDNF cleavage machinery. eNeuro 3(1). https://doi.org/10.1523/ENEURO.0020-15.2016
Thomas G (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nature reviews. Mol Cell Biol 3(10):753–766. https://doi.org/10.1038/nrm934
Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H (1993) Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 10(3):475–489. https://doi.org/10.1016/0896-6273(93)90335-o
Trang T, Koblic P, Kawaja M, Jhamandas K (2009) Attenuation of opioid analgesic tolerance in p75 neurotrophin receptor null mutant mice. Neurosci Lett 451(1):69–73. https://doi.org/10.1016/j.neulet.2008.12.032
Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K (2013) P2X4 receptors and neuropathic pain. Front Cell Neurosci 7:191. https://doi.org/10.3389/fncel.2013.00191
Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F (2008) Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci 28(44):11263–11268. https://doi.org/10.1523/JNEUROSCI.2308-08.2008
Vignoli B, Battistini G, Melani R, Blum R, Santi S, Berardi N, Canossa M (2016) Peri-synaptic glia recycles brain-derived neurotrophic factor for LTP stabilization and memory retention. Neuron 92(4):873–887. https://doi.org/10.1016/j.neuron.2016.09.031
Vignoli B, Canossa M (2017) Glioactive ATP controls BDNF recycling in cortical astrocytes. Commun Integr Biol 10(1):e1277296. https://doi.org/10.1080/19420889.2016.1277296
Vohra PK, Thompson MA, Sathish V, Kiel A, Jerde C, Pabelick CM, Singh BB, Prakash YS (2013) TRPC3 regulates release of brain-derived neurotrophic factor from human airway smooth muscle. Biochim Biophys Acta 1833(12):2953–2960. https://doi.org/10.1016/j.bbamcr.2013.07.019
Waterhouse EG, An JJ, Orefice LL, Baydyuk M, Liao G-Y, Zheng K, Lu B, Xu B (2012) BDNF promotes differentiation and maturation of adult-born neurons through GABAergic transmission. J Neurosci 32(41):14318–14330. https://doi.org/10.1523/JNEUROSCI.0709-12.2012
Wei Y, Chang L, Hashimoto K (2020) A historical review of antidepressant effects of ketamine and its enantiomers. Pharmacol Biochem Behav 190:172870. https://doi.org/10.1016/j.pbb.2020.172870
Wennerberg K, Rossman KL, Der CJ (2005) The Ras superfamily at a glance. J Cell Sci 118(Pt 5):843–846. https://doi.org/10.1242/jcs.01660
Werry EL, Bright FM, Piguet O, Ittner LM, Halliday GM, Hodges JR, Kiernan MC, Loy CT, Kril JJ, Kassiou M (2019) Recent developments in TSPO PET imaging as a biomarker of neuroinflammation in neurodegenerative disorders. Int J Mol Sci 20(13). https://doi.org/10.3390/ijms20133161
Wetmore C, Olson L (1995) Neuronal and nonneuronal expression of neurotrophins and their receptors in sensory and sympathetic ganglia suggest new intercellular trophic interactions. J Comp Neurol 353(1):143–159. https://doi.org/10.1002/cne.903530113
Wetmore C, Olson L, Bean AJ (1994) Regulation of brain-derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non-NMDA type glutamate receptors. J Neurosci 14(3 Pt 2):1688–1700
Wilson Horch H, Krüttgen A, Portbury SD, Katz LC (1999) Destabilization of cortical dendrites and spines by BDNF. Neuron 23(2):353–364. https://doi.org/10.1016/S0896-6273(00)80785-0
Wirth MJ, Patz S, Wahle P (2005) Transcellular induction of neuropeptide Y expression by NT4 and BDNF. Proc Natl Acad Sci U S A 102(8):3064–3069. https://doi.org/10.1073/pnas.0404712102
de Wit J, Toonen RF, Verhage M (2009) Matrix-dependent local retention of secretory vesicle cargo in cortical neurons. J Neurosci 29(1):23–37. https://doi.org/10.1523/JNEUROSCI.3931-08.2009
Wong YH, Lee CM, Xie W, Cui B, Poo MM (2015) Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc Natl Acad Sci U S A 112(32):E4475–E4484. https://doi.org/10.1073/pnas.1511830112
Xia X, Lessmann V, Martin TFJ (2009) Imaging of evoked dense-core-vesicle exocytosis in hippocampal neurons reveals long latencies and kiss-and-run fusion events. J Cell Sci 122(Pt 1):75–82. https://doi.org/10.1242/jcs.034603
Xue J, Dong JH, Huang GD, Qu XF, Wu G, Dong XR (2014) NF-kappaB signaling modulates radiationinduced microglial activation. Oncol Rep 31(6):2555–2560. https://doi.org/10.3892/or.2014.3144
Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA (1997) Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience 78(2):431–448. https://doi.org/10.1016/s0306-4522(96)00613-6
Yang J, Siao C-J, Nagappan G, Marinic T, Jing D, McGrath K, Chen Z-Y, Mark W, Tessarollo L, Lee FS, Lu B, Hempstead BL (2009) Neuronal release of proBDNF. Nat Neurosci 12(2):113–115. https://doi.org/10.1038/nn.2244
Yu C, Li CH, Chen S, Yoo H, Qin X, Park H (2018) Decreased BDNF release in cortical neurons of a Knock-in mouse model of Huntington’s disease. Sci Rep 8(1):16976. https://doi.org/10.1038/s41598-018-34883-w
Zagrebelsky M, Tacke C, Korte M (2020). BDNF signaling during the lifetime of dendritic spines. Cell Tissue Res.
Zakharenko SS, Patterson SL, Dragatsis I, Zeitlin SO, Siegelbaum SA, Kandel ER, Morozov A (2003) Presynaptic BDNF required for a presynaptic but not postsynaptic component of LTP at hippocampal CA1-CA3 synapses. Neuron 39(6):975–990. https://doi.org/10.1016/S0896-6273(03)00543-9
Zhang F, Lu Y-F, Wu Q, Liu J, Shi J-S (2012) Resveratrol promotes neurotrophic factor release from astroglia. Exp Biol Med (Maywood, NJ) 237(8):943–948. https://doi.org/10.1258/ebm.2012.012044
Zhang HT, Li LY, Zou XL, Song XB, Hu YL, Feng ZT, Wang TT (2007) Immunohistochemical distribution of NGF, BDNF, NT-3, and NT-4 in adult rhesus monkey brains. J Histochem Cytochem 55(1):1–19. https://doi.org/10.1369/jhc.6A6952.2006
Zhao L, Yeh ML-W, Levine ES (2015) Role for endogenous BDNF in endocannabinoid-mediated long-term depression at neocortical inhibitory synapses. eNeuro 2(2). https://doi.org/10.1523/ENEURO.0029-14.2015
Zheng Z, Zhang L, Zhu T, Huang J, Qu Y, Mu D (2016) Peripheral brain-derived neurotrophic factor in autism spectrum disorder: a systematic review and meta-analysis. Sci Rep 6:31241. https://doi.org/10.1038/srep31241
Zhou D, Ji L, Chen Y (2020) TSPO modulates IL-4-induced microglia/macrophage M2 polarization via PPAR-γ pathway. J Mol Neurosci 70(4):542–549. https://doi.org/10.1007/s12031-019-01454-1
Zhou LJ, Peng J, Xu YN, Zeng WJ, Zhang J, Wei X, Mai CL, Lin ZJ, Liu Y, Murugan M, Eyo UB, Umpierre AD, Xin WJ, Chen T, Li M, Wang H, Richardson JR, Tan Z, Liu XG, Wu LJ (2019) Microglia are indispensable for synaptic plasticity in the spinal dorsal horn and chronic pain. Cell Rep 27(13):3844–3859 e6. https://doi.org/10.1016/j.celrep.2019.05.087
Zimbone S, Monaco I, Gianì F, Pandini G, Copani AG, Giuffrida ML, Rizzarelli E (2018) Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 17(1). https://doi.org/10.1111/acel.12684
Funding
Open Access funding provided by Projekt DEAL. This work was funded by the Deutsche Forschungsgemeinschaft (DFG) project SFB 779, TP B6, the DFG grant LE 1020/2-1, project SFB 779, TP B6, the DFG grant LE 1020/2-1, SFB 779, TP B6, the EU Joint Programme—Neurodegenerative Disease Research (JPND) project CIRCPROT (jointly funded by BMBF and European Union’s Horizon 2020 research and innovation program (No 643417)), the Carl-Zeiss Stiftung (0563-2.8/515/2 and TelMa) and by the Ministerium für Wissenschaft, Weiterbildung und Kultur (52 171/40).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Brigadski, T., Leßmann, V. The physiology of regulated BDNF release. Cell Tissue Res 382, 15–45 (2020). https://doi.org/10.1007/s00441-020-03253-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-020-03253-2