
Abstract
Population studies suggest that atopic dermatitis (AD) is associated with an increased risk of obesity, however a causal relationship between these two conditions remains to be established. We therefore use Mendelian randomization (MR) to evaluate whether obesity and AD are causally interlinked. We used summary statistics extracted from genome wide association studies of Body Mass Index (BMI) and AD. MR analysis was performed in both directions to establish the direction of causality between BMI and AD. We find that genetically determined increase in adiposity is associated with increased risk of AD (odds ratio of AD 1.08 [95% CI 1.01 to 1.14; p = 0.015] per unit increase in BMI). Conversely, genetically determined increased risk of AD is not associated with a higher BMI (change in BMI attributable to AD based on genetic information: 0.00; 95% CI − 0.02 to 0.02; p = 0.862). There was no evidence for confounding of these genetic analyses by horizontal pleiotropy. Our results indicate that the association of AD with obesity is likely to reflect a causal role for adiposity in the development of AD. Our findings enhance understanding of the etiology of AD, and the basis for experimental studies to evaluate the mechanistic pathways by which adiposity promotes AD.
Similar content being viewed by others
Introduction
Atopic dermatitis (AD) is a common chronic inflammatory skin disease with significant patient and population burden. It currently affects 20% of children and 10% of adults in the developed world1. It is characterised by itch and skin inflammation2 reflecting the underlying epidermal barrier dysfunction3,4 and immune dysregulation of the skin5. Increasingly, it has been reported to be a systemic disease and observed to be associated with other chronic co-morbidities that have immunological basis, such as asthma and allergic rhinitis, as well as metabolic and psychological disturbances6.
AD has been reported to be associated with the presence of obesity in many observational epidemiological cohort studies. In a recent meta-analysis, it has been reported that patients who were obese had close to 1.5 times higher odds of having AD7. The rising global prevalence of AD, also closely parallels increasing global burden of obesity8,9. However, these predominantly cross sectional and observational studies may be limited by confounding factors such as adverse demographic or environmental exposures, and also cannot exclude reverse causation10,11. Whether BMI has a causal role in the development of AD therefore remains uncertain.
Causality between an exposure and outcome can be evaluated or estimated with a study design known as Mendelian randomization (MR)12,13. This approach investigates causal relationships by using inherited genetic variants as instrumental variables that influence exposure status. As these genetic variants are randomly allocated at point of conception, they are analogous to the randomization process in controlled trials and are less affected by problems of confounding factors and reverse causation. In this study, we therefore used genetic associations and concept of Mendelian randomization (MR) to evaluate the causal relationships between AD and obesity, as measured by Body Mass Index (BMI, a widely used measure of adiposity).
Results
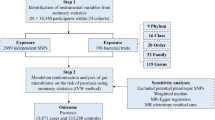
Our experimental design is summarized in Fig. 1. In brief, we carried out bi-directional MR analysis using genetic data from two recently published genome-wide association study (GWAS) datasets of BMI and AD in accordance to recent proposed guidelines for reporting MR analysis14,15,16,17.

Schematic diagram of Mendelian randomization (MR) experiment of exposure upon outcome of interest. Known genetic instruments (SNPs) for exposure were used as instrumental variables to assess the causal effect of exposure upon outcomes. Β1exposure is the estimated coefficient from the regression of exposure on the genetic variant(s) using the exposure GWAS. Β2(Observed) is the observed coefficient of the relationship between the exposure and outcome of interest. Β3outcome is the regression coefficient of outcome on the corresponding genetic variant using the outcome GWAS. MR estimate is the wald ratio of Β3outcome/Β1exposure. Significant (P < 1 × 10−8) and near-independent BMI SNPs (based on an approximate conditional and joint multiple-SNP (COJO) analysis that takes into account LD (linkage disequilibrium) between SNPs at a given locus) from BMI GWAS were used as instrumental variables to assess the causal effect of BMI upon AD while significant (P < 5 × 10−8) and independent AD SNPs (at least 4 MB(mega base pairs) apart) from AD GWAS were used as instrumental variables to assess the causal effect of AD upon BMI.
Effect of BMI upon AD
We identified genetic variants influencing BMI (BMI SNPs) for use as genetic instruments to evaluate the effect of adiposity on risk of AD in our MR experiment. (Supplementary Fig. S1a). We used the most recent published GWAS for BMI which comprises the meta-analysis between data from the GIANT consortium and the UK Biobank study (N = 681,275 participants). There were a total of 941 SNPs that were associated with BMI at P < 1 × 10–8(Supplementary Table S1). For the genetic effects on BMI, we used the results of the conditional and joint multiple SNP (COJO) analysis. We then evaluated the relationship between these BMI SNPs and AD using effect estimates from a recent GWAS of AD (N = 21,000 cases and 95,000 controls). Results for MR analyses are summarized in Table 1. The MR estimate of BMI on AD using inverse variance weighted (IVW) analysis yielded an odds ratio of 1.08 (95% CI 1.01 to 1.14; p = 0.015) for having AD per unit increase in BMI (kg/m2). The maximum-likelihood MR estimate gave a similar odds ratio for AD of 1.08 (95% CI 1.01 to 1.14; p = 0.015) per unit increase of BMI (kg/m2). The weighted median based method and MR-Egger regression analysis yielded odd ratios of 1.07 (95% CI 0.97 to 1.18) and 1.06 (95% CI 0.96 to 1.16) respectively. However, neither reached statistical significance (p = 0.195 and p = 0.248 respectively). The MR-Egger regression analysis gave no significant evidence of horizontal pleiotropy, with the intercept being not significantly deviated from zero (intercept: 0.000; 95% CI − 0.002 to 0.003; p = 0.663).
As part of sensitivity analyses, we repeated the MR studies removing SNPs with pleiotropic effects that might confound the relationship between BMI and AD (see “Methods” section). This modified IVW analysis yielded a similar odds ratio for AD per unit increase in BMI (kg/m2) of 1.12 (95% CI 1.04 to 1.21). As SNPs identified by the COJO analysis could potentially include non-independent variants in high linkage disequilibrium (LD), as a further sensitivity analysis, we systematically excluded SNPs across a range of r2 thresholds (r2 > 0.1, 0.2 and 0.5) followed by distance pruning at < 500 kb as well as SNPs with r2 thresholds (r2 > 0.001 and > 0.01) within 1000 kb apart (Supplementary Table S2a). MR estimates obtained were statistically significant and consistent with results obtained when all 941 GWAS BMI SNPs were analysed (Supplementary Table S2b).
Effect of AD upon BMI
We next identified genetic variants influencing AD (AD SNPs) for use as genetic instruments to assess whether AD has a causal effect on BMI. We identified 24 AD SNPs from the results of the EAGLE consortium dataset. (Supplementary Fig. S1b, Supplementary Table S3) MR results are summarized in Table 1. The MR estimate for the effect of AD on BMI using the IVW method yielded an effect estimate of 0.00 unit change in BMI (kg/m2) among those who had AD compared to controls (95% CI − 0.02 to 0.02; p = 0.862). The maximum-likelihood MR estimate gave a similar effect estimate of 0.00 (95% CI − 0.03 to 0.02; p = 0.868). The weighted median based method and MR-Egger regression analysis yielded effect estimates of − 0.01 (95% CI − 0.03 to 0.01) and 0.04 (95% CI − 0.02 to 0.11) respectively. None reached statistical significance (p = 0.312 and p = 0.189 respectively). The MR-Egger regression analysis gave no significant evidence of horizontal pleiotropy, as the intercept was not significantly deviated from zero (Intercept: − 0.00; 95% CI − 0.01 to 0.00; p = 0.152). Modified IVW analysis after excluding potential horizontal pleiotropic SNPs, based upon published associations (see “Methods” section), yielded a similar effect estimate of 0.00 (95% CI − 0.02 to 0.02).
No measurement error [NOME] assumption
To assess how measurement errors in SNP exposure estimates might affect our results, we estimated the attenuation of effect estimates in IVW and MR Egger MR methods using F statistics and I2GX respectively (see “Methods” section). Both statistics range from 0 to 100%, with values close to 100% suggestive of minimal attenuation.
For the IVW analyses, we find almost no attenuation of effect estimates (FGX − 1/FGX− = 99.6% and 93.1% for the BMI to AD and AD to BMI MR analyses respectively). The I2GX estimate for MR Egger analysis (BMI to AD) was 93.6%, suggesting that any measurement errors in the SNP BMI associations did not attenuate the effect estimates to a large degree. In contrast, the I2GX estimate for AD to BMI was 4%, suggesting that MR estimate derived from MR-Egger may be attenuated by measurement errors in AD, and is therefore less robust in evaluating MR estimates.
Discussion
Using Mendelian randomization, we showed that a higher BMI was causally associated with an increased risk of AD. In contrast, there was no evidence for a causal relationship of AD with an increase in BMI. We demonstrated that these findings are based upon valid primary MR assumptions and were also robust across different MR methods12,18,19.
Our results are consistent with the findings of prospective observational birth/infant cohort studies20,21. BMI at infancy or fat mass at birth is associated with an increased risk of a subsequent AD diagnosis, with other prospective observational studies of children also reporting that presence of obesity was associated with subsequent development of AD22,23,24,25. Although cross-sectional studies in adults support a relationship between AD and obesity, the direction of causality is uncertain26. Mendelian randomization thus provides a unique opportunity to examine this directionality.
Our mendelian randomization study suggested that BMI may have a causal role in the development of AD. The underlying mechanisms for this phenomenon could be secondary to the pro-inflammatory state and impaired epidermal barrier status of obesity27,28,29. The body adipose tissue contributes to the persistent low-grade inflammation by production of inflammatory cytokines27,28,30,31. Adipokines such as leptin and adiponectin play a role in the production of further inflammatory cytokines such as tumour necrosis factor-alpha (TNF-α) and interleukin (IL-) 6 from the adipose tissue30,31. Leptin further drives the T cell activation towards a Th1 phenotype with production of interferon γ28. These pro-inflammatory cytokines TNF, IL-6 and interferon γ in play a role in the inflammation of AD28,32. The adipose tissue is also the site for peripheral aromatization of androgens to oestrogen hormones such as oestrone and β-oestradiol30. The latter has been demonstrated to switch the initiating immune reaction from Th1 to Th2 type and also increase production of IL-4 and IL-13, characteristic of AD27. In addition to these inflammatory responses, obese individuals are observed to have an impaired epidermal barrier function as evidenced by an increased trans-epidermal water loss28.
The finding in our MR study that obesity has a causal relationship with AD is of great clinical importance as obesity could therefore be a modifiable risk factor for AD. Obesity itself has significant morbidity and mortality risks and therefore physicians should be cognizant of concomitant obesity and its related complications among AD patients. Although the importance of weight loss is widely appreciated by cardiovascular and metabolic medicine physicians, there is currently little appreciation of the importance of healthy weight in the management of dermatological conditions. Our results therefore have the potential to change current management guidelines for example, incorporating advisory regarding weight control through lifestyle modifications as a potential measure to alleviate AD.
Strengths of our study include the use of the two largest GWAS datasets for AD and obesity consisting of participants with European descent to date, with a total sample size of about 700,000 individuals and 21,000 AD cases and 95,000 controls respectively14,15. The genetic variants identified represent strong genetic instruments for evaluating causality. The correlation between genetic risk score based on the 941 SNPs and the BMI measurement in an independent validation cohort were 0.2214. The variance explained in this cohort from the SNPs were 6.0%. The AD GWAS dataset explain about 12.3% and 2.6% of variance for the previously established and newly identified SNPs15. This provided evidence that the MR assumption of relevance was valid in our study33.
We also assessed and supported the validity of the remaining two key MR assumptions of independence and exclusion restriction33. Modified IVW MR analysis, excluding potential pleiotropic factors based on published associations yielded effect estimates similar to our main analysis. Our MR-Egger intercept analysis also showed no evidence of confounding by horizontal pleiotropy19.
Various other MR methods (maximum likelihood, MR-Egger and weighted median method), in addition to the IVW method, were also performed as part of sensitivity analysis17. Results were consistent and similar in its direction and magnitude. Estimates from the MR-Egger and weighted median methods were not statistically significant and tend to have less precise confidence intervals.
Limitations include the use of GWAS datasets of participants of only European ancestry, which limits potential generalization to other populations such as Asians. Meta-analysis of studies from Asia has reported a significant relationship between AD and obesity7. However, as GWAS datasets from Asian participants have a much smaller sample size (weak instruments), they were not utilised in our current analyses34,35.
Conclusions
In this study, taking a MR approach, we provide evidence to suggest that a higher BMI causally increases the risk of AD in individuals of European descent. There was no evidence that the reverse direction is true. Obesity can therefore act as a modifiable risk factor for AD, potentially changing our current clinical dermatological practice for management of patients with AD who are obese. In addition, our results open up new avenues to better understand the mechanistic pathways of BMI driving the risk of AD. This could in turn lead to the development of novel therapeutics to maintain skin health and prevent AD.
Methods
Two-sample MR analysis with multiple genetic variants as instrumental variables using summarized data was performed in our study to assess the causal relationship and its strength between BMI and AD36. The MR analysis was performed in both directions to establish the direction of causality between BMI and AD. The validity of the instrumental variables are important for the MR analysis and are defined by three key assumptions: (1) genetic variants are associated with the exposure factor of interest (Relevance assumption); (2) genetic variants are independently associated with the outcome with no unmeasured confounders (Independence assumption); (3) genetic variants only affect the outcome through their effect on the exposure factors with no evidence of other horizontal pleiotropic factors (Exclusion restriction assumption)33. Data was extracted from the two largest GWAS reported to date on BMI and AD to fulfil the relevance assumption of MR. We carried out sensitivity analyses by excluding SNPs that have associations with possible confounders in the MR analyses, and performed an MR-Egger test of pleiotropy, to assess the validity of the independence and exclusion restriction assumptions respectively.
GWAS of BMI and AD
The following GWAS for BMI and AD provided genetic risk variants information for the MR analysis. Genetic risk variants for BMI were determined using the combined analysis of the Genetic Investigation of ANthropometric Traits (GIANT) consortium and the UK biobank dataset of about 700,000 individuals with European ancestry in total14. The GIANT consortium is a joint GWAS and metabochip meta-analysis of 114 studies that measured BMI as its phenotype of interest. Details of included studies (assessment and definitions of phenotypes, genotyping and quality control process) in the BMI GWAS analysis and UK biobank are provided in Supplementary Table S4. Meta-analysis of summary statistics from these two studies identified 941 near-independent single nucleotide polymorphisms (SNPs) associated with BMI at a revised genome-wide significance threshold of P < 1 × 10−8). These SNPs were identified using an approximate conditional and joint multiple-SNP (COJO) analysis that takes into account LD (linkage disequilibrium) between SNPs at a given locus. As part of sensitivity analyses, we did a between-SNPs LD r2 analysis and excluded SNPs across a range of r2 thresholds (> 0.1, > 0.2, > 0.5) followed by distance pruning at < 500 kb as well as SNPs with r2 thresholds (r2 > 0.001 and > 0.01) within 1000 kb apart (Supplementary Table S2a).
Similarly, genetic risk variants for atopic dermatitis were determined using the dataset of the EArly Genetics and Life course Epidemiology (EAGLE) Consortium15 of 21,000 cases and 95,000 controls. This GWAS meta-analysis analyzed AD case–control status in 22 individual cohorts of European ancestry. Details of included studies in the AD GWAS analysis are provided in Supplementary Table S5. The study reported 24 SNPs reaching genome wide significance (P < 5 × 10−8) for AD risk. These SNPs were independent with at least 4 Mb apart.
Statistical analysis
The two-sample MR strategy was adopted in our analysis37. The SNP-exposure effects and the SNP-outcome effects were obtained from separate GWAS datasets. First, summary level SNPs-exposure associations were extracted from the first GWAS dataset on exposure as MR instruments. These instruments SNPs-outcome associations were then extracted with the second GWAS dataset on outcome. Any missing exposure associated variants in the outcome GWAS dataset were replaced by linkage disequilibrium (LD) proxies of a minimum of 0.6. Only one SNP out of the 941 SNPs (rs11172702) in the BMI GWAS dataset had missing SNP-outcome effect measure in the outcome (AD) dataset that required a proxy SNP (rs11172644; LD: r2 = 1) for analysis. Comparatively, 10 out of the 24 SNPs (rs10199605, rs12730935, rs2227483, rs2592555, rs4809219, rs10791824, rs12188917, rs2212434, rs2918307 and rs6419573) had missing SNP-outcome effect measures in the BMI dataset and required proxy SNPs of LD r2 values between 0.67 to 1.00. Four missing SNPs (rs112111458, rs4713555, rs145809981 and rs61813875) had no proxy SNPs with adequate LD r2 values of greater than 0.6 and were therefore excluded from the analysis.
The exposure and outcome effects were then harmonized. This involved identification of SNP variants of unmatched effect and alternate alleles between the two datasets and correction by switching the direction of their effect estimates and corresponding effect allele frequencies. We searched for palindromic SNPs that might have inverted the direction of effect in the BMI and AD GWAS datasets, and found no evidence for such inversion. There was therefore no need to realign any of the palindromic SNPs used. Individual SNP estimates were first calculated using the Wald ratio method. The Wald ratio refers to the ratio estimate of the effect of the variant-outcome divided on the effect of variant-exposure. The standard error of the ratio estimate was approximated using the delta method38.
Inverse variance weighted MR
An MR estimate using multiple SNPs was obtained by performing a random effects inverse variance weighted (IVW) meta-analysis of each Wald ratio of corresponding SNP18. The IVW method assumes that all SNPs are valid instruments or any underlying horizontal pleiotropy is balanced across the SNPs39. In order to examine the robustness of the estimates, we also estimated the causal effect estimate of exposure on outcome using three other MR methods: maximum likelihood method, weighted median-based method and MR-Egger regression analysis.
Maximum likelihood method
The causal effect parameter was estimated from a model that assumed a linear relationship between exposure and outcome and a bivariate normal distribution for the genetic variants40. Standard errors for the maximum-likelihood estimates were obtained using the inverse Hessian matrix. This method made similar assumptions as the IVW approach but provides more reliable estimates in the presence of measurement error in SNP-exposure effects41.
Weighted median-based method
This approach examines the median effect of all available SNPs and only requires half the SNPs to be valid instruments for the effect estimate to be unbiased. 50% of the weights in the analysis will be from ratio estimates smaller or equal to the weighted median42.
MR-Egger regression analysis
The MR-Egger regression analysis is an adaptation of the IVW analysis by allowing a non-zero intercept that is estimated as part of the analysis43. This relaxed the assumption of no horizontal pleiotropy and allowed net horizontal pleiotropic effect across all SNPS to be unbalanced, or directional. However, any horizontal pleiotropic effect should not correlate with SNP-exposure effects. MR-Egger intercept test was also performed as part of the MR-Egger regression analysis to assess whether the genetic variants have directional horizontal pleiotropic effects on the outcome. It is a test of whether the intercept of the MR-Egger regression analysis significantly differs from zero.
Power calculation of MR analysis
Calculations for statistical power for our MR analyses were performed according to Brion et al. using their web-based application44. Power calculations of the MR analysis of BMI upon AD were provided for a range of true odds ratio of AD per unit increase of BMI (Supplementary Fig. S2)44. Calculations were based on a sample size of 116,000 (21,000 AD cases and 95,000 controls) in the outcome dataset (i.e. AD GWAS study) and a type 1 error rate of 0.05. Proportion of variance explained for the association of the included SNPs (n = 941) with BMI (exposure variable) was 0.06. Our study had 80% power to detect an odds ratio for AD of 1.09 per unit (kg/m2) increase of BMI in the MR analysis.
Power calculations of the MR analysis of AD upon BMI were provided according to a range of values for the true underlying causal association between AD and BMI (Supplementary Fig. S2)44. These was based on a sample size of 681,275 in the outcome dataset (i.e. BMI GWAS study) and a regression coefficient of 0.10 kg/m2 for the observational association between AD and BMI according to a recent cohort study by Shalom et al.45 The variances for AD (exposure) and BMI (outcome) used were 0.148 and 16 kg2/m4 respectively based on AD and BMI GWAS datasets14,15. Proportion of variance explained for the association of the included SNPs (n = 20) with AD (exposure variable) was 0.026. Our study had 80% power to detect a 0.220 kg/m2 increase in BMI arising from the presence of AD at a type 1 error rate of 0.05.
Sensitivity analysis
In order to improve the reliability of our MR results, we performed sensitivity analyses by excluding potential pleiotropic variants in our MR analyses. The associations of the genetic variants used as genetic instruments with potential confounders of BMI and AD were annotated using the PhenoScanner V2 database46,47. (Supplementary Tables S6, S7). Potential confounders considered for the BMI SNPs included education, behavioural factors such as tobacco smoking, alcohol ingestion and physical activity, psychiatric diseases and psychological well-being (anxiety, depression)48,49,50,51,52. Potential confounders considered for the AD SNPs included education, alcohol ingestion psychiatric diseases and psychological well-being (anxiety, depression)48,49,50,51,52.
Given that there might be a certain degree of measurement error in the SNP exposure associations, resulting in departure of the no measurement error (NOME) assumption, we also estimated the attenuation of effect estimates in IVW and MR-Egger methods using F statistics and I2 respectively. Both range from 0 to 100%, with values close to 100% suggestive of minimal attenuation53,54.
The attenuation of the effect size as a result of departure of NOME assumption in the IVW method can be estimated by measuring the instrument strength (F statistics) for the genetic variant used53. F statistic for each variant is calculated as the ratio of its effect size estimate to the variance of its SNP-exposure association, a weighted F statistic is then measured across all the SNPs used in the MR experiment. Degree of attenuation of effect estimates resulting from departure of NOME assumption is then estimated by:
In contrast, MR Egger estimates are not governed by F statistics and are better assessed using I2GX estimate54. I2GX represents the true variance of SNP-exposure associations divided by the variance of the SNP-exposure estimates.
TwoSampleMR (version 0.4.26) and Mendelian Randomization (version 0.4.1) packages in R statistical software (RStudio version 1.2.1335) were used to perform data clumping of SNPs and two-sample Mendelian randomization analysis respectively. The MR experiment is summarized in Fig. 1.
Data availability
All data generated or analysed during this study are included in this published article and its Supplementary Information files.
References
Bieber, T. Atopic dermatitis. Ann. Dermatol. 22, 125 (2010).
Williams, H. C., Burney, P. G., Pembroke, A. C. & Hay, R. J. The U.K. working party’s diagnostic criteria for atopic dermatitis. III. Independent hospital validation. Br. J. Dermatol. 131, 406–416 (1994).
Pellerin, L. et al. Defects of filaggrin-like proteins in both lesional and nonlesional atopic skin. J. Allergy Clin. Immunol. 131, 1094–1102 (2013).
Rawlings, A. V. & Harding, C. R. Moisturization and skin barrier function. Dermatol. Ther. 17(suppl 1), 43–48 (2004).
Kim, J. E., Kim, J. S., Cho, D. H. & Park, H. J. Molecular mechanisms of cutaneous inflammatory disorder: Atopic dermatitis. Int. J. Mol. Sci. 17, E1234 (2016).
Brunner, P. M. et al. Increasing comorbidities suggest that atopic dermatitis is a systemic disorder. J. Investig. Dermatol. 137, 18–25 (2017).
Zhang, A. & Silverberg, J. I. Association of atopic dermatitis with being overweight and obese: A systematic review and metaanalysis. J. Am. Acad. Dermatol. 72, 606–616 (2015).
Williams, H. C., Robertson, C. F., Stewart, A. W. & on behalf of the ISAAC Steering Committee. Worldwide variations in the prevalence of atopic eczema symptoms. J. Allergy Clin. Immunol. 103, 125–138 (1999).
Stevens, G. A. et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul. Health Metrol. 10, 22 (2012).
Violante, R. et al. Obesity risk factors in the ISAAC (International Study of Asthma and Allergies in Childhood) in Mexico City. Rev. Alerg Mex. 52, 141–145 (2005).
Sidoroff, V., Hyvarinen, M. K., Piippo-Savolainen, E. & Korppi, M. Overweight does not increase asthma risk but may decrease allergy risk at school age after infantile bronchiolitis. Acta Paediatr. 101, 43–47 (2012).
Smith, G. D. & Ebrahim, S. “Mendelian randomization”: Can genetic epidemiology contribute to understanding environmental determinants of disease?. Int. J. Epidemiol. 32, 1–22 (2003).
Lawlor, D. A., Harbord, R. M., Sterne, J. A., Timpson, N. & Davey Smith, G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Yengo, L. et al. Meta-analysis of genome-wide association studies for height and body mass index in ∼700,000 individuals of European ancestry. Hum. Mol. Genet. 27, 3641–3649 (2018).
Paternoster, L. et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 47, 1449–1456 (2015).
Lor, G. C. Y. et al. Reporting and guidelines for Mendelian randomization analysis: A systematic review of oncological studies. Cancer Epidemiol. 62, 101577 (2019).
Burgess, S. et al. Guidelines for performing Mendelian randomization investigations. Wellcome Open Res. 4, 186 (2020).
Didelez, V. & Sheehab, N. Mendelian randomization as an instrumental variable approach to causal inference. Stat. Methods Med. Res. 16, 309–330 (2007).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur. J. Epidemiol. 32, 377–389 (2017).
Berents, T. L. et al. Weight-for-length, early weight-gain velocity and atopic dermatitis in infancy and at two years of age: A cohort study. BMC Pediatr. 17, 141 (2017).
O’Donovan, S. M. et al. Neonatal adiposity increases the risk of atopic dermatitis during the first year of life. J. Allergy Clin. Immunol. 137, 108–117 (2016).
Byberg, K. K., Eide, G. E., Forman, M. R., Juliusson, P. B. & Oymar, K. Body Mass Index and physical activity in early childhood are associated with atopic sensitization, atopic dermatitis and asthma in later childhood. Clin. Transl. Allergy 6, 33 (2016).
Lei, Y., Yang, H. & Zhen, L. Obesity is a risk factor for allergic rhinitis in children of Wuhan (China). Asia Pac. Allergy 6, 101–104 (2016).
Silverberg, J. I. et al. Association between obesity and atopic dermatitis in childhood: A case-control study. J. Allergy Clin. Immunol. 127, 1180–1186 (2011).
Ali, Z., Suppli Ulrik, C., Agner, T. & Thomsen, S. F. Is atopic dermatitis associated with obesity? A systematic review of observational studies. J. Eur. Acad. Dermatol. Venereol. 32, 1246–1255 (2018).
Gregor, M. F. & Hotamisligil, G. S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445 (2011).
Danso, M. O. et al. TNF-alpha and Th2 cytokines induce atopic dermatitis like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J. Investig. Dermatol. 134, 1941–1950 (2014).
Guida, B. et al. The impact of obesity on skin disease and epidermal permeability barrier status. J. Eur. Acad. Dermatol. Venereol. 24, 191–195 (2010).
Bastard, J. P. et al. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 17, 4–12 (2006).
Loffreda, S. et al. Leptin regulates proinflammatory immune responses. FASEB J. 12, 57–65 (1998).
Martin-Romero, C., Santos-Alvarez, J., Gobernam, R. & Sanchez-Margalet, V. Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell Immunol. 199, 15–24 (2000).
Ali, Z. & Ulrik, C. S. Obesity and asthma: A coincidence or a causal relationship? A systematic review. Respir. Med. 107, 1287–1300 (2013).
Davies, N. M., Holmes, M. V. & Davey, S. G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ 362, k601 (2018).
Akiyama, M. et al. Genome-wide association study identifies 112 new loci for Body Mass Index in the Japanese population. Nat. Genet. 49, 1458–1467 (2017).
Sun, L. D. et al. Genome-wide association study identifies two new susceptibility loci for atopic dermatitis in the Chinese Han population. Nat. Genet. 43, 1458–1467 (2011).
Frayling, T. M. Genome-wide association studies: The good, the bad and the ugly. Clin. Med. (Lond.) 14, 428–431 (2014).
Hartwig, F. P., Davies, N. M., Hemani, G. & Davey Smith, G. Two-sample Mendelian randomization: Avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int. J. Epidemiol. 45, 1717–1726 (2016).
Thomas, D. C., Lawlor, D. A. & Thompson, J. R. Re: Estimation of bias in nongenetic observational studies using “Mendelian triangulation” by Bautista et al.. Ann. Epidemiol. 17, 511–513 (2007).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Pierce, B. L. & Burgess, S. Efficient design for Mendelian randomization studies: Subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 178, 1177–1184 (2013).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, e34408. https://doi.org/10.7554/eLife.34408 (2018).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Brion, M. A., Shakhbazov, K. & Visscher, P. M. Calculating statistical power in Mendelian randomization studies. Int. J. Epidemiol. 42, 1497–1501 (2013).
Shalom, G. et al. Atopic dermatitis and the metabolic syndrome: A cross-sectional study of 116 816 patients. J. Eur. Acad. Dermatol. Venereol. 33, 1762–1767 (2019).
Staley, J. R. et al. PhenoScanner: A database of human genotype-phenotype associations. Bioinformatics 32, 3207–3209 (2016).
Kamat, M. A. et al. PhenoScanner V2: An expanded tool for searching human genotype-phenotype associations. Bioinformatics 35, 4851–4853 (2019).
Shaw, T. E., Currie, G. P., Koudelka, C. W. & Simpson, E. L. Eczema prevalence in the United States: Data from the 2003 National Survey of Children’s Health. J. Investig. Dermatol. 131, 67–73 (2011).
Kantor, R., Kim, A., Thyssen, J. P. & Silverberg, J. I. Association of atopic dermatitis with smoking: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 75, 1119–1125 (2016).
Halling-Overgaard, A. S. et al. Atopic dermatitis and alcohol use—A meta-analysis and systematic review. J. Eur. Acad. Dermatol. Venereol. 32, 1238–1245 (2018).
Silverberg, J. I. et al. Atopic dermatitis is associated with less physical activity in US adults. J. Investig. Dermatol. 136, 1714–1716 (2016).
Rønnstad, A. T. M. et al. Association of atopic dermatitis with depression, anxiety, and suicidal ideation in children and adults: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 79, 448–456 (2018).
Bowden, J. et al. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med. 36, 1783–1802 (2017).
Bowden, J. et al. Assessing the suitability of summary data for two-sample mendelian randomization analyses using MR-Egger regression: The role of the I2 statistic. Int. J. Epidemiol. 45, 1961–1974 (2016).
Author information
Authors and Affiliations
Contributions
Y.W.Y., M.L., S.T.G.T. and J.C.C. wrote the main manuscript text and Y.W.Y. prepared the main and supplementary tables and figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yew, Y.W., Loh, M., Thng, S.T.G. et al. Investigating causal relationships between Body Mass Index and risk of atopic dermatitis: a Mendelian randomization analysis. Sci Rep 10, 15279 (2020). https://doi.org/10.1038/s41598-020-72301-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72301-2
This article is cited by
-
Investigating causal relationships between obesity and skin barrier function in a multi-ethnic Asian general population cohort
International Journal of Obesity (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
