
Abstract
The q11.2 region on chromosome 22 contains numerous low-copy repeats that lead to deleted or duplicated regions in the chromosome, thereby resulting in different syndromes characterized by intellectual disabilities or congenital anomalies. The association between patient phenotypes and 22q11.2 copy number abnormalities has been previously described in postnatal cases; however, these features have not been systematically evaluated in prenatal cases because of limitations in phenotypic identification in prenatal testing. In this study, we investigated the detection rate of 22q11.2 copy number abnormalities in 2500 fetuses using single nucleotide polymorphism (SNP) array and determined the common abnormal ultrasound findings in fetuses carrying the 22q11.2 copy number abnormalities. The 22q11.2 copy number abnormalities were identified in 13 fetuses with cardiovascular malformations (6/13), kidney malformations (3/13), isolated ultrasound markers (3/13), or high-risk Down syndrome based on maternal serum screening (1/13). Approximately 0.5% (13/2500) of the fetuses harbored 22q11.2 copy number abnormalities. The most frequent ultrasound findings in fetuses with these abnormalities were cardiovascular malformations, followed by kidney malformations and isolated ultrasound markers. Prenatal diagnosis of these genetic abnormalities allows for the delineation of differential diagnoses, characterization of a wide spectrum of associated malformations, and determination of associations that exist between prenatal diagnosis and obstetrical outcomes.
Similar content being viewed by others


Introduction
Chromosome 22 has long been implicated in genomic diseases, such as 22q11.2 deletion syndrome (22q11.2DS) and cat-eye syndrome (CES), which are associated with decreased and increased gene dosages, respectively [1]. Approximately 1 in every 4000 live births shows 22q11.2DS, also known as velocardiofacial or DiGeorge syndrome, making the most common microdeletion syndrome in humans [2]. The 22q11.2DS phenotype is highly variable with over 180 clinical features [3], including congenital conotruncal cardiac defects, characteristic facial features, immunodeficiency, palatal abnormalities, hypocalcemia, urogenital abnormalities, a spectrum of cognitive deficits, and psychiatric symptoms. On the contrary, CES is rare disorder caused by duplication of a part of chromosome 22 [4]. CES is characterized by preauricular tags or pits, ocular coloboma, dysmorphic facial features, and cardiac, urogenital, and anorectal anomalies [5].
The long arm (q region) of chromosome 22 is vulnerable to chromosomal rearrangement because of the large number of low-copy repeats (LCR) in this region, which mediate non-allelic homologous recombination resulting in unequal crossover rearrangements [6]. These mechanisms lead to deleted or duplicated regions of the chromosome, resulting in different syndromes characterized by intellectual disabilities and/or congenital anomalies [4]. More than 90% of the patients with 22q11.2DS have a classic 3 Mb deletion spanning LCR22A–D, including TBX1 [7]. A corresponding chromosomal duplication in 22q11.2 in LCR22A–D has also been described, which is associated with diverse phenotypic abnormalities. This duplication may result in phenotypes similar to those associated with 22q11.2DS; however, this also occurs in individuals with a normal phenotype [8,9,10,11]. Less than 10% of patients with either 22q11.2 deletions or duplications are affected in a smaller 1.5 Mb region spanning LCR22A–B [12,13,14].
The etiology in most patients is a classic 3 Mb recurrent deletion or duplication in 22q11.2. However, cases of atypical deletions and duplications with different sizes and locations have also been described, generally with a milder, slightly different phenotype for duplications; however, few studies have focused on 22q11.2 copy number abnormalities in fetuses. In this study, we report 13 cases with 22q11.2 copy number abnormalities (including atypical breakpoints), which were all diagnosed prenatally. In all cases, we delineated the differential diagnoses, illustrated the spectrum of associated malformations, and explored the association between prenatal diagnosis of 22q11.2 copy number abnormalities and obstetrical outcomes. Although few cases are included in this study, these results likely represent the intrauterine phenotypic profiles of 22q11.2 copy number abnormalities, thereby improving the understanding of intrauterine phenotypic heterogeneity associated with these abnormalities.
Materials and methods
Patient data
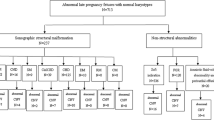
The study was conducted between January 2016 and December 2018 at the Prenatal Diagnosis Center of the Fujian Provincial Maternal and Children Health Hospital. A total of 2500 fetuses participated in this study, including 1821 fetuses with abnormal sonographic findings (classified as fetal structural malformations, ultrasound soft markers, hydrops fetalis, fetal growth restriction, and abnormal amniotic fluid volume), 432 fetuses with advanced maternal age (over the age of 35 years), and 247 fetuses with established high-risk Down syndrome based on maternal serum screening (cut-off for high-risk was 1:270). This study was approved by the ethics committee of Fujian Provincial Maternal and Child Health Hospital, and informed consent was obtained from the parents for invasive prenatal diagnosis. Amniotic fluid was collected by amniocentesis at 18–24 weeks of gestation and cord blood was collected at 24 weeks of gestation. The median gestational age for screening was 26 weeks (18–34 weeks).
SNP array
Our laboratory previously established the SNP array technology that was used in this study [15]. Genomic DNA was directly extracted from uncultured amniotic fluid and cord blood samples using the QIAamp DNA Blood Mini kits (Qiagen, Hilden, Germany). The genome-wide high-resolution SNP array CytoScan HD (Affymetrix Genome CytoScan 750 K, Affymetrix, Santa Clara, CA, USA), including SNPs and oligonucleotide probes, was used in this study. DNA (250 ng) was amplified, labeled, and hybridized. The copy number variant (CNV) reporting filter was set at > 100 kb with a minimum set of 50 marker counts.The results were analyzed using Chromosome Analysis Suite software (Affymetrix) and annotated based on GRCh37 (hg19).The CNVs identified using the SNP array were classified as pathogenic (P), variants of uncertain significance (VOUS), and benign (B) according to the American College of Medical Genetics guidelines [16]. Pathogenic CNV and VOUS were classified as abnormal. All annotated CNVs were experimentally validated by fluorescence in situ hybridization.
Results
Among 2500 fetuses subjected to SNP array analysis, 227 fetuses had abnormal CNVs. Among 1821 fetuses with abnormal sonographic findings, 165 fetuses had abnormal CNVs (9.1%, 165/1821). Among 432 fetuses with advanced maternal age, 26 fetuses had abnormal CNVs (6.0%, 26/432). Among 247 fetuses with established high-risk Down syndrome based on serum screening, 36 fetuses had abnormal CNVs (14.6%, 36/247). Among 227 fetuses with abnormal CNVs, the 22q11.2 copy number abnormalities were most common. The 22q11.2 copy number abnormalities were identified in 13 cases of 11 singleton pregnancies and 1 twin pregnancy, accounting for approximately 0.5% of the cases. Out of the 13 fetuses carrying 22q11.2 copy number abnormalities, seven were deletions and six were duplications, indicating cardiovascular malformations (6/13), kidney malformations (3/13), isolated ultrasound markers (3/13), or high-risk Down syndrome based on maternal serum screening (1/13). All parents were healthy and non-consanguineous.
Molecular characterization of 22q11.2 deletion in fetuses
Five cases were found to harbor a classic 22q11.2 deletion that encompassed TBX1and contributed to 22q11.2DS. For instance, a 36-year-old mother in case E2351 was pregnant with her second child. The fetus in this case presented with a ventricular septal defect, right-sided aortic arch, U-shaped vascular ring, aberrant left subclavian artery, and classic 3.1 Mb deletion. Furthermore, a 35-year-old mother in case E2503 had two previous healthy children. A fetal echocardiography at 20 weeks of gestation revealed tetralogy of Fallot. SNP results also revealed a classic 3.1 Mb deletion. Another case, P4643, was a 31-year-old primigravid mother. At 18 weeks of gestation, ultrasound examination revealed ventricular septal defect and pulmonary atresia. SNP analysis revealed a classic 2.8 Mb deletion. Genetic analysis of the parents revealed normal results. In case G9924, a 32-year-old mother was pregnant with her second child. The mother’s elder child had congenital heart disease and physical retardation. Ultrasound anomalies were accompanied with bilateral choroid plexus cysts. SNP results revealed a classic 3.1 Mb deletion. Genetic analysis of the parents showed that case G9924 arose from a maternal deletion on chromosome 22. The mother’s elder child was also found to carry this deletion. Lastly, case G9932 was a 27-year-old gravida 2, para 1 mother with an unremarkable personal history. Ultrasound examination at 23 weeks of gestation revealed an ectopic right kidney with polycystic dysplasia. SNP array detected a classic 2.8 Mb deletion. Ultrasound results for case G9924 and case G9932 revealed no abnormalities in the heart.
The results also revealed two cases with atypical deletions. In case W23, a 30-year-old mother had one previous healthy child. The family history was found to be non-significant. However, maternal serum screening resulted in a high Down syndrome risk estimate of 1:135 and the attempts to cultivate amniotic fluid from this patient failed. The remaining amniotic fluid was subjected to SNP array, which showed an atypical 2.0 Mb deletion. Similarly, the patient in case P4666 was a 31-year-old primigravida. Ultrasound anomalies were accompanied with nuchal cystichygroma and increased nuchal translucency in the fetus at 19 weeks of gestation. SNP array analysis detected an atypical 1.0 Mb 22q11.21–q11.22 deletion. Genetic analysis of the parents was non-significant (Table 1).
Molecular characterization of 22q11.2 duplication in fetuses
Microduplicationin 22q11.2 was detected in six separate cases by SNP analysis. Herein, we describe cases E2151, G8565, and Z18, all of which showed a duplicationin 22q11.2 with few overlapping features common to22q11.2DS. The patient in case E2151 was a 31-year-old gravida 2, para 1 with an unremarkable personal history. Ultrasound examination at 31 weeks of gestation revealed a complex cardiac defect with oval valve aneurysm, a small ascending aorta, and aortic arch associated with poor prognosis. SNP results revealed a classic 3.1 Mb duplication. In addition, the 37-year-old mother in case G8565 had one previous healthy child and two miscarriages. The family history was non-significant. Ultrasound examination at 18 weeks of gestation revealed a complex cardiac defect with ventricular septal defect, aortic dysplasia, and enhanced ventricular echo. SNP array detected a classic 1.7 Mb duplication that resulted in CES. Lastly, the patient in case Z18 was a 39-year-old primigravida. At 24 weeks of gestation, ultrasound examination revealed ventricular septal defect, aberrant subclavian artery, double superior vena cava, and a single umbilical artery. SNP array also showed a classic 1.7 Mb duplication resulting in CES.
Cases P4876 and P4877 were unique as they were dizygotic twin fetuses. The 28-year-old mother had three previous miscarriages. The mother had not conceived for 2 years and, therefore, used in vitro fertilization and embryo transfer to achieve her current pregnancy with dizygotic twins. Ultrasound examination at 24 weeks of gestation revealed that both the dizygotic fetuses had unilateral polycystic kidney dysplasia. SNP array detected an atypical 1.0 Mb duplication. Genetic analysis of the parents revealed normal results (Table 2).
The patient in case E3027 was a 23-year-old primipara. Ultrasound examination revealed an increased nuchal translucency and fetal growth restrictionin the fetus at 30 weeks of gestation. SNP array analysis detected a 2.8 Mb 22q11.2 duplication. Genetic analysis of the parents showed that the variation was inherited from the unaffected mother.
Inheritance analysis and obstetrical outcomes
Hereditary information was screened for seven families where in SNP array analysis showed 22q11.2 copy number abnormalities (parents from six cases refused the collection of peripheral venous blood). Parental analysis revealed that two of the fetuses had abnormalities inherited from an unaffected parent, while in the five remaining fetuses, the CNVs occurred de novo.
The decision to terminate 11 of the pregnancies, according to the mothers, was because of the presence of abnormal CNVs. However, the parents of the dizygotic twins continued to full-term delivery although the SNP results were VOUS.
Discussion
SNP array is an important prenatal genetic diagnostic tool for exploring the genetic causes of fetuses with ultrasound abnormalities. In this study, out of the 2500 fetuses subjected to SNP array analysis, 227 fetuses had abnormal CNVs. Among 227 fetuses with abnormal CNVs, 165 fetuses with abnormal sonographic findings had abnormal CNVs (9.1%, 165/1821), 26 fetuses with advanced maternal age had abnormal CNVs (6.0%, 26/432), and 247 fetuses with established high-risk Down syndrome based on serum screening had abnormal CNVs (14.6%, 36/247). Out of the 227 fetuses with abnormal CNVs, 22q11.2 copy number abnormalities were most common. Thirteen fetuses had CNVs in chromosome 22 but showed normal karyotype analysis results. Thus, SNP array is superior to karyotype analysis because of its ability to detect CNVs with high accuracy and resolution. Amniotic fluid cell culture was unsuccessful in one case and could not be analyzed further; however, SNP array detected genetic anomalies.
The primary causes of chromosome 22 abnormalities are LCR-mediated non-allelic homologous disorders and unequal intra- or inter-chromosomal recombination during meiosis [17]. In addition to the classic deletion/replication region (3 Mb) at the proximal and distal breakpoints, several atypical deletions or replications with variable breakpoints can be caused by different recombinants mediated by LCR22A and LCR22H. In this study, 22q11.2 copy number abnormalities in 13 cases were identified. Seven of these abnormalities were deletions and six were duplications. Nine cases exhibited classic deletion/replication within this region,while four cases were atypical with variable breakpoints likely caused by different recombinants mediated by LCR22A–H.
Our study revealed that cases E2351, E2503, and G9924 shared a common 3.1 Mb hemizygous 22q11.2 deletion, which is consistent with the results in the literature [18]. The sonogram findings for cases E2351 and E2503 revealed cardiac abnormalities; however, case G9924 showed only bilateral choroid plexus cysts. Additionally, case P4643 was characterized by a ventricular septal defect and pulmonary atresia and case G9932 by an ectopic right kidney with polycystic dysplasia. Both cases were accompanied by a 2.8 Mb deletion. These results demonstrate highly variable abnormalities in the heart, which is consistent with the highly variable phenotype associated with 22q11.2DS. Ultrasound technology may be inefficient in effectively screening the abnormalities and it is possible that the varying size of the deletions contributed to the variable phenotypes found to be associated with the 22q11.2 deletion cases [19].
Interestingly, in case W23, maternal serum screening revealed a high-risk estimate of 1:135 for Down syndrome; although analysis of the amniotic cell culture was unsuccessful, SNP analysis of the remaining amniotic fluid detected a 2.0 Mb deletion. Case P4666 was determined to have an atypical 1.0 Mb deletion of 22q11.21–q11.22 accompanied by a nuchal cystichygroma and increased nuchal translucency. Notably, genetic analysis of the parents showed normal results. According to the database, although this region is associated with a 22q11.2 deletion, the clinical significance is unclear.
Recently, new cases of 22q11.2 duplication have been described [20, 21]. In our study, six 22q11.2 duplications were identified via abnormal ultrasound. Cases E2151 and E3027 were identified with 3.0 and 2.8 Mb duplications, respectively, corresponding in size to the classical deletions observed in patients with 22q11.2DS. We also found that cases G8565 and Z18 had a tetrasomy of 22q11.1q11.21that included the proximal flanking region of chromosome 22, which is associated with CES [5, 22]. Cases P4876 and P4877 were dizygotic twin fetuses with unilateral polycystic kidney dysplasia accompanied by an atypical 1.0 Mb duplication. The region in which the duplication occurred includes 29 Online Mendelian Inheritance in Man genes. According to the database, although this region is associated with a 22q11.2 duplication, the clinical significance is unclear.
Out of the 22q11.2 copy number abnormalities present in the 13 cases, five cases were de novo, two cases were inherited from unaffected mothers, and six cases had an unknown inheritance because parental specimens were unavailable. Parental analysis showed that fetus G9924 inherited a maternal 22q11.2 deletion; the mother’s elder child, who also had congenital heart disease, showed an identical genetic abnormality. Interestingly, the phenotype of the mother was normal; however, an abnormal ultrasound indicated choroid plexus cysts in the fetus. The mother’s elder child had syndromic congenital heart disease. Clinical manifestations of 22q11.2DS vary significantly even within a single family and are not definitively associated with a single genotype. The parents were informed that the infant may show symptoms of disease, leading to their decision to terminate the pregnancy. Parental analysis determined that fetus E3027 inherited a maternal 22q11.2 duplication. The mother was a healthy 23-year-old primipara. Although, according to the database, the clinical significance of this region associated with a 22q11.2 duplication is unclear, the parents decided to terminate the pregnancy. The parents of case P4666 also terminated the pregnancy when genetic analysis revealed the presence of VOUS. Based on the informed decisions, 11 parents opted to terminate their pregnancies. Alternatively, the parents of the dizygotic twin fetuses continued the pregnancy to full term even after the SNP array was VOUS because they were unable to conceive and opted for in vitro fertilization and embryo transfer to achieve a successful pregnancy.
Common phenotypes associated with 22q11.2 deletion/duplication syndrome include congenital cardiac defects, characteristic facial appearance, immune deficiency, palatal abnormalities, hypocalcemia, urogenital abnormalities, and a varying degree of cognitive deficits and intellectual disability [23, 24]. However, our fetal ultrasound results presented variable abnormalities with few normal results. We suggest that any hypothetical correlation between the abnormal fetus and size and location of the 22q11.2 alterations may be masked by other genetic or epigenetic modifying factors. SNP technology identified atypical breakpoints that do not correlate with known regions, thereby suggesting a greater level of genomic complexity underlying the 22q11.2 region.
Conclusions
We prenatally diagnosed 13 cases, among 2500 fetuses, with 22q11.2 copy number abnormalities. Prenatal analysis via SNP array allows for effective diagnosis of genetic abnormalities, delineation of differential diagnoses, characterization of a wide spectrum of associated malformations, and determination of the associations that exist between prenatal diagnosis and obstetrical outcomes. Approximately 0.5% of the fetuses harbored 22q11.2 copy number abnormalities. The most frequent ultrasound findings in fetuses with these abnormalities were cardiovascular malformations, followed by kidney malformations and isolated ultrasound markers.
References
Shaikh TH, Kurahashi H, Saitta SC, O'Hare AM, Hu P, Roe BA, Driscoll DA, Mcdonaldmcginn DM, Zackai EH, Budarf ML (2000) Chromosome 22-specific low copy repeats and the 22q11.2 deletion syndrome: genomic organization and deletion endpoint analysis. Hum Mol Genet 9(4):489–501
Botto LD, Kristin M, Fernhoff PM, Adolfo C, Karlene C, Rasmussen SA, Merritt RK, O'Leary LA, Lee-Yang W, Marsha E (2003) A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics 112(1):101–107
Robin NH, Shprintzen RJ (2005) Defining the clinical spectrum of deletion 22q11.2. J Pediatr 147(1):90–96
Mcdermid HE, Morrow BE (2002) Genomic disorders on 22q11. Am J Hum Genet 70(5):1077–1088
Berends MJ, Tan-Sindhunata G, Leegte B, Essen AJ (2001) Phenotypic variability of cat-eye syndrome. Genet Couns 12(1):23–34
Rowe LR, Lee JY, Rector L, Kaminsky EB, Brothman AR, Martin CL, South ST (2009) U-type exchange is the most frequent mechanism for inverted duplication with terminal deletion rearrangements. J Med Genet 46(10):694–702
Lopez-Rivera E, Liu YP, Verbitsky M, Anderson BR, Capone VP, Otto EA, Yan Z, Mitrotti A, Martino J, Steers NJ (2011) Genetic drivers of kidney defects in the diGeorge syndrome. N Engl J Med 376(8):742–754
Ensenauer RE, Adeyinka A, Flynn HC, Michels VV, Lindor NM, Dawson DB, Thorland EC, Lorentz CP, Goldstein JL, McDonald MT (2003) Microduplication 22q11.2, an emerging syndrome: clinical, cytogenetic, and molecular analysis of thirteen patients. Am J Hum Genet 73(5):1027–1040
De LRC, Jolyhélas G, Goldenberg A, Durand I, Laquerrière A, Ickowicz V, Saugierveber P, Eurin D, Moirot H, Diguet A (2010) The intrafamilial variability of the 22q11.2 microduplication encompasses a spectrum from minor cognitive deficits to severe congenital anomalies. Am J Med Genet A 140A(14):1608–1613
Winnie C, Inge S, Annick L (2010) Microduplication 22q11.2: a benign polymorphism or a syndrome with a very large clinical variability and reduced penetrance? Report of two families. Am J Med Genet A 146A(6):758–763
Fernández L, Nevado J, Santos F, Heine-Suñer D, Martinez-Glez V, García-Miñaur S, Palomo R, Delicado A, Pajares IL, Palomares M (2009) A deletion and a duplication in distal 22q11.2 deletion syndrome region. Clinical implications and review. BMC Med Genet 10(1):48
Lindsay EA, Goldberg R, Jurecic V, Morrow B, Carlson C, Kucherlapati RS, Shprintzen RJ, Baldini A (2010) Velo-cardio-facial syndrome: frequency and extent of 22q11 deletions. Am J Med Genet 57(3):514–522
Carlson C, Sirotkin H, Pandita R, Goldberg R, Mckie J, Wadey R, Patanjali SR, Weissman SM, Anyane-Yeboa K, Warburton D (1997) Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet 61(3):620–629
Alberti A, Romano C, Falco M, Calì F, Schinocca P, Galesi O, Spalletta A, Benedetto D, Di Fichera M (2010) 1.5 Mb de novo 22q11.21 microduplication in a patient with cognitive deficits and dysmorphic facial features. Clin Genet 71(2):177–182
Cai M, Huang H, Su L, Lin N, Wu X, Xie X, An G, Li Y, Lin Y, Xu L, Cao H (2018) Chromosomal abnormalities and copy number variations in fetal ventricular septal defects. Mol Cytogenet 11:58. https://doi.org/10.1186/s13039-018-0408-y
Hanemaaijer NM, Birgit SR, Gerben VDV, Trijnie D, Roel H, Essen AJ, Van V-K, Herkert JC, Gerkes EH (2012) Practical guidelines for interpreting copy number gains detected by high-resolution array in routine diagnostics. Eur J Hum Genet 20(2):161–165
Shaikh TH, O'Connor RJ, Pierpont ME, Mcgrath J, Hacker AM, Nimmakayalu M, Geiger E, Emanuel BS, Saitta SC (2007) Low copy repeats mediate distal chromosome 22q11.2 deletions: sequence analysis predicts breakpoint mechanisms. Genome Res 17(17):482–491
Kurahashi H, Tsuda E, Kohama R, Nakayama T, Masuno M, Imaizumi K, Kamiya T, Sano T, Okada S, Nishisho I (1997) Another critical region for deletion of 22q11: a study of 100 patients. Am J Med Genet A 72(2):180
Bittel DC, Yu S, Newkirk H, Kibiryeva N, Holt Iii A, Butler MG, Cooley LD (2009) Refining the 22q11.2 deletion breakpoints in DiGeorge syndrome by aCGH. Cytogenet Genome Res 124(2):113–120
Lo-Castro A, Galasso C, Cerminara C, El-Malhany N, Benedetti S, Nardone AM, Curatolo P (2009) Association of syndromic mental retardation and autism with 22q11.2 duplication. Neuropediatrics 40(03):137–140
Portnoï MF, Lebas F, Gruchy N, Ardalan A, Biran-Mucignat V, Malan V, Finkel L, Roger G, Ducrocq S, Gold F (2010) 22q11.2 duplication syndrome: two new familial cases with some overlapping features with DiGeorge/velocardiofacial syndromes. Am J Med Genet A 137A(1):47–51
Schinzel A, Schmid W, Fraccaro M, Tiepolo L, Zuffardi O, Opitz JM, Lindsten J, Zetterqvist P, Enell H, Baccichetti C (1981) The "cat eye syndrome": dicentric small marker chromosome probably derived from a no.22 (tetrasomy 22pter to q11) associated with a characteristic phenotype. Report of 11 patients and delineation of the clinical picture. Hum Genet 57(2):148–158
Sullivan K (2007) Chromosome 22q11.2 deletion syndrome: diGeorge syndrome/velocardiofacial syndrome. Immunol Allergy Clin N Am 28(2):353–366
Shprintzen RJ (2010) Velo-cardio-facial syndrome: 30 years of study. Dev Disabil Res Rev 14(1):3–10
Acknowledgements
This study was supported by the Key Special Projects of Fujian Provincial Department of Science and Technology (No. 2013YZ0002-1), Key Clinical Specialty Discipline Construction Program of Fujian (No. 20121589), and Fujian Provincial Natural Science Foundation (No. 2017J01238).
Funding
This study was supported by the Key Special Projects of Fujian Provincial Department of Science and Technology (No. 2013YZ0002-1), Key Clinical Specialty Discipline Construction Program of Fujian (No. 20121589), and Fujian Provincial Natural Science Foundation (No. 2017J01238).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
Approval was obtained from the Ethics Committee of Fujian Provincial Maternal and Child Health Hospital to conduct the research on prenatal diagnosis (FJPMCHH 2027).
Informed consent
Patients undergoing invasive prenatal diagnosis provided informed consent at the Fujian Provincial Maternal and Child Health Hospital.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cai, M., Lin, N., Su, L. et al. Prenatal diagnosis of 22q11.2 copy number abnormalities in fetuses via single nucleotide polymorphism array. Mol Biol Rep 47, 7529–7535 (2020). https://doi.org/10.1007/s11033-020-05815-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05815-7