
Abstract
The genetic structure of populations at the edge of species distribution is important for species adaptation to environmental changes. Small populations may experience non-random mating and differentiation due to genetic drift but larger populations, too, may have low effective size, e.g., due to the within-population structure. We studied spatial population structure of pedunculate oak, Quercus robur, at the northern edge of the species’ global distribution, where oak populations are experiencing rapid climatic and anthropogenic changes. Using 12 microsatellite markers, we analyzed genetic differentiation of seven small to medium size populations (census sizes 57–305 reproducing trees) and four populations for within-population genetic structures. Genetic differentiation among seven populations was low (Fst = 0.07). We found a strong spatial genetic structure in each of the four populations. Spatial autocorrelation was significant in all populations and its intensity (Sp) was higher than those reported in more southern oak populations. Significant genetic patchiness was revealed by Bayesian structuring and a high amount of spatially aggregated full and half sibs was detected by sibship reconstruction. Meta-analysis of isoenzyme and SSR data extracted from the (GD)2 database suggested northwards decreasing trend in the expected heterozygosity and an effective number of alleles, thus supporting the central-marginal hypothesis in oak populations. We suggest that the fragmented distribution and location of Finnish pedunculate oak populations at the species’ northern margin facilitate the formation of within-population genetic structures. Information on the existence of spatial genetic structures can help conservation managers to design gene conservation activities and to avoid too strong family structures in the sampling of seeds and cuttings for afforestation and tree improvement purposes.
Similar content being viewed by others


Introduction
Genetic variation within populations is crucial for their survival in the changing environment. Populations of forest trees harbor generally high levels of genetic diversity due to the life cycle characteristics leading to large effective population size (e.g., Hamrick 2004). Marginal populations, on the other hand, can be small, subject to severe bottlenecks, scattered amongst suitable microenvironments, and they receive less gene flow than the central populations. According to the leading-edge model of colonization, populations from the colonization front account for most range expansions whereas long-distance dispersal events are rare, which explains the decreased intrapopulation genetic variation and increased interpopulation differentiation observed in northern marginal areas (Eckert et al. 2008).
In the future, climate change may lead to a situation where marginal populations of European trees experience intensified selection, at the southern margin (rear edge), due to higher temperatures and more severe drought (Hampe and Petit 2005) and at the northern margin (leading edge), due to the less predictable weather and rapidly rising mean temperatures. Thus, during a range shift, marginal populations at the rear edge and at the leading-edge face diverse challenges and play different roles in the evolution of populations and species (Hampe and Petit 2005). Northern marginal populations in Europe are of special importance, as they will serve as potential gene pools for expanding populations in areas where the climate becomes more favorable to the species. However, genetic characteristics and dynamics of leading-edge populations at local scales are still insufficiently known (Logan et al. 2019). Understanding the genetic structure of leading-edge populations at different hierarchical levels (individual-subpopulation and subpopulation-population levels) can help to assess conservation priorities and allocation of resources within peripheral regions.
In addition to reduced genetic variation, marginal populations may have different spatial genetic structure (SGS) compared with central populations, i.e., non-random distribution of alleles within populations. SGS can be caused by many factors, e.g., population density, seed dispersal patterns, colonization and disturbance history, and spatio-temporal patterns in seed establishment (Vekemans and Hardy 2004). Limited gene flow (pollen and seed dispersal) is assumed to increase SGS in marginal and fragmented populations (e.g., Wang et al. 2011). Spatial genetic structure within a population promotes non-random mating, and thus, the population in question is not a panmictic population. Spatial aggregation of any type of related genotypes can cause biparental inbreeding, which in turn can lead to a reduced fitness due to inbreeding depression (Levin 1984; Campbell 1986; Latta et al. 1998; Stacy 2001). SGS may be expressed as continuous spatial autocorrelation or discontinuous aggregation of distinct genetic groups, i.e., genetic patchiness. In the autocorrelation type, trees growing at a close distance tend to be genotypically more similar than trees farther away, and the pattern is approximately the same in the whole population. In the case of genetic patchiness, nearby trees within a group are also genotypically similar, but there may be an unpredictable step in the relatedness at the edge of the patch.
Presently, environmental changes and increased human activity have led to forest fragmentation and declining population sizes. Trees play a major role in terrestrial ecosystems and thus the adaptive potential of forest tree populations is highly important. The goal of genetic conservation is to capture much genetic variation in the target populations to ensure their adaptive capacity. Generally, forest genetic resources in Europe are conserved in dynamic in situ units, which are natural populations designated for this purpose (Koskela et al. 2013). As conservation programs are bounded by the constraints of limited resources, it is essential to apply the most efficient measures for genetic conservation. The information on spatial genetic structure can help to maximize diversity in a conservation sample of fixed size and to avoid making erroneous estimates of population diversity (Epperson 1989). Also, in practical gene conservation work, such information is important to define a minimum distance between trees to be used for collecting seed or cuttings for afforestation and tree improvement purposes or for establishing ex situ collections.
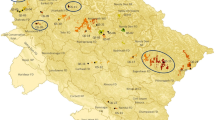
The populations of pedunculate oak (Quercus robur) in Finland are strongly fragmented and growing at the northern margin of the species’ European distribution. The distribution in Finland is presently limited to the southwestern part of the country (see Fig. 1). The cooling climate after the warm Atlantic period (4000 years before present) and the increasing demand for arable land (starting ca. 300 years ago) have led to the present distribution of small to medium–sized distinct populations (Alho 1990). The genetic diversity within Finnish pedunculate oak populations is slightly lower and among-population differentiation slightly higher than in more centrally located populations (analysis of 13 isozyme loci in 33 Finnish populations of Q. robur: He = 0.164, and Fst = 0.066, Vakkari et al. 2006, whereas analysis of 13 isozyme loci in seven populations of Q. robur from populations covering a much wider geographic area in the more central part of distribution He = 0.252 and Fst = 0.024; Zanetto et al. 1994).

Study populations of pedunculate oak (Quercus robur) in Southern Finland. Solid circles indicate populations for spatial analysis. Source of the background map: EUFORGEN 2009, www.euforgen.org
Climate change will shift bioclimatic envelopes for forest trees north-eastwards in Europe, enabling new suitable areas for oak in Finland (Buras and Menzel 2019). However, the success of colonization may depend on the genetic composition of founders—which originate from present marginal populations (Kremer et al. 2012). In this study, we investigate the genetic structure of pedunculate oak in leading-edge populations and pan-European trends in the genetic diversity of oak populations. Due to relatively recent (compared with the generation time of oak) fragmentation of oak populations, we expect (i) to see rather low interpopulation genetic differentiation. However, we expect that (ii) present population fragmentation may have an impact on intrapopulation distribution of genetic variability, causing strong spatial genetic structures within populations.
Materials and methods
Study populations
We sampled four distinct and isolated pedunculate oak stands Hästö, Näse, Torplandet, and Veitakkala in southern Finland (Fig. 1) for within-population spatial genetic analysis. Population sizes ranged from about 60 trees of reproductive age to about 300 trees. All trees with dbh > 10 cm (likely initiated flowering) were sampled for spatial genetic analysis. In Veitakkala, a narrow strip in the south-east corner was not sampled. The population in Hästö island is located on an island where fertile soil is restricted quite close to the sea level (0.5–15 m), and due to the land uplift process (for an overview of the process, see https://www.maanmittauslaitos.fi/en/research/interesting-topics/land-uplift), the maximum age of the population is less than 1000 years. The other three populations grow on higher ground, 20–45 m above sea level. When a forest fire during the Second World War destroyed parts of the Hästö stand, only 40–50 larger trees survived, and the present population is the result of a rapid re-growth. Three further populations were sampled to analyze population differentiation at a larger spatial scale (locations and other information in Table 1). All study stands have trees in several size classes and there is no indication that stands had been recently colonized.
DNA isolation and microsatellite amplification
DNA extraction from leaf samples and PCR conditions are described in Pohjanmies et al. (2015). Samples were genotyped for 15 microsatellite loci: ssrQpZAG9, ssrQpZAG15, ssrQpZAG1/5, ssrQpZAG16, ssrQpZAG36, ssrQpZAG104, ssrQpZAG110, ssrQrZAG7, ssrQrZAG11, ssrQrZAG87, ssrQrZAG101, ssrQrZAG108, ssrQrZAG112, MSQ4, and MSQ13 (Dow et al. 1995; Steinkellner et al. 1997; Kampfer et al. 1998). Three loci (ssrQpZAG16, ssrQpZAG1/5, and MSQ4) were excluded because the loci had odd and even alleles (resulting in a high frequency of scoring errors) or high frequency of null alleles (MSQ4) in at least one of the study populations.
Statistical analysis
Three different approaches were applied to analyze the spatial genetic structure of the populations: spatial autocorrelation analysis of genotypic relatedness, Bayesian group assignment analysis, and kinship reconstruction analysis. In addition, genetic diversity was investigated within and among populations.
Genetic variation
Genetic variation was explored estimating standard genetic parameters allelic richness (AR), observed (Ho), and expected (He) heterozygosity and inbreeding coefficient (Fis) for each population. Statistical significance of Fis was determined by randomization tests (1000 permutations). Genetic differentiation among populations was described by Fst-statistic. All calculations were performed using software FSTAT ver 2.9.4 (Goudet 2002).
Spatial genetic structure
Autocorrelation analysis
Analysis of spatial genetic structure expressed as spatial autocorrelation was performed using Spagedi v1.5a (Hardy and Vekemans 2002). The kinship between individual trees was estimated according to Loiselle et al. (1995). The distance classes were set at 10-m intervals up to 200 m, for longer distances class width was set separately in each population to keep the number of tree pairs per distance class at approximately constant level. The average number of pairs per distance class was 50 pairs in Näse, 130 pairs in Torplandet, 1100 pairs in Hästö, and 450 pairs in Veitakkala.
The intensity of spatial autocorrelation/SGS was measured by the Sp statistic. Sp is computed as Sp = bF/(F1–1), where bF is the regression slope of the kinship estimator Fij on geographical distances (logarithmic scale), and F1 is the average kinship coefficient between individuals of the first distance class. The standard error (SE) of Sp is estimated by the standard error of bF obtained by jackknifing over loci (Hardy et al. 2006; Jump et al. 2012). The statistical significance of F1 and bF was tested based on 1000 permutations of individual locations among individuals.
Bayesian grouping
Bayesian group assignment analysis was used as implemented in Structure 2.3.4 (Pritchard et al. 2000; Falush et al. 2003) in order to detect genetic structures within and among populations. To analyze the among-population structure, 30 trees were subsampled from larger populations (Ajo, Hästö, Näse, Torplandet, Veitakkala; random sampling without replacement) to achieve even sample sizes while accommodating for the size of two smallest samples (Kaapinmäki, Kylämäki), as uneven sample sizes may yield biased results (Puechmaille 2016). The hierarchical within-population analysis was started with full data for each population separately and continued with repeated analysis for each detected cluster separately until no more structure could be detected. Structure simulations were run for 100,000 repetitions with a burn-in phase of 100,000 repetitions and 30 iterations. The value of K (assumed number of groups) was set from 2 to 20 in large populations and from 2 to 10 in smaller subsets. Further settings applied were correlated allele frequency model and the admixture model. Online service Structure Harvester (Earl and von Holdt 2012) was applied to produce deltaK (Evanno et al. 2005) and meanLnProb–graphs and software CLUMPP (Jakobsson and Rosenberg 2007) with GREEDY option to calculate average Q-matrices over iterations for the group assignment of the samples. Each tree was then assigned to a single group in which it showed the highest average Q-score following the standard procedure (Balkenhol et al. 2014). The final level of hierarchical analysis was judged on the basis of the relative change in LnProb and the distribution of Q-scores. The group was considered final, when the graph of Lnprob was level or decreasing or the distribution of Q-scores did not indicate a dominant group even for a fraction of the samples.
Spatial aggregation
L-functions were estimated for all groups in the first and the final level of the hierarchical analysis. For r ≥ 0, L(r) is a monotonic function of the cumulative number of further trees within distance r from a typical tree (Illian et al. 2008, sec. 4.3). If the spatial pattern of trees is completely random, then the expected value of L(r) is equal to r, and values greater than r indicate spatial aggregation (clustering) for at least some distances between 0 and r. Within-group aggregation was contrasted to the overall aggregation in the population with the standard Monte Carlo approach (Illian et al. 2008, sec. 7.4): The original estimates Lg(r) for each group g were compared with the mean values L∗g(r) of estimates L∗g, t(r), t = 1, …, T, obtained from T = 2500 random permutations of the group labels between all trees in the population. Values of Lg(r) greater than L∗g(r) indicate stronger aggregation in group g trees than in a random subset of the same number of trees from the population. Statistical significance of the difference between Lg(r) and L∗g(r) was assessed with a one-tailed global rank envelope test (Myllymäki et al. 2017), where only positive values of Lg(r) − L∗g(r) were considered significant. The estimates of the L-functions were computed using spatstat (Baddeley et al. 2015) and the global envelope tests were implemented using package GET (Myllymäki and Mrkvička 2019), both in the R environment (R Core Team 2019).
Kinship analysis
To analyze pedigree relationships within populations, we used the software Colony 2.0 based on the methodology described in Wang (2004). Parameter settings applied were male and female polygamous, inbreeding allowed, and monoecious diploid species as basic biological descriptors. The run length was set to medium. In the case of sibship size prior, we followed the recommendations of the Colony manual (pp. 14–15) of a weak (instead of no) prior and average size of 1 for both maternal and paternal sibships to reduce false sibship assignments. The probability of finding the paternal or maternal parent in the population was set to 0.5 for both sexes, which we assume to describe the situation in fragmented Finnish oak populations reasonably well. Smallest sampled trees (dbh 10–15 cm, just reached reproductive size) were excluded from the list of candidate parents so that they could not appear as possible parents of the larger (dbh > 20 cm) and supposedly older trees. In practice, this did not have any effect on the analysis outcome.
The Monte Carlo test for assessing whether sib pairs are located significantly closer to each other than to other trees was based on random permutations of trees between all observed locations within the population. Random permutations of trees can also be viewed as random allocations of new coordinates to each tree from all observed locations in the population. In such permutations, distances between pairs of trees are independent of their genetic relationship. The distributions of distances over the permutations only depend on the distribution of all pairwise distances between the trees in the population and on the number of full sib, half sib, and unrelated pairs.
We also analyzed whether populations showed signs of having experienced bottleneck events using BOTTLENECK version 1.2.02 (Piry et al. 1999). The inference was based on the Wilcoxon sign test after analysis under the single-step (SMM) and two-phase (TPM) mutation model with 90% single-step mutations. In addition, mode-shift analysis was performed.
Results
In each of the four populations analyzed, spatial autocorrelation was significant, the average kinship being significantly above zero at short distances with a trend towards zero and negative values in longer distances (Fig. 2). The average kinship of the first distance interval, F1, is quite variable ranging from 0.045 in Veitakkala to 0.165 in Torplandet. The high F1-value in Torplandet is followed by a steep decline. Due to the different population sizes, the number of trees per distance class varies among populations, and consequently, also the width of confidence limits varies among populations. The zigzag pattern of the line of the observed relatedness values in Näse is regarded as random variation resulting from the fairly low number of trees per distance class. The intensity of SGS, Sp, ranges from 0.0193 to 0.0283 (Table 2).

Autocorrelograms of kinship coefficient (Loiselle et al. 1995) in populations of pedunculate oak in Näse (a), Torplandet (b), Hästö (c), and Veitakkala (d). Observed within-class average of the kinship coefficient is marked by blue diamonds, lower and upper 95% confidence limits by red squares and green triangles, respectively. X-axis value is the realized distance class mean
Structure analysis at the first level reveals two groups in Hästö, Näse, and Veitakkala, and four groups in Torplandet. The final number of groups after hierarchical analysis was 13 in Hästö, 12 in Veitakkala, eight in Torplandet, and five in Näse (Fig. 3a–d, Table 2). The assignment of the trees into the groups was clear with a few exceptions (Q-score bar plots in Fig. 4a–d). The spatial coverage of the groups is variable; extremes are found in Torplandet where group 30 (yellow in Fig. 3b) spreads over the whole stand area, whereas other groups occupy much smaller areas, e.g., group 42 (dark blue in Fig. 3b) being limited to a small patch of 25 by 75 m in the eastern corner of the stand. Groups in the highest hierarchy level were significantly aggregated in Veitakkala and also in other stands except in one case level group (Fig. S1, p < 0.05). The spatial aggregation of the lowest level groups is statistically significant for 10 groups in Hästö, 7 in Veitakkala, 6 in Torplandet, and 2 in Näse (Figs. 3 and S2).

Groups identified by Structure software (Falush et al. 2003) in Näse (a, 5 groups), Torplandet (b, 8 groups), Veitakkala (c, 12 groups), and Hästö (d, 13 groups). The groups where trees are aggregated statistically significantly (p < 0.05) are indicated by an asterisk. The L-functions of aggregation tests and exact p values for each group are shown in Supplementary Data (Figs. 1 and 2)

The relationship of Structure 2.3.4 (Pritchard et al. 2000; Falush et al. 2003) groups and Colony 2.0 (Wang 2004) sibship dyads in four pedunculate oak populations. In the dyad, plot trees are in the same order (matching the bar plot) on vertical and horizontal axis. Bar plots show Structure group assignments after first level analysis (left) and at the lowest level of hierarchical analysis (right). The bar plot illustrating final grouping is a composite; black lines denote boundaries of preceding level groups, second level in Hästö and Veitakkala and first level in Näse and Torplandet. Half sib pairs detected by Colony are marked by green squares above the diagonal and full sib pairs by red squares below the diagonal. Marker size is relative to the probability of the sib pair. Structure group boundaries are marked with the black lines in the sibship triangle plot. a Näse, b Torplandet, c Hästö, and d Veitakkala
The sibship reconstruction analysis yielded sib pairs at high frequency in Näse (0.252) and at lower frequencies in the other stands: Hästö 0.04, Torplandet 0.051, and Veitakkala 0.058. In line with the significant spatial autocorrelation, the sibs are located significantly closer to each other than unrelated/more distantly related trees (Fig. 5). The reconstructed sibship structure matches with the Structure group pattern (Fig. 4); the trees in the full-sib pairs are within the same Structure group with few exceptions, and also half-sib pairs cluster within the same group very strongly in some cases (e.g., Torplandet groups 30 and 42). The pattern of half-sib pairs gives also a refining view on the group structure of populations, as two groups identified by Structure may form one dense cluster of half sibs covering both of them; examples of this type are groups 11 and 12 in Näse, 231 and 232 in Hästö, and in Veitakkala groups 121 and 122 and also groups 221 and 222, linked with a few trees from group 213 (Fig. 4). The pedigree status of non-sib pairs included in the groups remains unresolved and they may be either virtually unrelated trees or more distant relatives not discernible in the kinship reconstruction analysis.

Average distance between full sibs, half sibs, and unrelated (or more distantly related) pedunculate oaks (colored bars) and boxplots of the distributions of these average distances over 1000 random permutations of trees between the observed locations
Genetic variability within populations was high, allelic richness (Ar, sample size 25) ranging from 6.7 to 12.2 and with expected heterozygosity (He) from 0.72 to 0.81 (Table 1). Highest estimates for Ar and He were found in the medium-sized stand of Torplandet, the two lowest He estimates from the two smallest populations. Heterozygote deficiency (positive Fis) was found only in Torplandet; otherwise, Fis was negative (in Hästö practically zero), indicating an excess of heterozygotes in all other populations (Table 1). At the lowest hierarchical level, heterozygote deficiency is found in only 2 groups (Hästö group 131 and Torplandet group 30). Highest heterozygote excess estimates for groups was found in Näse (Fis = − 0.405 and Fis = − 0.247 in groups 13 and 12, respectively), in Veitakkala (Fis = − 0.324 and Fis = − 0.291 in groups 221 and 124, respectively), and in Hästö (Fis = − 0.287 in group 232) (Table S1 in supplementary data). These groups are characterized by a high frequency of full sibs with one exception only, Veitakkala group 124 (Fig. 4).
Genetic differentiation among populations was modest (Fst = 0.07), pairwise Fst estimates ranging from 0.027 (Kaapinmäki-Torplandet) to 0.13 (Kylämäki-Näse) without any indication of isolation by distance structure. Differentiation among the groups within populations ranged from Fst = 0.09 to 0.13 (Table 2). Structure analysis of the seven populations did not reveal geographical patterns in genetic differentiation (Fig. 6 structure analysis, Fig. 1 for population locations). Nearby eastern populations Näse and Torplandet did not group together, also in the west, Kylämäki and Kaapinmäki populations were distinct from each other.

Clustering results of pedunculate oaks from seven Finnish populations. Results are shown for K values 3 and 5 applied in the Structure analysis
The bottleneck analysis did not reveal any signs for any recent reduction in population size. Wilcoxon test results were non-significant for all populations under both mutation models, SMM and TPM, and the mode-shift analysis also showed normal L-shaped distributions in all populations.
Discussion
We found strong within-population spatial genetic structure in the four studied pedunculate oak populations, expressed as spatial autocorrelation and spatially aggregated genetic groups (genetic patchiness), some of the groups including considerable amounts of full and half sibs. This kind of genetic patchiness is to our knowledge the only reported case for any oak species. Hampe et al. (2010), working with a French Q. robur population, report explicitly that they did not find genetic groups (patchiness); elsewhere, the existence of genetic groups is not reported. In other tree species, genetic patchiness has been reported only in few cases: e.g., in forest trees have so far been reported; e.g., Paffetti et al. (2012) found clear patchiness in an old-growth population of beech, and Troupin et al. (2006) found patchiness in a Pinus halepensis population. Genetic groups have been found within Populus trichocarpa (Slavov et al. 2010) and beech (Piotti et al. 2013) populations but without spatial aggregation. It seems to us that the phenomenon is either quite rare in forest tree populations or overlooked in spatial genetic studies. Interestingly, “chaotic genetic patchiness” is a common observation in the populations of marine organisms with potentially high dispersal capacity (Selwyn et al. 2016), a case which is to some degree analogous to trees with potentially high gene dispersal capacity (due, e.g., to wind-pollination). On the other hand, spatial genetic autocorrelation seems to be common in oak and beech populations, both wind-pollinated species but with limited seed dispersal (Hampe et al. 2010; Jump et al. 2012; Lind-Riehl and Gailing 2015; Streiff et al. 1998; Vornam et al. 2004).
The level of kinship in the genetic groups reveals considerable variation; some groups consist almost exclusively of full and half sibs (e.g., Näse 11 and 12, Torplandet group 20), whereas in other groups the frequency of sibs is quite low (e.g., Torplandet group 30). However, the low sib frequency does not mean that the trees in such a group are completely unrelated, as the kinship reconstruction process does not recognize more distant relatives than half sibs. In some cases, Structure software seems to be very sensitive and splits tight sibling networks into different groups, e.g., in Näse (groups 11 and 12) and in Veitakkala (groups 121 and 122). Hence, the assignment of individual trees into different groups is not necessarily an indication of their unrelatedness. Still further, several trees have their (half) sib pairs in another group; examples are seen e.g. in Näse population, where three trees assigned to groups 13 and 14 have their main half-sib partners in the group 12. In Veitakkala, four trees in group 213 have most of their half siblings in group 221. These cases highlight three topics: First, group assignment is not a universal fact and, e.g., with a larger (or smaller) set of loci, assignment in the first grouping could be different, leading to a quite different assignment of some trees at the lowest hierarchical level. Second, while Bayesian grouping is sensitive to the sib status of the trees, it is also sensitive to the more complex pedigree relations. Third and maybe biologically most important, the groups are connected to the other groups, being part of an interbreeding population. These group connections are probably visualized in the variability within Q-score bars observed even at the lowest hierarchical level of grouping (Fig. 4).
There is an indication of a more pronounced spatial genetic structure in the Finnish than in populations in more central part of distribution. The intensity of spatial autocorrelation SGS, Sp, ranges from 0.0193 to 0.0283 in our data and is in all populations higher than reported values for Q. robur: Sp = 0.0034 in Romania (Curtu et al. 2015) and Sp = 0.005 in France (Hampe et al. 2010). The number of populations is low and the estimates could be affected by the fact that different loci are applied in the different studies, but we suggest that there is an indication of stronger SGS in Finnish than populations in central areas of the distribution. Similar differences between core and marginal populations have been observed also in two North American tree species: spatial genetic structure was stronger in marginal than core populations in Sitka spruce (Gapare and Aitken 2005) and in Eastern white cedar (Pandey and Rajora 2012).
Genetic differentiation among studied Finnish populations of Q. robur is modest, Fst = 0.076, but higher than reported values in populations in central part of a distribution, Fst = 0.039 (Neophytou et al. 2010) and Fst = 0.051 (Ballian et al. 2010). A higher differentiation estimate Fst = 0.12 has been obtained at the southern edge of the distribution (Moracho et al. 2016). With isoenzyme markers, the difference between Finnish populations (Fst = 0.066, He = 0.164) and central populations (Fst = 0.024, He = 0.252) was even more pronounced (Vakkari et al. 2006). Thus, there is an indication of higher genetic drift in the northern range-edge populations, possibly a joint effect of lower population sizes and a lower level of gene flow between populations, leading to increased effects of genetic drift as compared with populations from the range center. The decreasing genetic variability in the northern oak populations can be observed also over a larger geographic scale assessment. We extracted metadata of pedunculate oak from the GD2 database (http://gd2.pierroton.inra.fr/) for estimates of genetic variation (expected heterozygosity, effective number of alleles) in microsatellite and isoenzyme markers and plotted those against latitude. There was a declining trend of variability towards the north (Fig. 7) that may suggest for long-term effects of small population sizes and genetic drift in the northern peripheral populations compared with the central populations. This trend and higher Fst among northern populations are consistent with the central-marginal hypothesis (e.g., Eckert et al. 2008).

Latitudinal trends of microsatellite (SSR) and isoenzyme genetic diversity in pedunculate oak populations based on the data in GD2 database (http://gd2.pierroton.inra.fr/). Coefficient of determination R2 was obtained as the square of Pearson correlation coefficient of expected heterozygosity and effective number of alleles on latitude for both microsatellite and isoenzyme data
The conditions leading to lower genetic diversity and higher differentiation in the northern marginal populations compared with the southern and central populations of Q. robur may also promote the stronger SGS in the North. The spatial genetic structure is a consequence of limited gene flow and limited seed dispersal in most cases of tree species (e.g., Wang et al. 2011), but restricted pollen flow also has an effect to the same direction. Geographical fragmentation is expected to decrease pollen flow among populations, if the distance between stands is large enough compared with the range of pollen dispersal. When the amount of incoming pollen is limited, non-random mating within a stand may be more common. Consequently, more pronounced spatial genetic structure is expected in the fragmented populations than in the area of continuous distribution. Supporting this hypothesis, a higher level of SGS has been found in fragmented populations (Browne and Karubian 2018; De-Lucas et al. 2009). The critical distance is difficult to judge in wind-pollinated trees, as the long-distance pollen dispersal, in particular the tail of leptokurtic dispersal distribution, is difficult to estimate. Mean within-stand pollen dispersal estimates for sessile and pedunculate oak reported by Chybicki and Burczyk (2010) and Moracho et al. (2016) are below 500 m, but there are observations of long-distance pollination events up to 10 and even 80 km (Buschbom et al. 2011, Moracho et al. 2016; for an overview, see Gerber et al. (2014)). Populations of pedunculate oak in our study are relatively isolated, and distance from the study populations to the nearest other stand ranges from 1.5 km (between study populations Näse-Torpland and Ajo-Kaapinmäki) to 14 km. These distances are well above the within-population pollen dispersal estimates but well below the observed long-distance events. Long-distance pollen movement in Finland is affected also by the Gulf of Finland, which increases the isolation from the more continuous oak distribution in Estonia and further Central Europe to more than 70 km. Although these distances do not rule out the long-distance pollination, we feel justified in assuming that the amount of immigrating pollen is low when compared to the local pollen cloud. The reduced genetic diversity and increased genetic differentiation among northern populations (see above) give some support to this assumption. Hence, we suggest that stronger SGS in our stands than in Central Europe is at least partly a result of limited pollen flow.
The existence of spatially aggregated genetic groups (genetic patchiness) within a population is a challenge for genetic conservation measures and also an early indicator of detrimental population fragmentation (Wang et al. 2011). In conservation units with aggregated structure, the census size gives much too optimistic estimates of the ability of the populations to retain genetic variability, and when fragmentation is emerging, measures are required to prevent the rapid loss of genetic diversity (Wang et al. 2011). Genetic patches, as in our case, can include a considerable amount of full and half sibs and, hence, pollination between nearby trees, which may result in biparental inbreeding and eventually inbreeding depression. The spatial genetic structure in local stands has to be considered when collecting forest reproductive material, for research or for ex situ conservation. Our results stress that without explicit knowledge of the structures in a stand, the basic strategy to extend sampling evenly spaced over the whole stand is the correct one, but does not ensure the unrelatedness of sampled trees. Similarly, when the natural reproduction of a target species in mixed stands requires opening the forest, the gaps need to be distributed evenly throughout the stand. In the analysis of genetic patchiness, the two different approaches, Bayesian structuring, and sibship reconstruction complement each other, as structuring does not infer pedigree relatedness and sibship reconstruction, on the other hand, does not identify more distant relations than sibs and parent-offspring. The recognition of genetic patches may be especially important in the marginal populations. Thus, further studies not only on spatial genetic structures but also on genetic patchiness are vital when assessing genetic viability of marginal populations.
References
Alho P (1990) Suomen metsittyminen jääkauden jälkeen. Summary: the history of forest development in Finland after the last ice age. Silva Fennica 24:9–19
Baddeley A, Rubak E, Turner R (2015) Spatial point patterns: methodology and applications with R. Chapman and Hall/CRC Press, London
Balkenhol N, Holbrook J, Onorato D, Zager P, White C, Waits L (2014) A multi-method approach for analyzing hierarchical genetic structures: a case study with cougars Puma concolor. Ecography 37:552–563
Ballian D, Belletti P, Ferrazzini D, Bogunic F, Kajba D (2010) Genetic variability of Pedunculate Oak (Quercus robur L.) in Bosnia and Herzegovina. Period Biol 112:353–362
Browne L, Karubian J (2018) Habitat loss and fragmentation reduce effective gene flow by disrupting seed dispersal in a neotropical palm. Mol Ecol 27:3055–3069
Buras A, Menzel A (2019) Projecting tree species composition changes of european forests for 2061–2090 under RCP 4.5 and RCP 8.5 scenarios. Front Plant Sci. https://doi.org/10.3389/fpls.2018.01986
Buschbom J, Yanbaev Y, Degen B (2011) Efficient long-distance gene flow into an isolated relict oak stand. J Hered 102:464–472
Campbell RB (1986) The interdependence of mating structure and inbreeding depression. Theor Pop Biol 30:232–244. https://doi.org/10.1016/0040-5809(86)90035-3
Chybicki I, Burczyk J (2010) Realized gene flow within mixed stands of Quercus robur L. and Q. petraea (Matt.) L. revealed at the stage of naturally established seedling. Mol Ecol 19:2137–2151
Curtu AL, Craciunesc I, Enescu CM, Vidalis A, Sofletea N (2015) Fine-scale spatial genetic structure in a multi-oak-species (Quercus spp.) forest. iForest 8:324–332. https://doi.org/10.3832/ifor1150-007
De-Lucas AI, Gonzalez-Martinez SC, Vendramin GG, Hidalgo E, Heuertz M (2009) Spatial genetic structure in continuous and fragmented populations of Pinus pinaster Aiton. Mol Ecol 18:4564–4576. https://doi.org/10.1111/j.1365-294X.2009.04372.x
Dow BD, Ashley MV, Howe HF (1995) Characterization of highly variable (GA/CT)n microsatellites in the bur oak, Quercus macrocarpa. Theor Appl Genet 91:137–141
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol Ecol 17:1170–1188. https://doi.org/10.1111/j.1365-294X.2007.03659.x
Epperson BK (1989) Spatial patterns of genetic variation within plant populations. In: AHD B, Clegg MT, Kahler AL, Weir BS (eds) Population Genetics and Germplasm Resources in Crop Improvement. Sinauer Associates, Sunderland, pp 229–253
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164:1567–1587
Gapare WJ, Aitken S (2005) Strong spatial genetic structure in peripheral but not core populations of Sitka spruce [Picea sitchensis (Bong.) Carr.]. Mol Ecol 14:2659–2667
Gerber S, Chadœuf J, Gugerli F, Lascoux M, Buiteveld J, Cottrell J, Dounavi A, Fineschi S, Forrest LL, Fogelqvist J, Goicoechea PG, Jensen JS, Salvini D, Vendramin GG, Kremer A (2014) High rates of gene flow by pollen and seed in oak populations across Europe. PLoS ONE 9(1):e85130. https://doi.org/10.1371/journal.pone.0085130
Goudet J (2002) FSTAT version 2.9.4. A program to estimate and test gene diversities and fixation indices. http://www2.unil.ch/popgen/softwares/fstat.htm
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467
Hampe A, El Masri L, Petit RJ (2010) Origin of spatial genetic structure in an expanding oak population. Mol Ecol 19:459–471
Hamrick JL (2004) Response of forest trees to global environmental changes. For Ecol Manag 197:323–335
Hardy OJ, Vekemans X (2002) Spagedi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hardy OJ, Maggia L, Bandou E, Breyne P, Caron H, Chevallier MH, Doligez A, Dutech C, Kremer A, Latouche-Hallé C, Troispoux V, Veron V, Degen B (2006) Fine-scale genetic structure and gene dispersal inferences in 10 Neotropical tree species. Mol Ecol 15:559–571
Illian J, Penttinen A, Stoyan H, Stoyan D (2008) Statistical analysis and modelling of spatial point patterns. Wiley, Chichester
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jump AS, Rico L, Coll M, Peñuelas J (2012) Wide variation in spatial genetic structure between natural populations of the European beech (Fagus sylvatica) and its implications for SGS comparability. Heredity 108:633–639
Kampfer S, Lexer C, Glössl J, Steinkellner H (1998) Characterization of (GA)n microsatellite loci from Quercus robur. Hereditas 129:183–186
Koskela J, Lefèvre F, Schueler S, Kraigher H, Olrik DC, Hubert J, Longauer R, Bozzano M, Yrjänä L, Alizoti P, Rotach P, Vietto L, Bordács S, Myking T, Eysteinsson T, Souvannavong O, Fady B, de Cuyper B, Heinze B, von Wühlisch G, Ducousso A, Ditlevsen B (2013) Translating conservation genetics into management: pan-European minimum requirements for dynamic conservation units of forest tree genetic diversity. Biol Conserv 157:39–49
Kremer A, Ronce O, Robledo-Arnuncio JJ, Guillaume F, Bohrer G, Nathan R, Bridle JR, Gomulkiewicz R, Klein EK, Ritland K, Kuparinen A, Gerber S, Schueler S (2012) Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecol Lett 15:378–392. https://doi.org/10.1111/j.1461-0248.2012.01746.x
Latta RG, Linhart YB, Fleck D, Elliot M (1998) Direct and indirect estimates of seed versus pollen movement within a population of ponderosa pine. Evolution 52:61–67. https://doi.org/10.1111/j.1558-5646.1998.tb05138.x
Levin DA (1984) Inbreeding depression and proximity-dependent crossing success in Phlox drummondii. Evolution 38:116–127. https://doi.org/10.1111/j.1558-5646.1984.tb00265.x
Lind-Riehl J, Gailing O (2015) Fine-scale spatial genetic structure of two red oak species, Quercus rubra and Quercus ellipsoidalis. Plant Syst Evol 301:1601–1612
Logan S, Phuekvilai P, Sanderson R, Wolff K (2019) Reproductive and population genetic characteristics of leading edge and central populations of two temperate forest tree species and implications for range expansion. For Ecol Manag 433:475–486
Loiselle B, Sork V, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Moracho E, Moreno G, Jordano P, Hampe A (2016) Unusually limited pollen dispersal and connectivity of pedunculate oak (Quercus robur) refugial populations at the species’ southern range margin. Mol Ecol 25:3319–3331. https://doi.org/10.1111/mec.13692
Myllymäki M, Mrkvička T (2019) GET: Global envelopes in R. arXiv:1911.06583
Myllymäki M, Mrkvicka T, Grabarnik P, Seijo H, Hahn U (2017) Global envelope tests for spatial processes. J R Stat Soc B 79:381–404. https://doi.org/10.1111/rssb.12172
Neophytou C, Aravanopoulos FA, Fink S, Dounavi A (2010) Detecting interspecific and geographic differentiation patterns in two interfertile oak species (Quercus petraea (Matt.) Liebl. and Q. robur L.) using small sets of microsatellite markers. For Ecol Manag 259:2026–2035
Paffetti D, Travaglini D, Buonamici A, Nocentini S, Vendramin GG, Giannini R, Vettori C (2012) The influence of forest management on beech (Fagus sylvatica L.) stand structure and genetic diversity. For Ecol Manag 284:34–44
Pandey M, Rajora OP (2012) Higher fine-scale genetic structure in peripheral than in core populations of a long-lived and mixed-mating conifer - eastern white cedar (Thuja occidentalis L.). BMC Evol Biol 12:48 http://www.biomedcentral.com/1471-2148/12/48
Piotti A, Leonardi S, Heuertz M, Buiteveld J, Geburek T, Gerber S, Kramer K, Vettori C, Vendramin GG (2013) Within-population genetic structure in beech (Fagus sylvatica L.) stands characterized by different disturbance histories: does forest management simplify population substructure? PLoS One 8:e73391
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Heredy 90:502–503
Pohjanmies T, Tack AJM, Pulkkinen P, Elshibli S, Vakkari P, Roslin T (2015) Genetic diversity and connectivity shape herbivore load within an oak population at its range limit. Ecosphere 6:101. https://doi.org/10.1890/ES14-00549.1
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puechmaille SJ (2016) The program Structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627. https://doi.org/10.1111/1755-0998.12512
R Core Team (2019) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna https://www.R-project.org/
Selwyn JD, Hogan JD, Downey-Wall AM, Gurski LM, Portnoy DS, Heath DD (2016) Kin-Aggregations explain chaotic genetic patchiness, a commonly observed genetic pattern, in a marine fish. PLoS ONE 11(4):e0153381. https://doi.org/10.1371/journal.pone.0153381
Slavov GT, Leonardi S, Adams WT, Strauss SH, DiFazio SP (2010) Population substructure in continuous and fragmented stands of Populus trichocarpa. Heredity 105:348–357
Stacy EA (2001) Cross-fertility in two tropical tree species: evidence of inbreeding depression within populations and genetic divergence among populations. Am J Bot 88:1041–1051
Steinkellner H, Fluch S, Turetschek E, Lexer C, Streiff R, Kremer A, Burg K, Glössl J (1997) Identification and characterization of (GA/CT)n - microsatellite loci from Quercus petraea. Plant Mol Biol 33:1093–1096
Streiff R, Labbe T, Bacilieri R, Steinkellner H, Glössl J, Kremer A (1998) Within-population genetic structure in Quercus robur L. and Quercus petraea (Matt.) Liebl. assessed with isozymes and microsatellites. Mol Ecol 7:317–328
Troupin D, Nathan R, Vendramin GG (2006) Analysis of spatial genetic structure in an expanding Pinus halepensis population reveals development of fine-scale genetic clustering over time. Mol Ecol 15:3617–3630
Vakkari P, Blom A, Rusanen M, Raisio J, Toivonen H (2006) Genetic variability of fragmented stands of pedunculate oak (Quercus robur) in Finland. Genetica 127:231–241
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13:921–935
Vornam B, Decarli N, Gailing O (2004) Spatial distribution of genetic variation in a natural beech stand (Fagus sylvatica L.) based on microsatellite markers. Conserv Genet 5:561–570
Wang J (2004) Sibship reconstruction from genetic data with typing errors. Genetics 166:1963–1979
Wang R, Compton SG, Chen XY (2011) Fragmentation can increase spatial genetic structure without decreasing pollen-mediated gene flow in a wind-pollinated tree. Mol Ecol 20:4421–4432. https://doi.org/10.1111/j.1365-294X.2011.05293.x
Zanetto R, Roussel G, Kremer A (1994) Geographic variation of inter-specific differentiation between Quercus robur L. and Quercus petraea (Matt.) Liebl. Forest Genet 1:111–123
Acknowledgments
Open access funding provided by Natural Resources Institute Finland (LUKE). Discussions with Antoine Kremer and Santiago Gonzalez-Martinez on oak genetics during the study have been very helpful. We also thank Annukka Korpijaakko for the laboratory work; Esa Ek, Elvi Pääkkönen, and Leena Yrjänä for the efficient field work; and Sonja Kujala for the valuable comments on the earlier version of the manuscript. Comments by Charlotte Angove improved the contents and the language of the manuscript. Insightful comments by Felix Gugerli and two anonymous reviewers helped us greatly to improve the MS.
Funding
Financial support from FORGER (FP7-KBBE-289119) and Luke is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Data archiving
Data have been stored in (GD)2 database (http://gd2.pierroton.inra.fr/).
Additional information
Communicated by F. Gugerli
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1079 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vakkari, P., Rusanen, M., Heikkinen, J. et al. Patterns of genetic variation in leading-edge populations of Quercus robur: genetic patchiness due to family clusters. Tree Genetics & Genomes 16, 73 (2020). https://doi.org/10.1007/s11295-020-01465-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11295-020-01465-9