
Abstract
Intellectual disability is a neurodevelopmental disorder in which genetic, epigenetic and environmental factors are involved. In consequence, the determination of its etiology is usually complex. Though many countries have migrated from conventional cytogenetic analysis to chromosomal microarrays as the first-tier genetic test for patients with this condition, this last technique was implemented in our country a few years ago. We report on the results of the implementation of chromosomal microarrays in a cohort of 133 patients with intellectual disability and dysmorphic features, normal karyotype and normal subtelomeric MLPA results in an Argentinean public health institution. Clinically relevant copy number variants were found in 12% of the patients and one or more copy number variants classified as variants of uncertain significance were found in 5.3% of them. Although the diagnostic yield of chromosomal microarrays is greater than conventional cytogenetics for these patients, there are financial limitations to adopt this technique as a first-tier test in our country, especially in the public health system.
Similar content being viewed by others


Introduction
Intellectual disability (ID) is a neurodevelopmental disorder characterized by significant impairment in cognitive functions and in one or more adaptive behaviors with onset before 18 years of age. There are genetic, epigenetic and environmental factors that can affect the development and functioning of the nervous system. In consequence, the determination of the ID etiology is usually complex and requires a multidisciplinary approach.
The prevalence of ID varies depending on the criteria used in the diagnosis as well as on the population's socioeconomic level. In developed countries, the prevalence is thought to be between 2 and 3% but it is higher in countries of lower socioeconomic status [1]. Severe ID is relatively stable and seems to be caused mostly by genetic factors in about 25% to 50% of ID patients [2]. This stresses the importance of genetic assessment for the diagnosis and prevention of the recurrence of ID, being this condition, therefore, a common reason for referral to a genetic clinic.
ID may occur either in isolation (non-syndromic) or in conjunction with other clinical manifestations as part of a recognizable syndrome (e.g. Down syndrome). Nevertheless, only in a handful of patients their clinical features indicate a defined chromosomic or monogenic condition and guide the choice of specific genetic tests.
Cytogenetic analysis (GTG-banding karyotyping) was for a long time the first step for genetic testing in patients with ID without a clinical presumptive diagnosis. Aneuploidies and unbalanced structural rearrangements visible on standard karyotypes account for approximately 3% of the patients with ID/developmental delay (DD) and/or multiple congenital anomalies (MCA) [3]. However, the resolution of this technique is limited to approximately 5 Mb, leading to smaller chromosomal aberrations to remain undetected. Over the last decades, molecular cytogenetic techniques such as FISH (fluorescent in situ hybridization) or MLPA (multiplex ligase-dependent probe amplification) have shown that cryptic chromosomal anomalies, particularly subtelomeric and interstitial rearrangements less than 3–5 Mb, are a significant cause of “idiopathic ID”.
The arrival of chromosomal microarray analysis (CMA) allowed the detection of smaller copy number variants (CNVs). This approach represents an integration of molecular techniques and conventional karyotyping and has a much higher diagnostic yield (10–25%) compared with GTG-banded karyotype analysis depending on the microarray platform and patient selection criteria [3,4,5]. This technique improves the diagnostic efficiency yield over cytogenetic analysis not only in patients with ID but also with DD, MCA, and autism spectrum disorders (ASD) [6]. Given this significant increase in the diagnostic yield in these groups of patients, CMA was recommended as the first-tier test for the postnatal evaluation of individuals with ID, ASD and/or MCA in 2010 [3] and guidelines were developed [7, 8].
Although many diagnostic laboratories over the world have already introduced CMA as the first-tier test for these groups of patients, in Argentina this has not been possible due to financial limitations. Without a clinical presumptive diagnosis, conventional cytogenetics is the first step in the search of the genetic cause of ID in the vast majority of patients. This work aims to report the diagnostic efficiency that resulted from the implementation of CMA at the clinical setting for patients with ID and dysmorphic features with or without ASD in an Argentinean referential public health center. We also provide clinical and molecular data of the patients with clinically relevant CNVs and with variants of uncertain significance (VUS). As far as we know, this is the first study in Argentina applying CMA for a series of individuals with ID.
Subjects and methods
Patients
A total of 133 patients (45 females and 88 males) with ID and dysmorphic features were included for the CMA study. All patients underwent the same protocol which included taking a prenatal/birth and family history, constructing a three-generation pedigree and both physical and neurological examinations. All patients included had normal G-banded karyotypes at 550-band resolution and no subtelomeric anomalies by MLPA (Multiplex Ligation-dependent Probe Amplification) testing. Patients exhibiting imbalances in the pericentromeric regions of the acrocentric chromosomes, as well as one patient that tested positive with only one of the two subtelomeric MLPA kits used (P036 and P070, MRC Holland, The Netherlands), were considered negative. Environmental etiologies, such as fetal alcohol syndrome, and common genetic etiologies, such as Fragile X syndrome (FXS), were generally ruled out by clinical geneticists and/or molecular testing.
When the determination of the de novo or inherited status of a detected anomaly was necessary, and parents were available, familial tests were performed. Eleven parents and one sister of patients with imbalances classified either as VUS or pathogenic were studied.
Chromosomal microarray analysis (CMA)
Patients and parents were studied with the ISCA v2 8 × 60 K (Agilent, USA) platform and/or with an in-house produced custom array design by INGEMM (KaryoArray®v3.0) [9, 10]. A cohort of patients was processed at INGEMM (Madrid, Spain), using the customized KaryoArray®v3.0 slides, and analyzed in Argentina as part of a collaborative project. These slides are not commercially available in our country; consequently, another design had to be selected. As the KaryoArray® is an Agilent-based 8 × 60 K design, the 8 × 60 K ISCA version was chosen. A total of 133 patients was analyzed by CMA: 80 with the customized slides, 17 with both and 36 with only the ISCA version.
Experiments were performed according to and with reagents from the array manufacturer (Agilent Technologies, Santa Clara, CA, USA). Briefly, DNA (500 ng) from the patient and a matching sex normal reference (Promega, Madison, WI, USA or Agilent Technologies, Santa Clara, CA, USA) were labeled by random priming (Genomic DNA Enzymatic Labeling Kit Agilent Technologies). After hybridization and washing steps, the arrays were scanned with the Agilent Microarray Scanner (#G2600D) and analyzed with the Agilent Cytogenomics software (v4.0, Agilent Technologies, Santa Clara, CA, USA). The aberration algorithm used was the Z-score ADM-2. A minimum of 3 consecutive oligonucleotides with the same polarity exceeding an absolute log2-ratio threshold of 0.35 was established to identify a CNV. Genomic imbalances were annotated based on the GRCh37/hg19 Genome Build (Feb 2009).
CNVs categorization
The ACMG and ClinGen technical standards for interpretation and reporting of constitutional CNVs [8] were followed for the categorization of the CNVs, sorting them into five categories: (1) pathogenic, (2) likely pathogenic, (3), VUS (4) likely benign, and (5) benign. For this purpose, the main databases used were the Database of Genomic Variants (DGV, https://projects.tcag.ca/variation/), DECIPHER (https://www.sanger.ac.uk/PostGenomics/decipher/), OMIM (https://www.omim.org/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), ClinGen (https://www.clinicalgenome.org/) and ORPHANET (https://www.orpha.net/). In addition, a literature search was performed in PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) to look for clinical and molecular information related to each detected imbalance. In some cases, familial samples were analyzed for a full interpretation of the proband’s array results and genetic counseling. For this report, the results were re-grouped into 3 classes: clinically relevant CNVs (categories 1 and 2), VUS (category 3) and normal (categories 4, 5 and patients with no CNVs detected).
The data produced in this study was deposited in ClinVar database (accession numbers provided in Tables 1 and 2).
Results
Considering that both platforms used are 8 × 60 K designs and that the patients studied with the two of them gave equivalent results, the results were analyzed as a whole (an example is illustrated in Fig. 1).

CMA image showing the 15q11.2 deletion of patient 20 using two different platforms: A) ISCA, B) Karyoarray
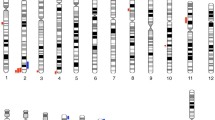
Clinically relevant CNVs were found in 16 of 133 patients (12%), 13 were categorized as pathogenic (CNV size ranged from 451.9 kb to 3.3 Mb) and 3 as probably pathogenic (size ranged from 2.3 to 3 Mb). These findings are illustrated in Fig. 2. Sex distribution was 9/88 for males and 7/45 for females, and 12 CNVs were deletions and 4 were duplications.

Ideogram showing the localization of the clinically relevant CNVs detected. In red are represented the deletions, in green the duplications and in yellow the CNV detected both in deletion and in duplication
One incidental finding was detected in patient 119, who presented a duplication of the region 17p12 (14111772_15442066), involving PMP22, among others, related to Charcot-Marie-Tooth disease type 1A (CMT1A). Given that this 35-year-old patient did not present a phenotype compatible with CMT1A, the CMA test was performed twice to confirm the results. To our knowledge, there is no record of an association between this duplication and ID; consequently, this CNV was not included in the detection rate calculation.
Table 1 summarizes the clinically relevant CNVs detected and the clinical features of the patients. The majority of the pathogenic CNVs corresponded to already known microdeletion syndromes: 1p36, 1q21.1, 3q13.2–13.31, 15q11.2, 15q25, 16p11.2, 17q11.2, 17q21.31 (Koolen-De Vries), 22q11.2 (DiGeorge); and for two of them (1q21.1 and 15q11.2) the reciprocal microduplication was also observed. In addition, the 8q22.1 Leri’s pleonosteosis chromosome duplication syndrome and a deletion in 12p12.1 associated with the Lamb-Shaffer syndrome (caused by the haploinsufficiency of the SOX5 gene) were found.
A particular situation was observed with the duplication detected in 16p13.11p12.3. This CNV partially overlaps with two syndromes described in ORPHANET, but not in OMIM: triplication 16p12.1p12.3 syndrome (ORPHA:485405) and microduplication 16p13.11 syndrome (ORPHA:261243). These syndromes, however, have been described in a few patients [11,12,13,14,15].
Eight CNVs classified as VUS were detected in 7 patients (5.3%), 7 were duplications and one a deletion (Table 2). The smallest had 25 kb and the largest one 451 kb.
Inheritance studies were performed for 9 patients and in 7 cases they helped establish the CNVs’ possible clinical significance. In 6 cases the CNV was inherited and in one case it was de novo. There were two cases where both parents could not be studied because the fathers were not available. Neither the CNV detected in 16p in patient 2 nor the VUS detected in patient 17, were inherited from their mothers.
Discussion
The advent of technologies that allowed whole-genome analysis and its inclusion into medical practice has contributed to the identification of genetic causes related to ID. The association of specific developmental disorders with chromosomal imbalances has led the CMA technique to become a routine test for patients with this condition in different parts of the world. However, in Argentina, it has not yet been extended so cytogenetic analysis would still be the first-tier genetic test for patients referred with ID. In this work, we report on our experience of implementing CMA for a group of patients referred with ID, dysmorphic features with or without ASD, and previous normal results from cytogenetic and subtelomeric MLPA analysis.
The diagnostic yield of the CMA, as a third-tier test, for a cohort of 133 patients from the Argentinian public health system was 12%. The percentage of clinically relevant (pathogenic and likely pathogenic) CNVs found in this cohort was in accordance with similar reports from other populations applying CMA in patients with normal karyotype. Koolen et al. [16] found a 9.1% pathogenic CNVs frequency in a cohort of 386 patients and also estimated, from a revision of results of several publications, an 11.2% detection frequency. Sagoo et al. [17] published a meta-analysis of the use of CMA in 13,926 subjects with ID and/or congenital malformations and reported that the average detection rate of causal genetic abnormalities in the 19 studies included was 10% (95% CI 8–12%). Nevertheless, the diagnostic yield of CMA depends on many factors including the resolution of the platform used, patient selection criteria, sample size, previous testing performed and the referring indication for testing [18,19,20].
Even though CMA is widely used, there are few publications from neighboring countries. Lay-Son et al. [21] performed CMA in a cohort of 40 Chilean patients selected by two or more criteria, including major congenital anomalies, facial dysmorphisms, DD, and ID, and they found 25% of pathogenic alterations. However, their cohort had the bias of analyzing selected patients, many of whom had remained without a diagnosis for a long time, and used a high-density platform. They also highlighted the elevated cost as a limitation for widespread use of the technique. In Brazil, however, CMA has been performed to a greater extent. Several groups studied cohorts of patients with similar inclusion criteria. Pratte-Santos et al. [22] investigated chromosomal abnormalities by CMA in 39 patients with dysmorphic features and ID with normal conventional karyotype retrospectively finding that 15.4% had pathogenic variants and 15% had VUS. Another Brazilian group found 17% causal CNVs in a total of 95 syndromic patients [23], and other described 22% of pathogenic CNVs in 15 patients with ID from Central Brazil [24]. Moreover, Vianna et al. [25] studied 200 patients from the Northeast of Brazil with ID and/or MCA, reaching a diagnosis rate of 16.5% (33/200), however, 10 (31.2%) of the pathogenic alterations that they found could have been detected by conventional karyotyping. Also, Ceroni et al. [26] recently published a CNVs study in 416 patients with MCA, ID and normal karyotype results by MLPA and only applied CMA to 14 selected patients, stressing the financial limitations. A larger cohort of 420 patients was studied by CMA in South Brazil, most of them without previous karyotyping, finding pathogenic CNVs in 18% of them and VUS in 12% [27]. To the best of our knowledge, the present report would be the first study of a cohort of patients with ID using CMA in Argentina.
Although the majority of the 16 clinically relevant CNVs detected in this work corresponded to already known imbalances, their detection was important for the medical managing and counseling of the carriers and their families. The information provided in Table 1 contributes to the description of the phenotypic characteristics of patients with these CNVs. This kind of contribution, giving clinical and molecular descriptions in different cohorts of patients, is essential for the adequate interpretation of CMA results.
Two reciprocal microdeletion/microduplication CNVs were detected at chromosomal bands 1q21.1 and 15q11.2. There are some genomic regions that have been described more frequently involved in reciprocal imbalances: 1p36, 7q11.23, 15q11-q13, 17p12, 17p11.27 and 22q11.2. These events are usually due to LCRs (Low Copy Repeats)-mediated non-allelic homologous recombination (NAHR) that originates the two related imbalances. Not detecting reciprocal imbalances for some regions has been related to a bias in patient assessment because of variable expressivity, incomplete penetrance, or the lethal effect that some rearrangement may present.
In seven patients, one or more CNVs were categorized as VUS. Seven out of the eight VUS detected were duplications. This observation is in line with other publications which highlighted that determining the clinical significance of duplications can be more challenging due to the more subtle and milder phenotypes associated with gains of genomic material [28, 29].
Deletions in the 4p16.3 region are a well-known cause of Wolf-Hirschhorn syndrome, but duplications of this region have been poorly described in the literature and databases. Bi et al. [30] proposed that disruption of the three genes involved in patient 17 duplication (Table 2) could confer seizure susceptibility, but this patient did not present seizures. Homozygous variants in the CPLX1 gene, involved in neuronal synaptic regulation and located at 4p16.3, have been associated to early infantile epileptic encephalopathy-63 (OMIM# 617976). Redler et al. [31] found homozygous CPLX1 variants in 3 patients (from 2 unrelated families) with ID, DD, and migrating myoclonic epilepsy.
Patient 39 presented a duplication of ELAC2. Compound heterozygous or homozygous variants in ELAC2 were found in patients with cardiomyopathy [32, 33], accompanied in most cases by lactic acidosis, DD, psychomotor delay, and/or other features. Many of these patients died as infants. Akawi et al. [34], on the other hand, found a homozygous variant in this gene in 5 members of a consanguineous family, affected with ID, psychomotor and DD and muscular hypotonia associated with subtle mild dysmorphic features, but with asymptomatic minimum septal hypertrophy. Kim et al. [35] also described a patient with encephalopathy, epilepsy, growth and developmental delay, and Tetralogy of Fallot, but presenting a single heterozygous ELAC2 variant. To our knowledge, ELAC2 duplications have not been described in the literature, so its relevance to the patient’s clinical features is uncertain.
Patient 125 carried a 9p24.3 duplication of 205 kb involving DOCK8. Similar duplications have been described in control populations and DGV database. Nevertheless, in a large-scale CNV meta-analysis across a spectrum of neurodevelopmental/psychiatric disorders (schizophrenia, bipolar disease, ASD, attention deficit hyperactivity disorder, and depression) a significant association of these disorders and DOCK8/KANK1 duplications was found [36]. Disruption of this gene has been associated to ID and speech and psychomotor delay with autosomal dominant inheritance pattern (OMIM #614113). Recently, it has also been proposed to be related to abnormalities in cognition and communication, aggressive behavior, and mood swings [37]. Deletions of DOCK8, or DOCK8 and KANK1, have been found in patients with ID, developmental and motor delay, and other variable clinical features [38, 39]. Patient 125 also presented a 132 kb deletion involving ZNF215 and olfactory receptors coding genes. Olfactory receptors have been associated to neuropsychiatric disorders such as ASD [40]. Although some of these receptors’ copy number is polymorphic, this CNV, in particular, has not been described previously.
Four patients (patient 131 in Table 1 and patients 5, 76 and 122 in Table 2) with duplications including the SHOX gene and/or its conserved non-coding elements (CNEs) have been found in this cohort. Duplications included one upstream CNE and the entire SHOX coding sequence in patient 131, all three upstream CNEs and part of the 5′ coding sequence in patient 5, and only downstream CNEs, without SHOX coding sequence, in patients 76 and 122. SHOX deficiency was associated with a phenotypic spectrum that ranges from Leri-Weill dyschondrosteosis (LWD) to idiopathic short stature. Also, cytogenetically visible deletions including SHOX have been found in patients with ID and short stature, among other clinical features [41, 42]. Duplications of SHOX and/or its CNEs have been reported in patients presenting different phenotypes. Regarding neurodevelopmental disorders (NDD), these imbalances have been found in patients with delayed speech and mild Asperger syndrome [43]; ID, congenital abnormalities and dysmorphic features [44]; ID and premature adrenarche [45]; mild ID, hypotonia, macrocephaly, facial dysmorphism, and short stature [46] and learning disability and immune deficiency [47]. However, in all cases, the imbalance had been inherited from an unaffected parent. Tropeano et al. [48] studied a large cohort of almost 19,000 patients with NDD and found a significant enrichment of SHOX microduplications in cases compared with 12,594 controls. These authors argue that microduplications at the SHOX locus are a low penetrance risk factor for ASD/NDD but a concomitant duplication of its enhancers may be required to trigger a NDD in females. Triplosensitivity for SHOX has been described as unlikely (https://dosage.clinicalgenome.org/), suggesting that duplications would not have an impact on phenotype. Nevertheless, even though it has been reported that these duplications are usually inherited from a healthy parent, these imbalances are not much represented in DGV database.
Although segregation could be established only in 7 out of the 9 analyzed cases, they helped to ascertain the CNVs’ possible clinical significance. In two patients, considering the nature of the genes involved in the imbalance and the fact that they had been inherited from an apparently normal parent, the detected CNV could be categorized as likely benign. In the case of patient 60, the analysis of his sister, who also had ID, revealed that they shared the CNV, nonetheless, it was categorized as probably benign due to the genes involved in the imbalance. The determination of the de novo nature of the detected CNV in patient 44 allowed counseling the family of low recurrence risk for future pregnancies. Inversely, in the case of patient 5, the CMA showed that his mother also had the SHOX gene duplication; this information was translated into counseling the family of a 50% transmission risk on future pregnancies. This is the same case as the deletion detected in patient 56, which was also inherited from his mother. However, in the latter case, his mother was also affected presenting mild ID and epilepsy. In patient 20, two relevant CNVs were found, one inherited from each parent, which was very important in order to provide appropriate genetic counseling to both branches of this family. Segregation studies for some of the remaining VUS may help to reclassify them as probably benign or probably pathogenic. However, inherited CNVs should not be excluded as causes of ID considering the possibility of variable expression and incomplete penetrance within families. Causal CNVs may be inherited from a healthy parent and still contribute to the child’s phenotype.
CMA has allowed the detection of a greater number of imbalances, optimizing the determination of the ID’s etiology in many patients. However, the categorization of certain CNVs can be challenging for different reasons. One is incomplete penetrance, which is thought to be related to epigenetic factors, environmental factors, or the genetic background, which can modulate or influence the phenotype. For example, as mentioned before, in patients 5 and 20 the detected imbalances were inherited from a phenotypically normal parent. Particularly, in the case of patient 20, where two imbalances were detected, the CNV classified as pathogenic was also present in their father, and it has been described that this CNV is associated to the patient’s cognitive characteristics. However, this patient had another CNV, the 15q13.3 recurrent duplication (D-CHRNA7 to BP5) involving the CHRNA7 and OTUD7A genes, inherited from their mother. This duplication has been considered of unknown clinical significance, or even benign, but there is literature suggesting that they may be pathogenic with decreased penetrance [49]. Besides, individuals with CNVs involving CHRNA7 manifest a range of phenotypes, mainly neurobehavioral or neuropsychiatric, with great variability in severity and expressivity [50]. So, this CNV could contribute to the patient’s phenotype.
This 15q13.3 CNV, as well as the common 15q11.2 duplication (involving TUBGCP5, CYF1P1, NIPA2, and NIPA1 genes) detected in another patient, have been classified differently in several publications and under the recent standards [8] they would be classified as benign. However, numerous reports have associated these CNVs with NDD, many describing them as low penetrant, risk factors and/or susceptibility loci. As a result, they should also be addressed as clinically relevant. Very recently, Maya et al. [51] have brought into discussion the difficulty in fitting low-penetrant susceptibility loci for NDD (such as ID, ASD, epilepsy, psychiatric and behavioral disorders) into one of the five categories proposed by the guidelines and followed by most laboratories to date. These authors argue that there is a lack of uniform categorization for these variants. Although the new ACMG/ClinGen technical standards [8] have provided a semiquantitative point-based scoring metric for CNV classification into the five categories, we agree with Maya et al. that the utilization of a new category, which they named “high-frequency low-penetrant variants”, would probably be better for addressing these type of variants. In our cohort, these two 15q duplications would fit this new category.
It is a possibility that the combination of the two CNVs detected in patient 20 was responsible for the observed cognitive phenotype. This situation, when phenotypic variability and/or severity is related to the presence of modifying factors has been described as the “two-hit model” by Girirajan et al. [52]. The dysmorphic features of this patient, however, remain molecularly unsolved and could be explained by another hit not detected by these technologies. Incomplete penetrance also makes genetic counseling more complicated because even though the recurrence risk is 50% it is very difficult to estimate the transmission of modifying factors. Fully penetrating syndromes are more straightforward in their categorization and impact on the associated phenotype. Also, in the cases of patients 5 and 20, where the mother is a carrier; there are certain publications that suggest that some protective factors exist, for example “being female”. However, the incomplete penetrance can be relative, given that some CNVs could have a different phenotypic expression in “healthy” parents such as psychiatric disorders, changes in body mass or low fertility [6]. The publication of the results of studies in different groups of patients allows a better interpretation of self-results. Many of the imbalances detected by this technique are rare, meaning that sharing molecular and clinical data of patients is of great value.
Although a CNV related to CMT1A was found in patient 119, he did not present the associated phenotype. This situation has been already described in other patients, who remained asymptomatic for this condition until the administration of chemotherapy drugs [53, 54]. Additionally, this disorder may appear later, in the fifth or sixth decade in life. Incidental findings stress the importance of genetic counseling and informed consent, especially the pre-test interview when patients and/or parents should be informed of the possibility that CMA could identify pathogenic CNVs unrelated to the referring indication for testing. Such findings could have implications for medical management for the patient and potentially other family members who may also carry the CNV.
The implementation of CMA contributed to the detection of clinically relevant CNVs in 12% of the patients, all of whom would have otherwise remained undiagnosed. In addition, it has been described that CMA detects, with better resolution, more than 99% of all pathogenic abnormalities detected by conventional cytogenetics [55] and may uncover additional genomic imbalances in patients with cytogenetically visible known syndromes [9]. These considerations are in agreement with implementing CMA as the first-tier genetic test for patients with ID. One of the major problems in effective and efficient management of patients with ID is the time that reaching a diagnosis takes, which can be several years. Implementing CMA would help reduce this time. Reaching an early diagnosis usually helps reduce the parents’ anxiety and avoid many medical appointments and unnecessary tests that sometimes are invasive and/or expensive. Besides, an accurate diagnosis would allow clinicians to perform preventive actions when possible, an adequate follow-up based on the published recommendations, and to improve family genetic counseling.
According to the obtained results and all the literature available, the use of CMA as the first-tier test for patients with ID would improve the karyotyping + subtelomeric MLPA detection rate by around threefold [3]. However, balancing costs is also important, especially in the public health system where, at present, CMA slides and reagents are approximately 8 times more expensive than those used for karyotyping + subtelomeric MLPA. Considering that these two require a greater time and human resources investment, cost–benefit studies are necessary to determine if this outweighs the CMA costs.
Although not being performed as first-tier, CMA is an indispensable test for patients with ID, elevating the diagnostic yield of conventional cytogenetic testing an additional ~ 10%. Maybe the best solution for many developing countries, at this moment, would be a consolidated network, where different laboratories perform conventional cytogenetics as the first-tier test and CMA remains restricted to a few laboratories for patients that fulfill specific criteria. Networking in genetic diseases has proven to be efficient in other countries like Brazil [56]. The challenge remains in deciphering which phenotypic features are the most suggestive of a pathogenic CNV. This would enhance the diagnostic yield of the technique and would allow a more cost-effective use of the resources.
Data availability
All relevant data are within the manuscript. In addition, the data produced in this study was deposited in ClinVar database.
References
Drews CD, Yeargin-Allsopp M, Decouflé P, Murphy CC (1995) Variation in the influence of selected sociodemographic risk factors for mental retardation. Am J Public Health 85:329–334. https://doi.org/10.2105/AJPH.85.3.329
Kaufman L, Ayub M, Vincent JB (2010) The genetic basis of non-syndromic intellectual disability: a review. J Neurodev Disord 2:182–209. https://doi.org/10.1007/s11689-010-9055-2
Miller DT, Adam MP, Aradhya S et al (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86:749–764. https://doi.org/10.1016/j.ajhg.2010.04.006
Beaudet AL (2013) The utility of chromosomal microarray analysis in developmental and behavioral pediatrics. Child Dev 84:121–132. https://doi.org/10.1111/cdev.12050
Vissers LELM, de Vries BBA, Veltman JA (2010) Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet 47:289–297. https://doi.org/10.1136/jmg.2009.072942
Rosenfeld JA, Patel A (2017) Chromosomal microarrays: understanding genetics of neurodevelopmental disorders and congenital anomalies. J Pediatr Genet. https://doi.org/10.1055/s-0036-1584306
Kearney HM, Thorland EC, Brown KK et al (2011) American College of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13:680–685. https://doi.org/10.1097/GIM.0b013e3182217a3a
Riggs ER, Andersen EF, Cherry AM et al (2020) Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22:245–257. https://doi.org/10.1038/s41436-019-0686-8
Vallespín E, Palomares Bralo M, Mori MÁ et al (2013) Customized high resolution CGH-array for clinical diagnosis reveals additional genomic imbalances in previous well-defined pathological samples. Am J Med Genet Part A 161:1950–1960. https://doi.org/10.1002/ajmg.a.35960
Rodríguez-Revenga L, Vallespín E, Madrigal I et al (2013) A parallel study of different array-CGH platforms in a set of Spanish patients with developmental delay and intellectual disability. Gene 521:82–86. https://doi.org/10.1016/j.gene.2013.02.043
Nimmo GAM, Guerin A, Badilla-Porras R et al (2016) Triplication of 16p12.1p12.3 associated with developmental and growth delay and distinctive facial features. Am J Med Genet A 170:712–716. https://doi.org/10.1002/ajmg.a.37483
Mefford HC, Cooper GM, Zerr T et al (2009) A method for rapid, targeted CNV genotyping identifies rare variants associated with neurocognitive disease. Genome Res 19:1579–1585. https://doi.org/10.1101/gr.094987.109
Ullmann R, Turner G, Kirchhoff M et al (2007) Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat 28:674–682. https://doi.org/10.1002/humu.20546
Ramalingam A, Zhou X-G, Fiedler SD et al (2011) 16p13.11 duplication is a risk factor for a wide spectrum of neuropsychiatric disorders. J Hum Genet 56:541–544. https://doi.org/10.1038/jhg.2011.42
Tropeano M, Ahn JW, Dobson RJB et al (2013) Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PLoS ONE 8:e61365. https://doi.org/10.1371/journal.pone.0061365
Koolen DA, Pfundt R, de Leeuw N et al (2009) Genomic microarrays in mental retardation: a practical workflow for diagnostic applications. Hum Mutat 30:283–292. https://doi.org/10.1002/humu.20883
Sagoo GS, Butterworth AS, Sanderson S et al (2009) Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta-analysis of 19 studies and 13,926 subjects. Genet Med 11:139–146. https://doi.org/10.1097/GIM.0b013e318194ee8f
Ho K, Wassman E, Baxter A et al (2016) Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders using an ultra-high resolution chromosomal microarray optimized for neurodevelopmental disorders. Int J Mol Sci 17:2070. https://doi.org/10.3390/ijms17122070
Shoukier M, Klein N, Auber B et al (2013) Array CGH in patients with developmental delay or intellectual disability: are there phenotypic clues to pathogenic copy number variants? Clin Genet 83:53–65. https://doi.org/10.1111/j.1399-0004.2012.01850.x
Vermeesch JR, Brady PD, Sanlaville D et al (2012) Genome-wide arrays: quality criteria and platforms to be used in routine diagnostics. Hum Mutat 33:906–915. https://doi.org/10.1002/humu.22076
Lay-Son G, Espinoza K, Vial C et al (2015) Chromosomal microarrays testing in children with developmental disabilities and congenital anomalies. J Pediatr (Rio J) 91:189–195. https://doi.org/10.1016/j.jped.2014.07.003
Pratte-Santos R, Ribeiro KH, Santos TA, Cintra TS (2016) Analysis of chromosomal abnormalities by CGH-array in patients with dysmorphic and intellectual disability with normal karyotype. Einstein (São Paulo) 14:30–34. https://doi.org/10.1590/S1679-45082016AO3592
Krepischi-Santos ACV, Vianna-Morgante AM, Jehee FS et al (2006) Whole-genome array-CGH screening in undiagnosed syndromic patients: old syndromes revisited and new alterations. Cytogenet Genome Res 115:254–261. https://doi.org/10.1159/000095922
Pereira RR, Pinto IP, Minasi LB et al (2014) Screening for intellectual disability using high-resolution CMA technology in a retrospective cohort from central Brazil. PLoS ONE 9:e103117. https://doi.org/10.1371/journal.pone.0103117
Vianna GS, Medeiros PFV, Alves AF et al (2016) Array-CGH analysis in patients with intellectual disability and/or congenital malformations in Brazil. Genet Mol Res. https://doi.org/10.4238/gmr.15017769
Ceroni JRM, Dutra RL, Honjo RS et al (2018) A multicentric Brazilian investigative study of copy number variations in patients with congenital anomalies and intellectual disability. Sci Rep 8:13382. https://doi.org/10.1038/s41598-018-31754-2
Chaves TF, Baretto N, de Oliveira LF et al (2019) Copy number variations in a cohort of 420 individuals with neurodevelopmental disorders from the South of Brazil. Sci Rep 9:17776. https://doi.org/10.1038/s41598-019-54347-z
Kaminsky EB, Kaul V, Paschall J et al (2011) An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med 13:777–784. https://doi.org/10.1097/GIM.0b013e31822c79f9
Weise A, Mrasek K, Klein E et al (2012) Microdeletion and microduplication syndromes. J Histochem Cytochem 60:346–358. https://doi.org/10.1369/0022155412440001
Bi W, Cheung SW, Breman AM, Bacino CA (2016) 4p16.3 microdeletions and microduplications detected by chromosomal microarray analysis: new insights into mechanisms and critical regions. Am J Med Genet A 170:2540–2550. https://doi.org/10.1002/ajmg.a.37796
Redler S, Strom TM, Wieland T et al (2017) Variants in CPLX1 in two families with autosomal-recessive severe infantile myoclonic epilepsy and ID. Eur J Hum Genet 25:889–893. https://doi.org/10.1038/ejhg.2017.52
Saoura M, Powell CA, Kopajtich R et al (2019) Mutations in ELAC2 associated with hypertrophic cardiomyopathy impair mitochondrial tRNA 3′-end processing. Hum Mutat 40:1731–1748. https://doi.org/10.1002/humu.23777
Shinwari ZMA, Almesned A, Alakhfash A et al (2017) The phenotype and outcome of infantile cardiomyopathy caused by a homozygous ELAC2 mutation. Cardiology 137:188–192. https://doi.org/10.1159/000465516
Akawi NA, Ben-Salem S, Hertecant J et al (2016) A homozygous splicing mutation in ELAC2 suggests phenotypic variability including intellectual disability with minimal cardiac involvement. Orphanet J Rare Dis 11:1–9. https://doi.org/10.1186/s13023-016-0526-8
Kim YA, Kim YM, Lee YJ, Cheon CK (2017) The first Korean case of combined oxidative phosphorylation deficiency-17 diagnosed by clinical and molecular investigation. Korean J Pediatr 60:408–412. https://doi.org/10.3345/kjp.2017.60.12.408
Glessner JT, Li J, Wang D et al (2017) Copy number variation meta-analysis reveals a novel duplication at 9p24 associated with multiple neurodevelopmental disorders. Genome Med 9:1–11. https://doi.org/10.1186/s13073-017-0494-1
Krgovic D, Vokac NK, Zagorac A, Kumperscak HG (2018) Rare structural variants in the DOCK8 gene identified in a cohort of 439 patients with neurodevelopmental disorders. Sci Rep. https://doi.org/10.1038/s41598-018-27824-0
Tassano E, Accogli A, Pavanello M et al (2016) Interstitial 9p24.3 deletion involving only DOCK8 and KANK1 genes in two patients with non-overlapping phenotypic traits. Eur J Med Genet 59:20–25. https://doi.org/10.1016/j.ejmg.2015.11.011
Wang JC, Mahon LW, Ross LP et al (2016) Enrichment of small pathogenic deletions at chromosome 9p24.3 and 9q34.3 involving DOCK8, KANK1, EHMT1 genes identified by using high-resolution oligonucleotide-single nucleotide polymorphism array analysis. Mol Cytogenet 9:1–8. https://doi.org/10.1186/s13039-016-0291-3
Schuch JB, Paixão-Côrtes VR, Longo D et al (2019) Analysis of a protein network related to copy number variations in autism spectrum disorder. J Mol Neurosci 69:140–149. https://doi.org/10.1007/s12031-019-01343-7
Boycott KM, Parslow MI, Ross JL et al (2003) A familial contiguous gene deletion syndrome at Xp22.3 characterized by severe learning disabilities and ADHD. Am J Med Genet 122A:139–147. https://doi.org/10.1002/ajmg.a.20231
Doherty MJ, Glass IA, Bennett CL et al (2003) An Xp; Yq translocation causing a novel contiguous gene syndrome in brothers with generalized epilepsy, ichthyosis, and attention deficits. Epilepsia 44:1529–1535. https://doi.org/10.1111/j.0013-9580.2003.61702.x
Thomas NS, Harvey JF, Bunyan DJ et al (2009) Clinical and molecular characterization of duplications encompassing the human SHOX gene reveal a variable effect on stature. Am J Med Genet A 149:1407–1414. https://doi.org/10.1002/ajmg.a.32914
Roos L, Nielsen KB, Tümer Z (2009) A duplication encompassing the SHOX gene and the downstream evolutionarily conserved sequences. Am J Med Genet A 149:2900–2901. https://doi.org/10.1002/ajmg.a.33118
Benito-Sanz S, Barroso E, Heine-Suñer D et al (2011) Clinical and molecular evaluation of SHOX/PAR1 duplications in léri-weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metab 96:404–412. https://doi.org/10.1210/jc.2010-1689
Hirschfeldova K, Baxova A, Kebrdlova V et al (2011) Cryptic chromosomal rearrangements in children with idiopathic mental retardation in the Czech population. Genet Test Mol Biomark 15:607–611. https://doi.org/10.1089/gtmb.2010.0218
Roberts JL, Hovanes K, Dasouki M et al (2014) Chromosomal microarray analysis of consecutive individuals with autism spectrum disorders or learning disability presenting for genetic services. Gene 535:70–78. https://doi.org/10.1016/j.gene.2013.10.020
Tropeano M, Howley D, Gazzellone MJ et al (2016) Microduplications at the pseudoautosomal SHOX locus in autism spectrum disorders and related neurodevelopmental conditions. J Med Genet 53:536–547. https://doi.org/10.1136/jmedgenet-2015-103621
Szafranski P, Schaaf CP, Person RE et al (2010) Structures and molecular mechanisms for common 15q13.3 microduplications involving CHRNA7: benign or pathological? Hum Mutat 31:840–850. https://doi.org/10.1002/humu.21284
Gillentine MA, Schaaf CP (2015) The human clinical phenotypes of altered CHRNA7 copy number. Biochem Pharmacol 97:352–362. https://doi.org/10.1016/j.bcp.2015.06.012
Maya I, Basel-Salmon L, Singer A, Sagi-Dain L (2020) High-frequency low-penetrance copy-number variant classification: should we revise the existing guidelines? Genet Med. https://doi.org/10.1038/s41436-020-0795-4
Girirajan S, Rosenfeld JA, Cooper GM et al (2010) A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet 42:203–209. https://doi.org/10.1038/ng.534
Kourie HR, Mavroudakis N, Aftimos P, Piccart M (2017) Charcot-Marie-tooth hereditary neuropathy revealed after administration of docetaxel in advanced breast cancer. World J Clin Oncol 8:425–428. https://doi.org/10.5306/wjco.v8.i5.425
Jariwal R, Shoua B, Sabetian K et al (2018) Unmasking a case of asymptomatic Charcot-Marie-tooth disease (CMT1A) with vincristine. J Investig Med High Impact Case Rep 6:232470961875834. https://doi.org/10.1177/2324709618758349
Hochstenbach R, van Binsbergen E, Engelen J et al (2009) Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. Eur J Med Genet 52:161–169. https://doi.org/10.1016/j.ejmg.2009.03.015
Vieira TA, Trapp FB, De Souza CFM et al (2019) Information and diagnosis networks—tools to improve diagnosis and treatment for patients with rare genetic diseases. Genet Mol Biol 42:155–164. https://doi.org/10.1590/1678-4685-gmb-2018-0214
Tardivo A, Masotto B, Espeche L et al (2017) Microdeleción 16p11.2: primeros casos reportados en Argentina. Arch Argent Pediatr 115:e449–e453. https://doi.org/10.5546/aap.2017.e449
Acknowledgements
We are grateful to the families that participated in this study, to the clinicians that assessed and counseled the patients, to Noemí Buzzalino, Marisol Delea and Javier Espeche for their critical revision of the manuscript, to Tania Castro, Belén Benavides Mori, and Patricia Pastore for their technical assistance, to Edid Andrada for the administrative assistance, and to the cytogeneticists that performed the patients karyotypes.
Funding
This research was supported by the Argentinean National Ministry of Health via institutional budget, by a grant from the Administración Nacional de Laboratorios e Institutos de Salud (FOCANLIS) and a fellowship “Carrillo-Oñativia”, and by a grant from REDES/FIBHULP08 of the Fundación para la Investigación Biomédica del Hospital Universitario La Paz, Madrid, Spain.
Author information
Authors and Affiliations
Contributions
SR and LDE designed the project, LDE, JN, PL and SR conceived and designed the experiments, RM and LDE performed the experiments, LDE, MP, MAM, LBD and SR analyzed the results, MP revised the karyotype results, APS, CM, VL, MS and RA assessed and counseled the patients and families with clinically relevant results.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study was approved by the Ethics Committee from the Centro Nacional de Genética Médica, Buenos Aires, Argentina (CE Acta14/11, Folio #28). All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki and its later amendments.
Informed consent
Written informed consent (from patients, parents or guardians) was obtained for all the individuals involved in this study. All the authors have read and approved the submission to the journal.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Espeche, L.D., Solari, A.P., Mori, M.Á. et al. Implementation of chromosomal microarrays in a cohort of patients with intellectual disability at the Argentinean public health system. Mol Biol Rep 47, 6863–6878 (2020). https://doi.org/10.1007/s11033-020-05743-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-020-05743-6