
Abstract
Increasing evidence suggests that miRNAs play important regulatory roles in the nervous system. However, the molecular mechanisms of how specific miRNAs affect neuronal development and functions remain less well understood. In the present study, we provide evidence that the conserved microRNA miR-210 regulates lipid metabolism and prevents neurodegeneration in the Drosophila retina. miR-210 is specifically expressed in the photoreceptor neurons and other sensory organs. Genetic deletion of miR-210 leads to lipid droplet accumulation and photoreceptor degeneration in the retina. These effects are associated with abnormal activation of the Drosophila sterol regulatory element-binding protein signaling. We further identify the acetyl-coenzyme A synthetase (ACS) as one functionally important target of miR-210 in this context. Reduction of ACS in the miR-210 mutant background suppresses the neurodegeneration defects, suggesting that miR-210 acts through regulation of the ACS transcript. Together, these results reveal an unexpected role of miR-210 in controlling lipid metabolism and neuronal functions.
Similar content being viewed by others


Introduction
microRNAs have emerged as crucial regulators in the nervous system by modulating neuronal development, physiology, and plasticity [1,2,3,4]. These small regulatory noncoding RNAs control gene expression through posttranscriptionally repression of the target mRNAs [5, 6]. Dysfunction of miRNAs and their regulatory gene networks has been linked to neuronal structural and functional changes and contributes to the onset of neurodegenerative diseases [7,8,9,10,11]. Mutant studies from fly and mouse models have revealed the neuroprotective roles of specific miRNAs, including miR-8, miR-132, miR-218, miR-263a, and miR-1000 [12,13,14,15,16,17]. Identifying the function of novel miRNAs and their regulatory mechanisms in adult neurons should lead to a better understanding of both fundamental biological processes as well as neuronal degeneration [4].
miR-210 is an evolutionarily conserved miRNA [18]. It was initially reported as a hypoxia-induced miRNA, known to regulate various processes under hypoxic conditions [18, 19]. Previous studies have shown that miR-210 is highly expressed in various types of cancers, such as breast cancer, osteosarcoma, and lung cancer [20,21,22,23,24]. Mechanistic studies have revealed that miR-210 regulates many cellular processes, including apoptosis, cell growth and differentiation, cell cycle progression, DNA damage, and mitochondrial metabolism [18, 23]. While the roles of miR-210 during cancer progression have been widely studied, the physiological functions of miR-210 are unclear [18, 23]. Interestingly, emerging evidence indicates that miR-210 might have important roles in the nervous system. In adult mice, overexpression or knockdown of miR-210 has been shown to promote or impair sensory axon regeneration, respectively [25]. Moreover, miR-210 is able to promote sensory hair cell formation [26]. miR-210 has also been reported to regulate neural plasticity and is involved in honeybee learning and memory [27]. Two recent studies have demonstrated that miR-210 regulates circadian rhythms in Drosophila [28, 29]. Thus, miR-210 appears to act in both cancer cells and neurons.
Here we report that miR-210 regulates lipid metabolism and prevents neurodegeneration in the Drosophila retina. miR-210 is expressed in the photoreceptor neurons and other sensory organs. The loss of miR-210 leads to progressively photoreceptor degeneration and lipid droplet accumulation. We provide evidence that these abnormalities are caused by abnormal activation of the sterol regulatory element-binding protein (SREBP) signaling pathway. Furthermore, we show that acyl-CoA synthetase (ACS), the Drosophila homolog of acetyl-coenzyme A synthetase 2 (ACSS2), is a miR-210 target. Genetic manipulation to reduce the level of ACS in miR-210 mutants results in partial rescue of the photoreceptor degeneration defects. Overall, our study uncovers a novel role of miR-210 in modulating lipid metabolism and neuronal functions.
Results
Expression of miR-210 in photoreceptor neurons and other sensory organs
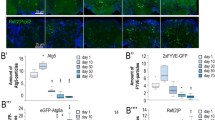
miR-210 is one of highly expressed miRNAs in the Drosophila adult head [30]. We began our study by examining the spatial and temporal expression of miR-210 throughout Drosophila development. To this end, a miR-210-Gal4 knock-in strain, miR-210KO, was used [31]. In this strain, the miR-210 hairpin was replaced by the Gal4 transcription factor [31]. As Gal4 is placed under the control of endogenous regulatory elements of miR-210, the Gal4 expression pattern indicates miR-210 activity in vivo. We crossed the miR-210-Gal4 knock-in strain with a UAS-mCD8::GFP reporter line, and first assessed the GFP expression pattern in adult flies. We assayed fluorescence under a dissecting microscope and observed GFP signals present in the eye, ocelli, antenna, maxillary palp, hot/cold cells, Johnston’s organ, and proboscis (Fig. 1a–d). Upon dissection of the brain and ventral nerve cord (VNC), we confirmed the expression of GFP in the photoreceptor neurons, HB eyelet, ocellar photoreceptors, and olfactory receptor neurons (Fig. 1e). In addition, expression in mushroom bodies and subsets of neurons in the VNC was apparent (Fig. 1e). Co-staining with the 24B10 antibody (which marks photoreceptor neurons) further confirmed the expression of miR-210 in photoreceptor neurons (Fig. 1f–f‴). We conclude that miR-210 exhibits strong expression in most adult sensory organs, including the eye (Fig. 1g).

a, a′ miR-210-Gal4 drives GFP (UAS-mCD8::GFP) expression in the adult eye. The same eye was shown in (a′). b, b′ miR-210-Gal4 drives GFP expression in the adult eye, ocelli, antenna, and maxillary palp. c, d miR-210-Gal4 drives GFP expression in the adult Johnston’s organ, hot/cold cells (located in the arista) and proboscis. e A confocal image showing miR-210-Gal4-driven GFP expression in the photoreceptors, ocellar photoreceptors, HB eyelets, olfactory receptor neurons, mushroom body and a subset of neurons in the VNC. g Schematic of the Drosophila adult and head, illustrating visual, olfactory, auditory, thermosensory, and gustatory organs (Modified from Ref. [48]). f–f‴ Confocal images showing miR-210-Gal4-driven GFP expression in the photoreceptor neurons, costained with anti-chaoptin (24B10). Scale bars: 100 μm.
As in adults, we also determined the expression of miR-210 in embryonic and larval stages. During late embryonic stages, strong GFP expression was present in the bilateral cluster of Bolwig’s organ cells (Fig. 2a). In the larvae, GFP expression was found in the chordotonal organs, larval brains and neurons at the anterior end of the larva (Fig. 2b–b″). After larval dissection, GFP expression was present in the eye disc, the axonal projections of photoreceptor neurons in the brain optic lobes, Bolwig’s organ with its nerve (costained with 24B10), dorsal organ (co-labeled with Or83b-RFP), larval antennal lobe, and optic neuropile (Fig. 2c–f). With various costained markers (Elav or 24B10), expression of miR-210 in all eight photoreceptor cells was further confirmed in the third instar eye imaginal disc, pupal, and adult retina (Fig. S1a–c″). Examination of GFP expression from the UAS-mCD8::GFP transgene confirmed that the observed signals were not due to the leaky expression of this transgene (Figs. S2 and S3).

a miR-210 drives GFP expression in the embryonic Bolwig’s organ. b–b″ miR-210 drives GFP expression in the chordotonal organs, larval brains, and neurons at the anterior part of the larva. c–c″ Confocal images showing miR-210-Gal4-driven GFP expression in the larval Bolwig’s organ and nerve, costained with anti-chaoptin (24B10). d–d″ Confocal images showing miR-210-Gal4-driven GFP expression in the larval dorsal organ, co-labeled by Or83b-RFP. e–e″ Confocal images showing miR-210-Gal4-driven GFP expression in the larval optic neuropile and antennal lobe. f Schematic of the Drosophila larval head, illustrating visual, olfactory, and chordotonal organs. Scale bars: 100 μm.
In summary, miR-210 is expressed in the developing and mature photoreceptor neurons as well as other sensory organs throughout development.
Retinal degeneration in miR-210 mutants
To further explore the function of miR-210, we generated a miR-210 loss-of-function mutant allele, miR-210Δ130, through the CRISPR/Cas9 system (Fig. 3a). miR-210Δ130 contains a 130 bp deletion, which removes the entire miR-210 hairpin, leading to the complete loss of miR-210 (Fig. 3a). This deletion was verified by genomic PCR using two primers spanning the miR-210 locus (Fig. 3b). Furthermore, miRNA qPCR also confirmed the absence of mature miR-210 transcript in mutant fly heads (Fig. 3c). miR-210Δ130 mutant flies were viable and appeared to be morphologically normal.

a Diagrams of the miR-210 locus and miR-210 mutant allele. All exons are shown as black boxes. Two sgRNA targeting sequence are indicated in red. Two PAM sequences are shown in blue. The region deleted in the miR-210 mutant strain is indicated by the bracket. The chromatogram of partial genomic DNA sequence around the target site from the miR-210 mutant is shown in the middle. Diagrams of the miR-210 genomic rescue construct are shown in the bottom. b PCR analysis of genomic DNA around the target sites in wild type, miR-210 heterozygous, and homozygous mutants. c qRT-PCR analysis of mature miR-210 expression levels in both wild type and miR-210 homozygous mutants. d–f″ Confocal images showing the phalloidin stained retinas from flies with the indicated genotypes at different ages. One-day-old wild-type, miR-210 mutant, and genomic rescued flies are shown in the upper panel; 5-day-old wild-type, miR-210 mutants, and genomic rescued flies are shown in the middle panel; 30-day-old wild-type, miR-210 mutants, and genomic rescued flies are shown in the bottom panel. Ommatidia in miR-210 mutant retinas at day 1 and at day 5 were slightly affected, but showed severe degeneration at day 30. These defects were rescued by introducing the miR-210 genomic rescue transgene. g Quantification of the number of intact rhabdomeres in each ommatidium from flies with the indicated genotypes. Five retinas were used for each genotype, and nine ommatidia from each retina were counted. h–j‴ Transmission electron microscopy images showing the ultrastructure of retinas from flies with the indicated genotypes at different ages. The cross sectional view of retinas was shown for 1-day-old flies, and a high magnification view of a single ommatidium was shown for each genotype at different ages. The morphology and number of miR-210 mutant rhabdomeres at day 1 and at day 5 were mildly affected, but they were severely affected at day 30. Arrows indicate the lipid droplet structure. k Western blot analysis of three photoreceptor-specific proteins in head extracts from 5-day-old flies with the indicated genotypes. The protein levels of Rh1, INAD, and PLC were reduced in miR-210 mutants, and this reduction was restored in the rescue flies. Tubulin was used as a loading control. Data are presented as mean ± SD. **p < 0.001, ***p < 0.001, by Student’s t test. Scale bars: 10 μm in (d–f″). 4 μm in (h). 2 μm in (h′–h‴). p = 9.11371E−11 in (c), p = 0.0034, 0.0054, and 0.0006 in (g).
As we have shown that miR-210 was highly expressed in the eye and other sensory organs, we therefore first focused our study in the fly visual system. The Drosophila eye consists of about 800 ommatidia, each of which contains six outer photoreceptors (R1–R6) and two inner photoreceptors (R7 and R8). Only seven photoreceptors are visible in each cross-section. We examined the morphology of photoreceptors using the fluorescently conjugated phalloidin to label actin rich rhabdomeres, the microvillar structure of stacked membranes of each photoreceptor. We found that miR-210Δ130 mutant flies exhibited age-dependent retinal degeneration (Fig. 3d–e″). In miR-210Δ130 mutants, photoreceptor morphology and number were mildly affected in 1-day- and 5-day-old flies (Fig. 3d, e, d′, e′, quantified in g). However, these phenotypes, including abnormal morphology and loss of some photoreceptors, became progressively worse by day 30 (Fig. 3d″, e″, quantified in g). Rescue with a 2.5 kb genomic DNA fragment containing the miR-210 hairpin restored these observed photoreceptor defects in the retina (Fig. 3f–f″, quantified in g).
To gain further insights into the degeneration mechanism, we also examined the ultrastructure of photoreceptor cells by transmission electron microscopy (TEM) in 1-day-, 5-day-, and 30-day-old flies. The disorganized microvilli structures were observed in miR-210Δ130 mutant rhabdomeres, and photoreceptor loss started at 1-day-old flies and increased progressively with age (Fig. 3h–i‴). The rhabdomeres were severely damaged and significantly smaller in size in 30-day-old miR-210Δ130 mutants (Fig. 3h‴, i‴). These defects were also recovered in the rescued flies (Fig. 3j–j‴).
As miR-210Δ130 mutants exhibited photoreceptor degeneration defects, we then asked whether the loss of miR-210 leads to the loss or misexpression of known photoreceptor enriched proteins. For this purpose, we checked for the presence of phototransduction proteins in both 5-day-old wild-type and miR-210Δ130 mutant adult head extracts. Western blot analysis revealed strong reduced levels of three phototransduction proteins, Rh1, INAD, and PLC in miR-210Δ130 mutants, as compared to the controls (Fig. 3k). In addition, the residual Rh1 was detected in the immature form (40 kDa) (Fig. 3k).
To test if miR-210 has a role during retinal development, we performed immunostaining analysis with two markers, F-actin and Eys, to monitor the structure and morphology of the developing rhabdomere at the pupal stage. The size of rhabdomere appeared to be slightly smaller in miR-210Δ130 mutants as compared to the controls at 48 h after pupariation (APF), suggesting that the loss of miR-210 leads to a developmental delay for the pupal retina (Fig. S4a–c″). At 96 h APF, the staining of F-actin and Eys revealed a mild defect in the morphology and organization of rhombomeres in miR-210Δ130 mutants (Fig. S4d–f″). Suppression of these defects was observed in the rescued flies (Fig. S4a–f″). Thus, miR-210 mutation mildly affects retinal development.
Taken together, these data strongly suggest that the loss of miR-210 leads to a progressive loss of photoreceptors, indicating that miR-210 is required for preventing photoreceptor degeneration.
Lipid droplet accumulation and alterations in lipid metabolism in miR-210 mutants
While examining photoreceptor morphology by TEM, we noticed that abundant lipid droplet structures were accumulated in the pigment cells of 1-day-old miR-210Δ130 mutant retina (Fig. 3i, i′, indicated by arrows). Importantly, these defects were restored in the rescue flies, indicating that the increase of lipid droplets was due to the specific loss of miR-210 (Fig. 3j, j′). To further confirm the lipid droplet structure observed in our TEM analysis, we next used Nile red to stain the retina and examined the distribution of lipid droplet in 1-day-old flies. The loss of miR-210 indeed resulted in lipid droplet accumulation in the retina, as revealed by the increased staining of Nile red (Fig. 4a, b, quantified in d). This was also rescued with the expression of miR-210 (Fig. 4c, quantified in d). Thus, we conclude that miR-210 mutations cause lipid droplet accumulation in the Drosophila eye.

a–c Confocal images showing the Nile red stained retinas from 1-day-old wild-type, miR-210 mutant, and the rescue flies. White arrowheads indicate lipid droplets. d Quantification of the number of lipid droplets for each ommatidium from flies with the indicated genotypes. Eight retinas were used for each genotype, and two ommatidia were counted for each retina. e Heatmap showing the levels of lipid species with a two times significant difference in the results of lipid profiling in both wild type and miR-210 mutants. Only the major lipid species were shown. The color of each bar indicates the levels of corresponding lipid species. PA phosphatidic acid, PC phosphatidylcholine, PE phosphatidylethanolamine, PS phosphatidylserine, DG diacylglycerol, TG triacylglycerol. Data are presented as mean ± SD. ****p < 0.0001, by Student’s t test. Scale bar: 10 μm. p = 5.12066E−10 in (d).
As the loss of miR-210 results in lipid droplet accumulation, we therefore asked whether miR-210Δ130 mutant flies may show abnormalities in lipid metabolism. To test this possibility, we performed the lipidomics analysis and measured the lipid levels using liquid chromatography/mass spectrometry analysis in extracts from wild-type and miR-210Δ130 mutant fly heads (Table S1). Consistent with the finding of the accumulation of lipid droplets in miR-210Δ130 mutants, we found that the levels of many species of triacylglycerols (TG), the major storage lipids of lipid droplets, were increased in miR-210Δ130 mutants compared to the control (Fig. 4e). In addition, the levels of some PA, PC, PE, and PS species were higher in miR-210Δ130 mutants (Fig. 4e). In contrast, the levels of diacylglycerols were reduced in the mutants (Fig. 4e). These results suggest that miR-210 mutation leads to the change of lipid levels in the retina.
Together, our data demonstrate that miR-210Δ130 mutants show dysregulated lipid metabolism.
Activation of SREBP signaling contributes to lipid accumulation and retinal degeneration in miR-210 mutants
Previous studies have shown that Drosophila SREBP, one of highly conserved proteins involved in controlling lipid metabolism, functions to promote lipid droplet accumulation in the eye, and other tissues [32,33,34,35,36,37,38,39]. We next tested the potential role of SREBP in mediating the effects of miR-210 on lipid droplet accumulation and retinal dysfunction. We first determined whether the activity of SREBP is altered by examining SREBP processing by western blot analysis. In wild-type fly heads, SREBP proteins exist as both full-length, membrane-bound precursors (p-SREBP) and cleaved and mature form (m-SREBP) (Fig. 5a). Compared to the wild-type control, activated mature SREBP protein levels were elevated in miR-210Δ130 mutants, suggesting that the loss of miR-210 leads to hyperactivation of SREBP signaling (Fig. 5a).

a Western blot analysis of membrane-bound (precursor, p-SREBP) and nuclear (mature, m-SREBP) forms of SREBPs in both 5-day-old wild-type and miR-210 mutant flies. The level of nuclear form of SREBP was increased in miR-210 mutants. Tubulin was used as a loading control. b–e Confocal images showing the Nile red stained retinas from 1-day-old wild -type, miR-210▵130, miR-210▵130; SREBP▵189/+, and SREBP▵189/+ flies. Scale bars: 10 μm. f Quantification of the number of lipid droplets for each ommatidium from flies with the indicated genotypes. Eight retinas were used for each genotype, and two ommatidia were counted for each retina. Removing one copy of SREBP reduced the number of lipid droplets in the miR-210 mutant background. g–j Confocal images showing the phalloidin stained retinas from 1-day-old miR-210▵130, 30-day-old miR-210▵130, miR-210▵130; SREBP▵189/+, and SREBP▵189/+ flies. Scale bars: 10 μm. k Quantification of the number of intact rhabdomeres in each ommatidium from flies with the indicated genotypes. Removing one copy of SREBP increased the number of intact rhabdomeres in the miR-210 mutant background. Five retinas were used for each genotype, and nine ommatidia from each retina were counted. l Western blot analysis of three photoreceptor-specific proteins in head extracts from 30-day-old flies with the indicated genotypes. Removing one copy of SREBP increased the protein levels of Rh1, INAD, and PLC in the miR-210 mutant background. Tubulin was used as a loading control. Data are presented as mean ± SD. ****p < 0.0001, by Student’s t test. Scale bar: 10 μm. p = 1.11994E−14 in (f), p = 5.78726E−05 in (k).
We then asked whether activation of SREBP signaling may be responsible to the accumulation of lipid droplets and retinal defects in miR-210 mutants. To this end, we reduced SREBP activity in the miR-210Δ130 mutant background to see if the accumulation of lipid droplets and the severity of retinal dysfunction are ameliorated. Introducing one copy of the SREBP mutant allele, SREBPΔ189, was able to suppress accumulation of lipid droplets and improve retinal morphogenesis (Fig. 5b–e, quantified in f; g–j, quantified in k). Furthermore, western blot analysis revealed that reduction of SREBP activity partially restored the levels of three previously examined phototransduction proteins, including Rh1, INAD, and PLC, in miR-210Δ130 mutants (Fig. 5l). Most of the Rh1 remained in the immature form (40 kDa) in these flies, which suggests that defects in rhodopsin processing in miR-210Δ130 mutants were not rescued when SREBP levels were reduced (Fig. 5l). These results together indicate that increased SREBP signaling contributes to lipid accumulation and retinal defects in miR-210Δ130 mutant flies.
The loss of miR-210 disrupts normal expression of photoreceptor genes and lipid metabolic genes
To further explore the molecular mechanisms underlying the photoreceptor degeneration of miR-210 mutants, we performed gene-expression profiling using genome-wide RNA sequencing analyses. For this analysis, total RNAs were prepared from both 5-day-old wild-type and miR-210Δ130 mutant fly heads. A total of 479 genes showed significant differences in expression levels in miR-210Δ130 mutants as compared with the controls, with a cutoff at 1.5-fold and a false discovery rate of <0.05 (Table S2). We identified 205 genes that were significantly downregulated and 274 genes that were significantly upregulated in miR-210Δ130 mutants (Fig. 6a). KEGG analysis for these genes further revealed that the downregulated genes were mostly enriched for known pathways implicated in phototransduction and phosphatidylinositol signaling and the upregulated genes were mostly enriched for the pathways involved in fatty acid metabolism and glyoxylate and dicarboxylate metabolism (Fig. 6b, c).

a Volcano plot of differentially expressed genes between wild-type and miR-210 mutants. b KEGG gene enrichment analysis for downregulated genes in miR-210 mutants. c KEGG gene enrichment analysis for upregulated genes in miR-210 mutants. d qRT-PCR analysis of selected downregulated genes in miR-210 mutants. e qRT-PCR analysis of selected upregulated genes in miR-210 mutants. Data are presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns not significant, by Student’s t test. p = 0.0134, 0.00014, 0.0023, 0.00013, 7.01185E−06, 0.0393, 0.0786, 0.0094, 0.0023, 0.1202, 0.1328, 0.0037, 0.2243, 0.0035, and 0.1199 in (d), p = 0.0022, 0.0433, 0.1157, 0.0472, 0.1203, 0.1899, 0.1134, 0.0197, 0.0169, 0.1706, 0.0227, and 0.002 in (e).
As phototransduction-related genes were enriched in the downregulated gene list, we further determined whether additional photoreceptor genes were also present in this list. To this end, we made a comparison between our downregulated gene list and two published data sets on photoreceptor or eye enriched genes in Drosophila [40, 41]. In total, we found that 45 photoreceptor or eye enriched genes, including phototransduction or retinal degeneration, structural/morphological maintenance, synaptic function, transcription factors and/or nucleic-acid-binding protein, signaling, posttranslational modifications, intermediary metabolism and apoptosis, were downregulated in miR-210Δ130 mutants (Table S3). Fifteen genes were selected for qRT-PCR analysis, and many of them showed the same trend of change as RNA-Seq analysis (Fig. 6d). These results are consistent with the above phenotypic data indicating that the loss of miR-210 leads to photoreceptor degeneration.
Of genes increasing expression, many function in fatty acid metabolism. These genes include several key lipogenic genes, such as ATP-citrate lyase (ATPCL), ACS short chain family member, acetyl-coA carboxylase (ACC), and fatty acid synthase (FASN1) (Table S4). ATPCL and ACS generate cytoplasmic acetyl-CoA for de novo fatty acid synthesis [42]. These genes are also known targets of SREBP signaling [32].
We noticed that the expression levels of several other genes related to de novo fatty acid synthesis were also increased in miR-210Δ130 mutants, including the SLC25 family of mitochondrial transporter (CG1628), malic enzyme, isocitrate dehydrogenase, and mitochondrial pyruvate carrier (Mpc1) (Table S4). A few glucose handling enzymes are also elevated, including trehalose/glucose/fructose transporter sut1, sut2, and CG8249, and organic ions transporter CG8654 (Table S4). We also observed upregulation of the fatty acid desaturase Desat1, one of downstream lipid-processing enzymes (Table S4). Moreover, two TG synthesis-related genes also showed increased expression in miR-210Δ130 mutants, including GK2 and Mino (Table S4). We also selected 12 genes for qRT-PCR, and most genes showed the same changing trend as RNA-Seq (Fig. 6e).
The increases in both de novo lipid and TG synthesis pathway genes further suggest that the loss of miR-210 triggers a global remodeling of lipid landscape.
Identification of ACS as a target of miR-210
miRNAs normally repress target expression. To identify the potential target of miR-210, we compared the upregulated genes after miR-210 knockout with in silico predicted miR-210 targets by TargetScan, PicTar, and miRanda (Fig. 7a). Of those computationally predicted overlapping targets, only three candidate targets, ACS, Ady43A, and ATPCL, were found among upregulated genes in miR-210 mutants (Fig. 7a). In the present study, we focused on ACS, as its mammalian homolog ACSS2 has been reported to regulate histone acetylation and gene expression in addition to its enzymatic role in converting acetate to acetyl-CoA [43, 44]. Moreover, ACSS2 has recently been shown to selectively regulate genes related to lipid metabolism, including ACC and FASN, to promote fat storage and utilization [45]. Thus, we hypothesized that Drosophila ACS could be one relevant target of miR-210 to mediate lipid signaling and retinal function. The qRT-PCR analysis confirmed upregulation of ACS expression in miR-210Δ130 mutants (Fig. 6e). Moreover, an increase of ACS protein level in miR-210Δ130 mutants was further verified by western blot analysis (Fig. 7b).

a A venn diagram shows the overlaps of the miR-210 target genes predicted by three algorithms, TargetScan, PicTar, and miRanda and the genes upregulated in miR-210 mutants. b Western blot analysis of ACS in head extracts from 5-day-old wild-type and miR-210▵130 mutant flies. c Diagram of the pairing of miR-210 to the predicted target site in the ACS 3′UTR. Mutated nucleotides in the ACS 3′UTR for the experiment (d) are shown in red with the underline. d Luciferase reporter assays in S2 cells transfected with pTub-miR-210 or the empty vector. ACS 3′UTR and mutated 3′UTR indicate the reporter with the intact or mutated target sites. e–h Confocal images showing the phalloidin stained retinas from 1-day-old miR-210▵130, 30-day-old miR-210▵130, miR-210▵130; ACSCB-6224-3/+, and ACSCB-6224-3/+ flies. Scale bars: 10 μm. i Quantification of the number of intact rhabdomeres in each ommatidium from flies with the indicated genotypes. Removing one copy of ACS increased the number of intact rhabdomeres in the miR-210 mutant background. Five retinas were used for each genotype, and nine ommatidia from each retina were counted. j The upper panel shows the western blot analysis of three photoreceptor-specific proteins in head extracts from 30-day-old flies with the indicated genotypes. Removing one copy of ACS increased the protein levels of Rh1, INAD, and PLC in the miR-210 mutant background. The lower panel shows the western blot analysis of ACS in head extracts from 5-day-old flies with the indicated genotypes. Tubulin was used as a loading control. k–n Confocal images showing the Nile red stained retinas from 1-day-old wild-type, miR-210▵130, miR-210▵130; ACSCB-6224-3/+, and ACSCB-6224-3/+ flies. Scale bars: 10 μm. o Quantification of the number of lipid droplets for each ommatidium from flies with the indicated genotypes. Removing one copy of ACS reduced the number of lipid droplets in the miR-210 mutant background, but the difference was not significant. Eight retinas were used for each genotype, and two ommatidia were counted for each retina. p–s Confocal images showing the phalloidin stained retinas from 1-day-old miR-210▵130, 30-day-old miR-210▵130, miR-210▵130; UAS-ACSRNAi/+; Rh1-Gal4/+ and UAS-ACSRNAi/+; Rh1-Gal4/+ flies. Scale bars: 10 μm. t Quantification of the number of intact rhabdomeres in each ommatidium from flies with the indicated genotypes. Knockdown of ACS by Rh1-Gal4 increased the number of intact rhabdomeres in the miR-210 mutant background. Five retinas were used for each genotype, and nine ommatidia from each retina were counted. u–x Confocal images showing the Nile red stained retinas from 1-day-old miR-210▵130, 30-day-old miR-210▵130, miR-210▵130; UAS-ACSRNAi/+; Rh1-Gal4/+ and UAS-ACSRNAi/+; Rh1-Gal4/+ flies. Scale bars: 10 μm. y Quantification of the number of lipid droplets for each ommatidium from flies with the indicated genotypes. Knockdown of ACS by Rh1-Gal4 slightly reduced the number of lipid droplets in the miR-210 mutant background, but the difference was not significant. Seven retinas were used for each genotype, and two ommatidia were counted for each retina. Data are presented as mean ± SD. **p < 0.01, ***p < 0.001, ****p < 0.001, ns not significant, by Student’s t test. p = 0.0062 and 0.001 in (d), p = 4.65778E−05 in (i), p = 0.2311 in (o), p = 0.0002 in (t), p = 0.4064 in (y).
We next assessed the direct miR-210-ACS repression. The 3′UTR of ACS mRNA contains one potential miR-210 binding site (Fig. 7c). To determine whether miR-210 regulates ACS expression through this binding site, we prepared a luciferase reporter containing the ACS 3′UTR. Luciferase assays showed that miR-210 was able to downregulate the reporter activity (Fig. 7d). This suppression effect was relieved by mutating the predicted target site of ACS 3′UTR to compromise seed pairing with miR-210 (Fig. 7d). Thus, ACS appears to be a direct target of miR-210.
miRNA mutant phenotypes can be attributed to upregulation of the target gene [6]. To further investigate whether upregulation of ACS contributes to the defects observed in the miR-210 mutant, we reduced ACS activity in the miR-210Δ130 mutant background. Removing one copy of ACS using an insertion ACS mutation, ACSCB-6224-3, was able to partially suppress the photoreceptor degeneration defects of miR-210Δ130 mutants at day 30 (Fig. 7e–h, quantified in i). Consistently, western blot analysis revealed that the levels of three phototransduction proteins, Rh1, INAD, and PLC, were recovered in miR-210Δ130 mutant flies heterozygous for ACS mutant allele as compared to miR-210Δ130 mutant itself (Fig. 7j). Although the level of Rh1 was increased, the majority of Rh1 was still present in the immature form (40 kDa) (Fig. 7j). We also measured ACS protein levels in these flies and found that introducing one copy of ACS mutant allele abrogated upregulation of ACS protein levels in miR-210Δ130 mutants (Fig. 7j). The protein level of ACS in the ACS heterozygous mutants was not obviously reduced (Fig. 7j). The reason for this was unknown. Moreover, reduction of ACS decreased the number of lipid droplets in miR-210Δ130 mutants, but the difference was not significant (Fig. 7k–n, quantified in o).
As miR-210 is expressed in photoreceptor neurons, it is expected that the regulation of ACS by miR-210 occurs in these neuronal cells. To test this, we further used a photoreceptor neuron-specific Gal4, Rh1-Gal4, to knockdown ACS in miR-210Δ130 mutants and examined the phenotypes. Our results revealed that RNAi of ACS by Rh1-Gal4 was able to partially rescue the photoreceptor degeneration defects of miR-210Δ130 mutants at day 30 (Fig. 7p–s, quantified in t). The accumulation of lipid droplets was not significantly suppressed (Fig. 7u–x, quantified in y). Together, these data suggest that ACS is a functional target of miR-210, and also point out that additional unidentified targets are involved in the regulation of lipid droplet formation.
Discussion
Our study has revealed that the loss of miR-210 disrupts lipid homeostasis and induces age associated degeneration in Drosophila photoreceptor neurons. We provide evidence that SREBP signaling is responsible for the lipid accumulation and retinal degeneration phenotypes in miR-210 mutants. Furthermore, ACS was identified as one of miR-210 functional targets to mediate neuronal degeneration defects in the mutants. We have shown that miR-210 is widely expressed in all major sensory organs. Thus, it is conceivable that miR-210 might also function to regulate lipid homeostasis and prevent neurodegeneration in other sensory organs.
In miR-210 mutants, we observed accumulated lipid droplets in the glial cells of the retina. This is similar to a previous study in which mitochondria defects in the photoreceptor neuron promote lipid droplet accumulation in these glial cells [35]. Consistent with the lipid accumulation phenotypes, our lipidomics analysis has revealed that TG levels were upregulated in miR-210 mutants. One of the central regulators involved in this process was the transcription factor SREBP. SREBP plays a crucial role in controlling lipogenic gene expression and promoting fatty acid synthesis [32, 37]. The lipid accumulation phenotype we observed in miR-210 mutant was consistent with increased SREBP activity. The western blot analysis also showed the hyperactivation of SREBP in miR-210 mutants. Furthermore, the RNA-Seq experiment uncovered significant increases in the mRNA levels of many SREBP downstream genes that regulate lipid metabolism. From the increases we observed in the lipid species and the key lipogenic genes after miR-210 knockout, we would predict profound effects on lipogenesis in these mutant animals. Lipid accumulation in the glial cells is an early step in degeneration and appears to predispose to subsequent severe neurodegeneration changes [35]. Consistent with this idea, reduction of SREBP activities led to the suppression of lipid droplet accumulation and neurodegeneration in the miR-210 mutants. Our findings further support the notion that SREBP activation and lipid synthesis are important causes for the neuronal degeneration.
From the western blot analysis and RNA-Seq data, we also found that expression of many phototransduction genes decreased in miR-210 mutants. Interestingly, the decrease was not limited to the phototransduction cascade genes, but also applied to many other genes known to be specifically expressed in the photoreceptor. For example, decreased expression of one key transcription factor, pph13, was found in miR-210 mutants. As overexpression of SREBP has recently been shown to reduce the expression of photoreceptor enriched genes, it is possible that the expression changes of these photoreceptor genes are the secondary effect due to upregulation of SREBP in miR-210 mutants [39].
We have identified ACS as one miR-210 target through the bioinformatics and RNA-Seq analysis. Several lines of evidence support the regulation of ACS by miR-210. First, luciferase assay in S2 cells confirmed that miR-210 regulate ACS 3′-UTR activity through the predicated binding site. Second, ACS expression level was upregulated in miR-210 mutants. Third, removing half of ACS activity was able to partially rescue neuronal degeneration defects in miR-210 mutant retina. How ACS might regulate lipid metabolism-related gene expression remains unknown and will be our focus for further studies. One possibility is that ACS can function to promote SREBP acetylation and stabilize the nuclear SREBP. Alternatively, ACS plays a role in regulating histone acetylation and gene expression, which are required for SREBP activation and lipid metabolism-related gene expression. As miRNAs normally have many targets, it is also likely that other unidentified targets mediated alterations in lipid metabolism caused by miR-210 deletion.
While this paper was being prepared for publication, an independent study of Drosophila miR-210 was published [46]. In this study, Weigelt et al. reported that the loss of miR-210 led to photoreceptor degeneration in Drosophila [46]. The similarities and differences between their study and ours are discussed in the Supplementary text.
In summary, the generation of miR-210 mutant flies has allowed us to identify the in vivo relevance of this conserved miRNA in lipid metabolic regulation in the photoreceptor neurons. As miR-210 is highly conserved, further studies on the roles of miR-210 in mammalian adult sensory neurons are warranted. Such investigations will provide important insights into the neuronal function of miR-210 and its role in regulating lipid metabolism. Together, these studies will contribute to identify promising therapeutic targets for the neurodegenerative diseases.
Materials and methods
Fly stocks
Flies were maintained on a standard cornmeal diet at 25 °C and 70% relative humidity under a 12/12 h light/dark cycle. The following Drosophila stocks were used: w1118, miR-210KO (miR-210-Gal4 knock-in allele, Bloomington Drosophila Stock Center BS58899), Rh1-Gal4 (BS8691), UAS-mCD8::GFP, UAS-nGFP, SREBPΔ189 (BS38392), ACSCB-6224-3 (Kyoto Stock Center 123983), UAS-ACSRNAi (Vienna Drosophila Resource Center, 330367), w1118; PBac{y[+mDint2]=vas-Cas9}VK00027 (BS51324).
Generation of the miR-210▵130 mutant and pattB-miR-210 genomic rescue transgenic flies
We initially characterized the mutant phenotype of miR-210 Gal4 knock-in allele (miR-210KO), and found that this allele contained a background mutation, which prevented the rescue of the homozygous/hemizygous mutant phenotype by introducing the wild-type miR-210 transgene. An additional miR-210 deletion strain was then generated with the CRISPR/Cas9 system. To do this, two sgRNAs were designed to target two different sites at the 5′- and 3′-ends of the miR-210 gene using the online CRISPR design tool (http://www.flyrnai.org/crispr/). The target sequences were the following: 5′-GTGATTCACCGACCACGTC-3′ and 5′-GCCATTGTTCCATCATCG-3′.
To generate sgRNA templates for in vitro transcription, one common sgRNA scaffold primer (5′-AAAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC-3′) and one sgRNA gene-specific primer that contains the T7 promoter (sgRNA1: 5′-TAATACGACTCACTATAGTGATTCACCGACCACGTCGTTTTAGAGCTAGAAATAGC-3′; sgRNA2: 5′-TAATACGACTCACTATAGCCATTGTTCCATCATCG GTTTTAGAGCTAGAAATAGC-3′) were used for PCR with Ex-taq polymerase (TAKARA, RR001A). The PCR product was purified by AxyPrep DNA gel extraction kit (Axygen, AP-GX-50). The sgRNAs were transcribed with the T7 High Yield RNA Transcription Kit (Vazyme, TR101-01) and then purified with RNeasy Mini Kit (QIAGEN, 74104). Both sgRNAs were mixed and injected into PBac{y[+mDint2]=vas-Cas9}VK00027 embryos. The newly emerged F0 flies were individually balanced over FM6 to establish stable stocks. The F1 progenies of these flies were used for PCR screening and sequencing to determine the potential knockout strain. One mutant line was recovered and named as miR-210△130, which contains a 131-bp deletion followed by an additional 1 bp insertion (from ChX:18128204 to ChX:18128334). This deletion leads to the removal of the entire miR-210 stem loop. To get rid of the background effect, miR-210△130 mutant line was outcrossed with w1118 flies for three generations.
To generate the miR-210 genomic rescue transgene, a 2.5 kb DNA fragment containing the miR-210 stem loop was amplified with primers (5′-GGATCCAATCTGTCACTTGATACGCG-3′ and 5′-GGATCCATTTGATTTTATTTGAAACT-3′) and then subcloned into the pattB vector. The obtained construct was inserted at the 25C6 site on the second chromosome using site-specific phiC31 integrase according to standard germ-line transformation procedures.
Quantitative RT-PCR
Total RNA was extracted from ~20 fly heads using TRIzol reagent (Sangon Biotech, B610409). cDNA was synthesized using a cDNA reverse transcription kit and oligo(dT) primers (Vazyme, R211-01). Quantitative RT-PCR was performed using the SYBR Green PCR master mix (Vazyme, Q311-02) on an Applied Biosystems 7900HT system. A list of primers used for the experiments is provided in Table S5. Rp49 was used as an internal control.
For the quantification of mature miRNA, small RNA was isolated using a mirVana™ miRNA Isolation Kit (InvitrogenTM, AM1561) and reversed to cDNA by a TaqMan™ MicroRNA Reverse Transcription Kit (Applied BiosystemsTM, 4366596). Real-time quantitative PCR was performed using a TaqMan MicroRNA Assay kit (Applied BiosystemsTM, 4427788) on an Applied Biosystems 7900HT system. The U14 small RNA was used as an internal control.
Nile red and phalloidin staining
Nile red and phalloidin staining of fly retinas were performed with whole-mount preparations as previously described [35]. Heads were removed from adult flies and fixed in phosphate buffered saline (PBS) containing 4% formaldehyde for 1 h at room temperature. After removing the head cuticle and brain tissues, retinas were fixed for additional 30 min. Fixed retinas were then washed three times (20 min each time) with PBS and incubated with 1 mg/ml Nile red (Sigma 72485-100MG, 1:1000) for 20 min at room temperature or with FITC-conjugated Phalloidin (Thermo Fisher, A12379) overnight at 4 °C. After incubation, retinas were washed three more times (20 min each time) and mounted in Vectashield mounting medium (Vector Labs). Images were obtained with an Olympus FV1000 confocal microscope.
Immunofluorescent staining
Dissection and immunofluorescent staining of larval eye imaginal discs, pupal retinas, adult retinas, and brains followed a previous protocol [47]. Briefly, samples were dissected and fixed in PBS with 4% paraformaldehyde for 20 min at room temperature. After three washes with PBT (0.1% Triton X-100 in PBS), samples were blocked in PBT containing 3% BSA for 1 h. Next, samples were incubated with primary antibodies overnight at 4 °C. Samples were then washed three more times with PBT before incubating with secondary antibodies for 2 h at room temperature. Primary antibodies used were: chicken anti-GFP (1:2000; ab13970; Abcam), mouse anti-chaoptin (1:100; 24B10; DSHB), mouse anti-Eys (1:100; 21A6; DSHB), and rat anti-Elav (1:50). Secondary antibodies used were: Alexa Fluor 488 Goat anti-Chicken, Alexa Fluor 594 Goat anti-Mouse or Rat (1:500; 103-545-155/115-585-003/112-585-003; Jackson ImmunoRsesearch). DAPI was added for the last 15 min (1:2000; 62247; Thermo Fisher). After three washes with PBT, samples were mounted in Vectashield mounting medium (Vector Laboratories). Images were acquired using an Olympus FV1000 confocal microscope.
Plasmids
To generate the pTub-miR-210 plasmid, a 340 bp fragment containing miR-210 hairpin was amplified from wild-type genomic DNA with primers (5′-GATCAGATCCGCGGCCGCCGGTTCGGCAATGTCCGTTG-3′ and 5′-ATTATGATCTAGCTCGAGTGCTGAACAAATCCTTCGAT-3′), and subcloned into the pCaspeR4-Tub-SV40-PolyA vector. For construction of ACS 3′UTR luciferase reporter, a 496 bp fragment containing the 3′UTR of ACS was amplified from wild-type genomic DNA with primers (5′-ATGGTAGCTGTTGGACCAAC-3′ and 5′-AAATTGTGGCGACAATTGG-3′), and subcloned into the pCaspeR4-Tubulin firefly luciferase vector. Mutations of the miR-210 binding site in the ACS 3′UTR were introduced by performing overlapping PCR with primers (5′-GTAGTTGACATGCTCACCCATAGGTCATTTTCCAGTTC-3′ and GGTGAGCATGTCAACTAC).
Cell culture and transfection
Drosophila S2 cells were cultured at 25 °C in Schneider’s medium (Sigma, S0146-500ML) supplemented with 10% fetal bovine serum (Gemini, 900-108 500ML). S2 cells were transfected using X-tremeGene HP (Roche, 6366236001) in 24-well plates with 25 ng of the firefly luciferase reporter, 25 ng of the Renilla luciferase control plasmid, and 250 ng of the miRNA expression plasmid or empty vector. Transfections were done in triplicate in three independent experiments. Sixty hours after transfection, luciferase activity was assayed using the Dual-luciferase reporter system (Promega™, E1960) according to the manufacturer’s instruction.
Transmission electron microscopy
TEM was performed according to standard procedures. Fly eyes were dissected and fixed with 2.5% glutaraldehyde in 1x PBS (pH7.4) for 2 days. Fixed samples were washed three times with PBS and then postfixed with 1% OsO4 (SPI-Chem, 20816-12-0) in phosphate buffer for 2 h. After three washes with PBS, samples were dehydrated in a series of ethanol dilution (30, 50, 70, 80, 90, and 95%) and embedded in Spurr’s resin (SPI-Chem, 02680-AB). Thin sections were obtained with a LEICA EM UC7 ultratome and collected on copper grids. Samples were stained with uranyl acetate and alkaline lead citrate and observed using a Hitachi Model H-7650 transmission electron microscope.
Western blot
Fly heads were homogenized in a modified RIPA buffer (250 mM Tris, 750 mM NaCl, 5% Triton X-100, 5 mM EDTA, 0.5% SDS, 2.5% Sodium deoxycholate) supplemented with the protease inhibitor cocktail (Roche, 4693132001) and PhosStop phosphatase inhibitor cocktail (Roche, 4906845001). The lysate was centrifuged and the supernatant was collected and mixed with an equal amount of 2x protein loading buffer. Samples were resolved by SDS-PAGE, transferred to Immobilon-P PVDF membranes (Millipore, IPVH00010), and probed with the primary and secondary antibodies. The following primary antibodies were used: mouse anti-Rh1 (1:100; 4C5; DSHB), rabbit anti-INAD (1:2000), rabbit anti-PLC (1:2000), mouse anti-SREBP (1:2000; 557036; BD Biosciences), and mouse anti-Tubulin (1:2000; AT819; Beyotime Biotechnology). Anti-ACS was raised in rabbit against a peptide corresponding to the C-terminal amino acids 564-577 of ACS (CAESAVVSRPHPVKG) (1:2000; GeneScript). Appropriate HRP conjugated secondary antibodies were used (A0216 and A0208; Beyotime Biotechnology). Blots were developed using the ECL detection system, and bands were visualized with the Chemiluminescence Imaging System.
Lipid profiling
Fly heads were dissected and snap-frozen in liquid nitrogen. Samples from six biological replicates for each genotype were homogenized in lysis buffer (MeOH:H2O, 1:1, v/v) using a motorized pestle. The lysate was centrifuged at 13,000 rpm for 20 min at 4 °C. After removing the supernatants, the lysis buffer (DCM:MeOH, 3:1, v/v) was added and the mixture was vortexed for 1 min. Samples were then centrifuged at 13,000 rpm for 20 min. After centrifugation, the lower phase was collected and diluted by adding equal amount of lysis buffer (DCM:MeOH, 3:1, v/v). Samples were subsequently used for lipidomic analysis. LC-MS analysis was performed on a Waters Acquity™ UPLC system with a C18 CSH column equipped with a Waters Xevo™ QT of mass spectrometer. Samples were analyzed both in positive and negative ionization modes. Lipid identification was performed using Progenesis QI and other online tools, including Lipidmaps (http://www.lipidmaps.org), HMDB (http://www.hmdb.ca), and NIST (http://chemdata.nist.org).
RNA-Seq analysis
Total RNA was extracted from fly heads using TRIzol reagent. Sequencing libraries were generated using NEBNext UltraTM RNA Library Prep Kit (NEB, E7530L) for Illumina according to the manufacturer’s protocol. Libraries from three biological replicates for each sample were sequenced on an Illumina NovaSeq 6000 platform, with paired-end 150 bp reads. RNA-Seq reads were mapped to the Drosophila genome (dm6) using Hisat2 and quantified using FeatureCounts. Differential gene expression was analyzed using DESeq2, and differentially regulated genes were determined using a cutoff significance level of false discovery rate of <0.05 and fold change > 1.5. KEGG analysis was performed using clusterProfiler in R. Raw sequencing data are available through Gene Expression Omnibus (accession number GSE146335).
References
McNeill E, Van Vactor D. MicroRNAs shape the neuronal landscape. Neuron. 2012;75:363–79.
Alberti C, Cochella L. A framework for understanding the roles of miRNAs in animal development. Development. 2017;144:2548–59.
Dubes S, Favereaux A, Thoumine O, Letellier M. miRNA-dependent control of homeostatic plasticity in neurons. Front Cell Neurosci. 2019;13:536.
Esteves M, Serra-Almeida C, Saraiva C, Bernardino L. New insights into the regulatory roles of microRNAs in adult neurogenesis. Curr Opin Pharm. 2019;50:38–45.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205.
Abe M, Bonini NM. MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol. 2013;23:30–6.
Kye MJ, Goncalves, Ido C. The role of miRNA in motor neuron disease. Front Cell Neurosci. 2014;8:15.
Hawley ZCE, Campos-Melo D, Droppelmann CA, Strong MJ. MotomiRs: miRNAs in motor neuron function and disease. Front Mol Neurosci. 2017;10:127.
Quinlan S, Kenny A, Medina M, Engel T, Jimenez-Mateos EM. MicroRNAs in neurodegenerative diseases. Int Rev Cell Mol Biol. 2017;334:309–43.
Chen TH, Chen JA. Multifaceted roles of microRNAs: from motor neuron generation in embryos to degeneration in spinal muscular atrophy. Elife. 2019;8:e50848.
Karres JS, Hilgers V, Carrera I, Treisman J, Cohen SM. The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell. 2007;131:136–45.
Amin ND, Bai G, Klug JR, Bonanomi D, Pankratz MT, Gifford WD, et al. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science. 2015;350:1525–9.
Verma P, Augustine GJ, Ammar MR, Tashiro A, Cohen SM. A neuroprotective role for microRNA miR-1000 mediated by limiting glutamate excitotoxicity. Nat Neurosci. 2015;18:379–85.
Salta E, Sierksma A, Vanden Eynden E, De Strooper B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol Med. 2016;8:1005–18.
Aw SS, Lim IKH, Tang MXM, Cohen SM. A Glio-protective role of mir-263a by tuning sensitivity to glutamate. Cell Rep. 2017;19:1783–93.
El Fatimy R, Li S, Chen Z, Mushannen T, Gongala S, Wei Z, et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018;136:537–55.
Huang X, Le QT, Giaccia AJ. MiR-210-micromanager of the hypoxia pathway. Trends Mol Med. 2010;16:230–7.
Ivan M, Huang X. miR-210: fine-tuning the hypoxic response. Adv Exp Med Biol. 2014;772:205–27.
Camps C, Buffa FM, Colella S, Moore J, Sotiriou C, Sheldon H, et al. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14:1340–8.
Lulla RR, Costa FF, Bischof JM, Chou PM, de FBM, Vanin EF, et al. Identification of differentially expressed microRNAs in osteosarcoma. Sarcoma. 2011;2011:732690.
Puissegur MP, Mazure NM, Bertero T, Pradelli L, Grosso S, Robbe-Sermesant K, et al. miR-210 is overexpressed in late stages of lung cancer and mediates mitochondrial alterations associated with modulation of HIF-1 activity. Cell Death Differ. 2011;18:465–78.
Dang K, Myers KA. The role of hypoxia-induced miR-210 in cancer progression. Int J Mol Sci. 2015;16:6353–72.
Qin Q, Furong W, Baosheng L. Multiple functions of hypoxia-regulated miR-210 in cancer. J Exp Clin Cancer Res. 2014;33:50.
Hu YW, Jiang JJ, Yan G, Wang RY, Tu GJ. MicroRNA-210 promotes sensory axon regeneration of adult mice in vivo and in vitro. Neurosci Lett. 2016;622:61–6.
Riccardi S, Bergling S, Sigoillot F, Beibel M, Werner A, Leighton-Davies J, et al. MiR-210 promotes sensory hair cell formation in the organ of corti. BMC Genomics. 2016;17:309.
Cristino AS, Barchuk AR, Freitas FC, Narayanan RK, Biergans SD, Zhao Z, et al. Neuroligin-associated microRNA-932 targets actin and regulates memory in the honeybee. Nat Commun. 2014;5:5529.
Cusumano P, Biscontin A, Sandrelli F, Mazzotta GM, Tregnago C, De Pitta C, et al. Modulation of miR-210 alters phasing of circadian locomotor activity and impairs projections of PDF clock neurons in Drosophila melanogaster. PLoS Genet. 2018;14:e1007500.
Niu Y, Liu Z, Nian X, Xu X, Zhang Y. miR-210 controls the evening phase of circadian locomotor rhythms through repression of Fasciclin 2. PLoS Genet. 2019;15:e1007655.
Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–64.
Chen YW, Song S, Weng R, Verma P, Kugler JM, Buescher M, et al. Systematic study of Drosophila microRNA functions using a collection of targeted knockout mutations. Dev Cell. 2014;31:784–800.
Seegmiller AC, Dobrosotskaya I, Goldstein JL, Ho YK, Brown MS, Rawson RB. The SREBP pathway in Drosophila: regulation by palmitate, not sterols. Dev Cell. 2002;2:229–38.
Lim HY, Wang W, Wessells RJ, Ocorr K, Bodmer R. Phospholipid homeostasis regulates lipid metabolism and cardiac function through SREBP signaling in Drosophila. Genes Dev. 2011;25:189–200.
Song W, Veenstra JA, Perrimon N. Control of lipid metabolism by tachykinin in Drosophila. Cell Rep. 2014;9:40–7.
Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–90.
Meltzer S, Bagley JA, Perez GL, O’Brien CE, DeVault L, Guo Y, et al. Phospholipid homeostasis regulates dendrite morphogenesis in Drosophila sensory neurons. Cell Rep. 2017;21:859–66.
Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology—divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710–30.
Senos Demarco R, Uyemura BS, D’Alterio C, Jones DL. Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat Cell Biol. 2019;21:710–20.
Tsai JW, Kostyleva R, Chen PL, Rivas-Serna IM, Clandinin MT, Meinertzhagen IA, et al. Transcriptional feedback links lipid synthesis to synaptic vesicle pools in Drosophila photoreceptors. Neuron. 2019;101:721–37.e4.
Xu H, Lee SJ, Suzuki E, Dugan KD, Stoddard A, Li HS, et al. A lysosomal tetraspanin associated with retinal degeneration identified via a genome-wide screen. EMBO J. 2004;23:811–22.
Xu Y, Wang T. CULD is required for rhodopsin and TRPL channel endocytic trafficking and survival of photoreceptor cells. J Cell Sci. 2016;129:394–405.
Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–21.
Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee JH, et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol Cell. 2017;66:684–97.e9.
Mews P, Donahue G, Drake AM, Luczak V, Abel T, Berger SL. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017;546:381–6.
Huang Z, Zhang M, Plec AA, Estill SJ, Cai L, Repa JJ, et al. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc Natl Acad Sci USA. 2018;115:E9499–506.
Weigelt CM, Hahn O, Arlt K, Gruhn M, Jahn AJ, Esser J, et al. Loss of miR-210 leads to progressive retinal degeneration in Drosophila melanogaster. Life Sci Alliance. 2019;2:e201800149.
Hsiao HY, Johnston RJ Jr., Jukam D, Vasiliauskas D, Desplan C, Rister J. Dissection and immunohistochemistry of larval, pupal and adult Drosophila retinas. J Vis Exp. 2012;69:4347.
Depetris-Chauvin A, Galagovsky D, Grosjean Y. Chemicals and chemoreceptors: ecologically relevant signals driving behavior in Drosophila. Front Ecol Evol. 2015;3:41.
Acknowledgements
We thank Zhefeng Gong, John R. Carlson, Junhai Han, the Developmental Studies Hybridoma Bank, the Vienna Drosophila Resource Center, the Kyoto and Bloomington Drosophila stock center for fly stocks and antibodies; Caodi Xu for the initial characterization of miR-210 expression pattern; Feng He for the help with the generation of heatmap. We are grateful to Jun Ma, Ting Gang Chew, Pushpa Sharma, Chao Tong, Feng He, Tao Wang, Zhefeng Gong, Jia Huang, and Nan Liu for their suggestions and help for various experiments. This work was funded by the National Key Research and Development Program of China (2018YFC1003200), and also by a grant (31970668) from the National Natural Science Foundation of China.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: E. Baehrecke
Rights and permissions
About this article

Cite this article
Lyu, J., Chen, Y., Yang, W. et al. The conserved microRNA miR-210 regulates lipid metabolism and photoreceptor maintenance in the Drosophila retina. Cell Death Differ 28, 764–779 (2021). https://doi.org/10.1038/s41418-020-00622-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00622-w
This article is cited by
-
p53-driven lipidome influences non-cell-autonomous lysophospholipids in pancreatic cancer
Biology Direct (2022)
-
Abundant small RNAs in the reproductive tissues and eggs of the honey bee, Apis mellifera
BMC Genomics (2022)
-
Profiling ATM regulated genes in Drosophila at physiological condition and after ionizing radiation
Hereditas (2022)
