

Abstract
Leptin signaling within the nucleus of the solitary tract (NTS) contributes to the control of food intake, and injections of leptin into the NTS reduce meal size and increase the efficacy of vagus-mediated satiation signals. Leptin receptors (LepRs) are expressed by vagal afferents as well as by a population of NTS neurons. However, the electrophysiological properties of LepR-expressing NTS neurons have not been well characterized, and it is unclear how leptin might act on these neurons to reduce food intake. To address this question, we recorded from LepR-expressing neurons in horizontal brain slices containing the NTS from male and female LepR-Cre X Rosa-tdTomato mice. We found that the vast majority of NTS LepR neurons received monosynaptic innervation from vagal afferent fibers and LepR neurons exhibited large synaptic NMDA receptor (NMDAR)-mediated currents compared with non-LepR neurons. During high-frequency stimulation of vagal afferents, leptin increased the size of NMDAR-mediated currents, but not AMPAR-mediated currents. Leptin also increased the size of evoked EPSPs and the ability of low-intensity solitary tract stimulation to evoke action potentials in LepR neurons. These effects of leptin were blocked by bath applying a competitive NMDAR antagonist (DCPP-ene) or by an NMDAR channel blocker applied through the recording pipette (MK-801). Last, feeding studies using male rats demonstrate that intra-NTS injections of DCPP-ene attenuate reduction of overnight food intake following intra-NTS leptin injection. Our results suggest that leptin acts in the NTS to reduce food intake by increasing NMDAR-mediated currents, thus enhancing NTS sensitivity to vagal inputs.
SIGNIFICANCE STATEMENT Leptin is a hormone that critically impacts food intake and energy homeostasis. The nucleus of the solitary tract (NTS) is activated by vagal afferents from the gastrointestinal tract, which promotes termination of a meal. Injection of leptin into the NTS inhibits food intake, while knockdown of leptin receptors (LepRs) in NTS neurons increases food intake. However, little was known about how leptin acts in the NTS neurons to inhibit food intake. We found that leptin increases the sensitivity of LepR-expressing neurons to vagal inputs by increasing NMDA receptor-mediated synaptic currents and that NTS NMDAR activation contributes to leptin-induced reduction of food intake. These findings suggest a novel mechanism by which leptin, acting in the NTS, could potentiate gastrointestinal satiation signals.
Introduction
The nucleus of the solitary tract (NTS) receives viscerosensory information carried by vagal afferent fibers from gastrointestinal (GI), cardiovascular, and respiratory systems (Andresen and Kunze, 1994; Berthoud, 2008; Zoccal et al., 2014). Food-related mechanical and chemical stimuli activate GI vagal afferents, which transmit this information to NTS neurons contributing to satiation (Berthoud, 2008; Grill and Hayes, 2012). The NTS also receives projections from the forebrain, including the hypothalamus, and these projections also participate in the control of food intake (Berthoud, 2008; Grill and Hayes, 2012). Parts of the NTS contain fenestrated capillaries (Gross et al., 1990) and receive inputs from the area postrema, a highly vascularized circumventricular organ (Price et al., 2008), potentially allowing NTS neurons to respond to circulating hormones and nutrients. Finally, NTS neurons have extensive projections, including to key nuclei that influence feeding, such as the hypothalamus, basolateral amygdala, mesolimbic system, parabrachial nucleus, and dorsal motor nucleus of the vagus (Travagli et al., 1991; Schwartz et al., 2000; García-Medina and Miranda, 2013; Maniscalco and Rinaman, 2014; Richard et al., 2015; Palmiter, 2018). The NTS is therefore ideally positioned to integrate viscerosensory information with metabolic conditions and motivational state in the control of food intake.
Vagal afferents release glutamate to activate NTS neurons (Talman et al., 1980; Bailey et al., 2006a; Peters et al., 2008) via both AMPA-type and NMDA-type glutamate receptors (Andresen and Kunze, 1994; Aylwin et al., 1997; Yen et al., 1999; Jin et al., 2003; Baptista et al., 2005; Balland et al., 2006, 2008; Zhao et al., 2015). The contribution of AMPA receptors to the activation of NTS neurons has been studied extensively (Andresen and Kunze, 1994; Doyle and Andresen, 2001; Appleyard et al., 2005). NMDA receptors are also critical for maintaining the fidelity of synaptic transmission at the vagus–NTS synapse during periods of high-frequency stimulation, as occurs following ingestion of a meal (Zhao et al., 2015). Many NTS neurons receive direct monosynaptic inputs from the vagus and are themselves projection neurons that relay vagal signals to higher brain structures (Andresen et al., 2004; Bailey et al., 2006a). Therefore, changes in the fidelity of transmission at the level of the vagus–NTS synapse likely have pronounced effects on how much visceral information downstream targets receive.
Leptin is an adipokine secreted by white adipose tissue that has been shown to act at multiple sites in the CNS to influence food intake and energy balance (Schwartz et al., 2000; Moran et al., 2006; Allison and Myers, 2014; Friedman, 2014). Intra-NTS leptin reduces meal size, promotes weight loss, and stimulates gene transcription (Grill et al., 2002; Maniscalco and Rinaman, 2014; Matheny et al., 2014). The long form of the leptin receptor (LepR) is expressed in vagal afferents (Buyse et al., 2001; Burdyga et al., 2002), and leptin potentiates vagal firing in response to cholecystokinin (CCK; Emond et al., 1999; Peters et al., 2006). Selective knockdown of LepRs in vagal afferent neurons increases food intake and body weight (de Lartigue et al., 2014). In addition, selective knockdown of LepRs in NTS neurons is associated with hyperphagia, weight gain, and attenuation of responses to satiety signals such as CCK and intraluminal nutrients (Hayes et al., 2010; Kanoski et al., 2012). Furthermore, activation of LepR-expressing neurons inhibits food intake and reduces body weight (Cheng et al., 2020). Together, this suggests that both presynaptic and postsynaptic LepRs at the vagal–NTS synapse are important for the control of food intake, yet the underlying mechanisms by which leptin influences NTS neuronal activity are not well understood.
LepRs and NMDARs are both expressed by subpopulations of NTS neurons (Elmquist et al., 1998; Aicher et al., 1999), and leptin has been shown to increase NMDAR function in the hippocampus (Shanley et al., 2001; Bland et al., 2020). The aim of this study is therefore to determine whether leptin increases NTS sensitivity to vagal input by potentiating NMDAR-mediated currents in NTS LepR neurons and whether NMDARs are required for leptin-induced inhibition of food intake at the level of the NTS.
Materials and Methods
Animals.
All mice and rats had free access to standard chow and water and were maintained on a 12 h light/dark cycle at room temperature in the Department of Integrative Physiology and Neuroscience vivarium. Transgenic mice on a C57BL/6J background were bred by crossing leptin receptor-Cre animals (LepR-Cre; provided by Martin Myers, University of Michigan, Ann Arbor, MI) with a fluorescent reporter mouse line (Lox-P-Rosa tdTomato, Allen Brain Institute). All animal procedures were conducted with the approval of the Institutional Animal Care and Use Committees at Washington State University and in accordance with the US Public Health Service Policy on Humane Care and Use of Laboratory Animals (PHS Policy) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Guide).
Horizontal NTS slices.
Adult male and female transgenic mice (8–16 weeks of age) were used in all electrophysiological experiments. Mice were anesthetized with isoflurane, and then the hindbrain was removed and placed for 1 min in cold (0–4°C) artificial CSF (aCSF) composed of the following (in mm): 125 NaCl, 3 KCl, 1.2 KH2PO4, 1.2 MgSO4, 25 NaHCO3, 2 CaCl2, and 10 dextrose, bubbled with 95% O2/5% CO2. The final osmolarity was adjusted to 305–307 mOsm/L using sucrose. For Mg2+-free experiments, MgSO4 was omitted from the aCSF used for recording (osmolarity was kept within the same range with extra sucrose). The medulla was trimmed, and a wedge was removed from the ventral surface to align the solitary tract (ST) with the NTS in the same cutting plane when mounted in a vibrating microtome (catalog #VT-1000S, Leica). Hindbrain slices (250 µm) were cut with a sapphire knife (Delaware Diamond Knives) and contained a long section of the ST. Slices were submerged in a perfusion chamber and were allowed to recover for 1 h in oxygenated aCSF flowing at a rate of ∼2 ml/min. All recordings were performed at a near-constant temperature (31.5–32°C) and pH 7.4.
Whole-cell electrophysiology.
LepRCre-tdTom neurons were visually selected for recordings using a fluorescence microscope (model BX51WI, Olympus). NTS neurons were recorded within 200 µm rostral or caudal of the obex and medial to the ST, and recording electrodes were guided to neurons one to three cell layers deep using differential interference contrast (DIC) optics. Recording electrodes (3–4 MΩ) were filled with an internal solution composed of the following (in mm): 10 NaCl, 130 potassium gluconate, 11 EGTA, 1 CaCl2, 2 MgCl2, 10 HEPES, 2 NaATP, and 0.2 NaGTP, pH 7.3 and 297–300 mOsm/L. For the depolarized “flip” protocol (neurons held at +40 mV), internal NaCl was replaced with CsCl, potassium gluconate was replaced with cesium methanesulfonate, and the pH was adjusted with CsOH. Whole-cell voltage-clamp and current-clamp (CC) recordings were made with an Axopatch 700 B amplifier, a Digidata 1440 A digitizer, and pCLAMP 10 software (all from Molecular Devices). Solutions flowing to the recording chamber were heated using a HPRE2 preheater (Cell Micro Controls), and bath temperature was monitored through a probe positioned next to the slice. Neurons with holding currents exceeding ±100 pA at a Vh (holding potential) of −60 mV during the initial 10 min control period were excluded from further study. For current-clamp experiments, only neurons with all action potentials (APs) reaching >0 mV were included in the study. Series resistance was monitored throughout recordings, and neurons were excluded from analysis if they were >20 MΩ or varied >5 MΩ. Synaptic responses were evoked with an ultrafine concentric bipolar stimulating electrode (inner diameter, 50 µm; FHC) placed on the ST 1–5 mm from the recording electrode. Electrical stimuli were delivered from an isolated programmable stimulator (Isoflex stimulator with Master-8, A.M.P.I.) triggered to deliver a burst of stimuli (5–50 Hz). Neurons were included only if electrical stimulation of the ST recruited one or more afferents contributing to an EPSC. In the voltage-clamp stimulus train experiments, the average size of the sustained current was measured as the baseline immediately after the fifth shock in the train, where the current normally reached a plateau. In the synaptic throughput experiments, stimulus intensity was slowly increased until a basal throughput of ∼0.3 was reached for each cell (∼6 of 20 possible evoked action potentials), and that intensity was used for the duration of that recording. All drugs were obtained from Tocris Cookson, Sigma-Aldrich, or Peprotech.
Overnight food intake study.
Ten male rats weighing between 298 and 400 g were anesthetized with isoflurane and implanted with 33 ga stainless steel guide cannulas stereotaxically aimed for the dorsal vagal complex (DVC) as previously described (Treece et al., 1998; Wright et al., 2011). Except during intracranial injections, the cannulas were occluded with 33 ga stainless steel obturators. The rats were allowed 2 weeks to recover from cannula implantation surgery, during which time they were weighed and handled daily. The animals were housed singly in suspended stainless steel mesh cages in a vivarium on a 12 h light/dark cycle (lights off at 7:00 P.M. and on at 7: A.M.). They had ad libitum access to water and pelleted laboratory rodent diet (catalog # 5001, Lab Diet) in food hoppers attached to their cages. At 4:30 P.M. on experimental days, the rats received intracranial injections of sterile 0.9% NaCl, leptin (200 ng) in NaCl, DCPP-ene (DCP; 20 ng) in NaCl, or leptin plus DCPP-ene in NaCl. All injections were 100 nl in volume and were made over 60 s via a 33 ga injector, using a microprocessor-controlled nanoliter infusion pump. The rats were tested in two separate subgroups on different days with different injection orders, and all injections were separated by at least 48 h such that each animal received all injection combinations within a ∼6 d period. Immediately following each intracranial injection, the rats were returned to their home cages and a weighed amount of fresh food was placed in each of their food hoppers. At 9:00 A.M. the following morning, the rats were reweighed, as was the remaining food and spillage, and the amount of overnight food intake was determined. After all 10 rats had received each of the four injection combinations, the rats were deeply anesthetized with isoflurane, exsanguinated, and perfused with phosphate-buffered NaCl, followed by 4% paraformaldehyde, pH 7.4. The brains were subsequently removed and postfixed for 12–15 h in 4% paraformaldehyde then cryoprotected by refrigerated storage in 25% sucrose for at least 24 h. Cannula tip placements were determined by microscopic examination of 30 μm hindbrain sections stained with cresyl violet. All rats included in our analysis had cannula tips in the DVC, and all reduced their overnight food intake by at least 1 g following the DVC injection of leptin alone.
Experimental design and statistical analyses.
All data are presented as the mean ± SEM. Both male and female animals were used for electrophysiological experiments with a roughly equal distribution of male and female mice for all conditions tested, and n refers to the numbers of independent cells recorded per condition. We found no significant differences in responses between male and female mice, therefore data were combined for further analysis. Male rats were used for the feeding study. Statistical comparisons were made using Mann–Whitney, Student's t test, or two-way ANOVA with Bonferroni post hoc analysis where appropriate (SigmaPlot 14.0). Differences were considered statistically significant for p values <0.05, with the actual p values provided in the Results section. Food intake data were analyzed using a repeated-measures ANOVA and Dunnett's test for multiple comparisons. The confidence limit was set for 0.05, and actual p values are provided in the Results section.
Results
Solitary tract stimulation evokes both monosynaptic and polysynaptic excitatory responses in NTS LepR neurons
Cell bodies of NTS LepR neurons in horizontal brain slices were easily identified for recordings through using a microscope equipped with DIC optics and epifluorescence illumination to detect expression of tdTomato under high magnification (Fig. 1A). Consistent with a previous report (Garfield et al., 2012), we found that LepR neurons were distributed diffusely throughout the NTS. LepR-positive (n = 40) and negative (n = 8) neurons had similar whole-cell capacitance (31.7 ± 1.2 pF vs 30.43 ± 3.9, 214 p = 0.769, Student's t test) and could not be easily distinguished by any visual comparison.

Solitary tract stimulation evokes both monosynaptic and polysynaptic excitatory responses in NTS LepR neurons. A, Horizontal brain slice preparation including the ST, fourth ventricle (4V), and NTS. Top left, dotted circle, General region of the NTS where LepR neurons were recorded. The stimulating electrode was placed 1–3 mm away from recorded neurons at the base of the ST. LepR neurons were identified by tdTomato fluorescence (bottom panels) and visualized using DIC. B, Voltage-clamp traces from separate LepR neurons showing compound EPSCs evoked by electric shocks to the ST. LepR neurons received monosynaptic inputs with low-variance latencies (circles) and polysynaptic input with moderate failure rates and high-variance latencies (triangles) following ST stimulation. The top trace shows an example of a neuron receiving one monosynaptic input and one polysynaptic input. The lower trace shows an example of a neuron receiving three polysynaptic inputs. C, D, Scatterplot and distribution of ST-driven excitatory input to LepR neurons. All neurons with at least one discernable monosynaptic connection (circles) also received polysynaptic input, while six neurons (triangles) did not display a monosynaptic connection based on our criteria.
We first determined whether LepR neurons in the NTS receive direct or indirect synaptic inputs from vagal afferent fibers. The vagus enters the NTS in the ST and stimulation of the ST-evoked compound EPSCs in all NTS LepR neurons between 2 and 7.5 ms after stimulation (Fig. 1B). To confirm that these EPSCs were the result of ST activation and not direct stimulation of local interneurons, during initial recordings we placed the stimulating electrode off the ST and verified that we were unable to elicit evoked EPSCs (data not shown). As previously described, NTS neurons were considered to have direct, monosynaptic connections to the solitary tract if ST-evoked EPSCs had low variability in latency to onset (jitter, <200 μs) and ST stimulation never failed to elicit EPSCs (Doyle and Andresen, 2001). ST-evoked EPSCs with multiple failures or jitter >200 µs were considered polysynaptic (Fig. 1B). Based on the presence of at least one discernable monosynaptic response to ST stimulation, 85% of recorded NTS LepR neurons (34 of 40 neurons) were identified as second-order neurons with a direct connection to the ST (Fig. 1C).
Every LepR neuron also received one or more polysynaptic responses following ST stimulation. Therefore, all evoked EPSCs examined were compound EPSCs comprised of both monosynaptic (direct) and polysynaptic (indirect) inputs. The number of polysynaptic EPSCs was striking, approximately three per neuron, which is higher than has been reported in other NTS populations, such as TH-EGFP (catecholamine; Appleyard et al., 2007), POMC-EGFP (Appleyard et al., 2005), and GAD67-EGFP (GABA) neurons (Bailey et al., 2008). It is possible that the proportion of LepR neurons receiving direct input from the ST is >85%, as concomitant polysynaptic EPSCs could introduce enough variability to push an input outside the bounds of our criteria (i.e., an apparent jitter >200 µs). Furthermore, some LepR neurons may have lost direct fiber connections during slice preparation (ST fibers above the plane of the slice). Plotting the approximate distribution of LepR neurons within the NTS that receive discernable monosynaptic ST inputs and those without did not reveal an obvious correlation between anatomic location and direct versus indirect ST connectivity (Fig. 1D). We also did not observe any significant sex difference in the total number of polysynaptic inputs per cell (male: n = 25, 2.6 ± 0.2; vs female: n = 15, 2.9 ± 0.2, p = 0.166; Mann–Whitney test). Together, these data show that the vast majority of NTS LepR neurons are second-order neurons that receive both direct input from the solitary tract as well as indirect inputs, suggesting that LepR neurons are well positioned to integrate information from multiple afferent fibers entering the NTS via the solitary tract.
To assess whether GABAA inputs altered the ST-evoked EPSCs, we recorded in the presence of the GABAA receptor antagonist gabazine. Bath application of gabazine did not affect the amplitude of evoked EPSCs (control/ACSF: 130.14 ± 17.02 pA; vs 10 μm gabazine: 128.61 ± 17.60 pA; t(7) = 0.349, p = 0.738, paired t test) or the latency of the monosynaptic inputs (EPSC latency: ACSF: 3.89 ± 0.58 ms; vs gabazine: 3.92 ± 0.59 ms; t(7) = 1.001, p = 0.35; EPSC jitter: ACSF: 188.78 ± 39.39 µs vs gabazine: 177.75 ± 35.92 µs; t(7) = 1.284, p = 0.24, paired t tests). It is important to note however, that we record under conditions that minimize the size of GABAA currents, and it is therefore possible that GABA does have important effects on these neurons under some conditions.
NTS LepR neurons express larger NMDAR-mediated currents than non-LepR neurons
In Figure 1, ST-EPSCs were recorded from cells held at −60 mV, a potential at which NMDA receptors are typically blocked by extracellular Mg2+. To evaluate the contribution of NMDA receptors to ST-EPSCs in LepR and non-LepR neurons, we performed voltage-clamp recordings under conditions that would remove the Mg block and allow us to measure NMDAR-mediated currents. We first recorded in 1.2 mm [Mg2+] at −60 mV and then again at +40 mV (to remove the Mg2+ block), a protocol that has been used to study the relative contributions of AMPA-type and NMDA-type receptors to evoked EPSCs (Baptista et al., 2005; Balland et al., 2008; Zhao et al., 2015; Fig. 2A,B). These recordings were taken in the presence of 5 μm gabazine (to block GABAA receptor currents) and with CsCl in the pipette to block potassium channels. At −60 mV, ST stimulation produced fast-decaying EPSCs that were blocked by the AMPAR antagonist NBQX (20 μm), and there was no significant difference in EPSC peak amplitude between LepR neurons (n = 9) and non-LepR neurons (n = 8; 173.8 ± 44 vs 171.4 ± 36 pA; p = 0.81, Mann–Whitney test; Fig. 2C). During ST stimulation at +40 mV holding conditions, all LepR neurons tested (n = 17) and the majority of non-LepR neurons (n = 9 neurons) displayed a slow-rising, slow-decaying EPSC that was blocked by the NMDAR antagonist DCP. The average peak NMDAR current amplitude was significantly larger in LepR neurons than in non-LepR neurons (150.2 ± 15.2 vs 95.9 ± 17.8 pA; t(24) = 2.203, p = 0.037, Student's t test; Fig. 2D). When comparing charge transfer (area under the curve), LepR neurons displayed an approximately twofold larger charge transfer through NMDARs than non-LepR neurons (23.9 ± 3.5 vs 11.3 ± 2.6 nA/ms; p = 0.015, Mann–Whitney test; Fig. 2E). Although peak NMDAR current amplitude was usually smaller than the AMPAR EPSC within each cell, the slow decay of the NMDAR current resulted in significantly more charge being transferred through these receptors in response to ST stimulation.

NTS LepR neurons express larger NMDAR-mediated currents than non-LepR neurons. A, B, Voltage-clamp traces of LepR+ and LepR– NTS neurons following a single ST stimulation. Recordings were made in 1.2 mm [Mg2+] at −60 mV (gray) then again at +40 mV (black) in the presence of NBQX, gabazine, and cesium internal to isolate NMDAR currents. C, Mean peak amplitude and distribution of AMPAR-mediated evoked EPSCs at −60 mV. D, Mean peak amplitude and distribution of NMDAR-mediated EPSCs at +40 mV. E, NMDAR-mediated charge transfer (area under the curve) at +40 mV. F, G, Example voltage-clamp traces from LepR+ and LepR− neurons following 50 Hz stimulation of the ST. Recordings were taken at −60 mV in the absence of magnesium (black) and then again in the presence of 10 μm DCP (gray). AMPAR-mediated current was measured at the peak of each evoked EPSC in the presence of DCP, and NMDAR-mediated current was calculated as the difference in the maximum sustained current before and after NMDAR antagonism (as labeled in F). H, Mean (±SEM) AMPAR current amplitude across the 50 Hz stimulus train. I, Mean (±SEM) NMDAR current amplitude across the 50 Hz stimulus train. J, Mean (±SEM) relative charge transfer comparing AMPAR current (empty circles) and NMDAR current (half-filled circles) in LepR+ neurons. Relative contribution to charge transfer shifted from predominantly AMPAR currents at the beginning of the stimulus train to NMDAR current at the end of the train. Example traces of EPSC1 and EPSC10 from the same neuron before and after DCP are shown on the right. †p < 0.05, Student's t test; ‡p < 0.05, Mann–Whitney test; *p < 0.05 ANOVA.
Previous studies from our laboratory demonstrated that NMDAR-mediated currents are evident during high-frequency stimulation of the solitary tract in the absence of extracellular Mg2+ (Zhao et al., 2015). Ten stimulations of the ST at 50 Hz in 0 [Mg2+] produced a series of compound EPSCs in LepR and non-LepR neurons that each had a large, fast-decaying component (AMPAR-mediated) and a slower component blocked by DCP (NMDAR-mediated; Fig. 2F,G). In both cell types, AMPAR currents displayed frequency-dependent depression (FDD), while NMDAR current continued to develop over the course of the stimulus train, typically reaching a plateau after the fifth ST stimulation. Glutamate release from ST afferents has been shown to have a very high probability of release (Bailey et al., 2006b); therefore, NTS neurons that display significant FDD in evoked EPSCs most likely receive direct ST input. When plotting the average AMPAR and NMDAR current amplitude at each point in the stimulus train, we saw no significant difference in AMPAR current between LepR neurons (n = 6) and non-LepR neurons (n = 8; F(1,9) = 0.603, p = 0.439, ANOVA; Fig. 2H), which suggests that both LepR and non-LepR neurons receive direct input from a similarly high probability of release vagal afferents. In contrast, the NMDAR current was significantly larger in LepR neurons at all but three points in the stimulus train (F(1,9) = 46.52, p < 0.001, ANOVA; Fig. 2I). LepR neuron NMDARs made a significant contribution to charge transfer despite having much lower EPSC peak amplitudes compared with AMPAR currents in the stimulus train. When comparing the relative charge transfer between AMPARs and NMDARs in LepR neurons, we observed a shift from a predominantly AMPAR-mediated current at the beginning of the stimulus train (EPSC 1: t(5) = 9.97, p < 0.001; EPSC 2: t(5) = 2.72, p = 0.007) to greater current transferred through NMDARs at the end of the train (EPSCs 8; t(5) = 2.55, p = 0.012; EPSC 9: t(5) = 2.65, p = 0.009; EPSC 10: t(5) = 2.35, p = 0.02; Bonferroni t test; Fig. 2J). These data suggest that processes conducive to high NMDAR activity in LepR neurons could prolong the responsiveness of these cells to vagal input, particularly during periods of high-frequency vagal firing as would occur following food intake (Schwartz et al., 1991).
Leptin increases ST-evoked NMDAR currents in NTS LepR neurons
We next examined the effects of exogenous leptin on evoked NMDAR-mediated currents. Similar to our previous study, we recorded under conditions that would maximize the contribution of NMDAR currents to ST-evoked EPSCs. High-frequency stimulation of the ST in the absence of extracellular Mg2+ produced a series of compound EPSCs containing both AMPAR- and NMDAR-mediated currents, and we estimated peak NMDAR current amplitude as the plateau on or after the fifth stimulation (Fig. 3A). We repeated this stimulus train (10 50 Hz stimuli every 6 s) for 25 min, comprising a 10 min control period before bath applying 100 nm leptin for 15 min. When comparing peak NMDAR current amplitude between the last 3 min of control and leptin treatment, we found that a majority of LepR neurons (8 of 13 neurons) displayed a significant increase in NMDAR current (1.30 ± 0.06- vs 0.99 ± 0.03-fold increase for nonresponders; t(11) = 3.95, p = 0.002, Student's t test; Fig. 3B). When comparing NMDAR current over time between the “responder” and “nonresponder” groups, we found that 100 nm leptin significantly increased the peak NMDAR current as early as 3–4 min into the drug treatment, and this effect continued to climb for the remainder of the recording (F(1,24) = 74.09, p < 0.001, ANOVA; Fig. 3C). Notably, leptin did not appear to have a significant effect on EPSC1 peak amplitude, when current is predominantly transferred through AMPA receptors (F(1,24) = 0.0923, p = 0.761, ANOVA; Fig. 3D). While there was a trend for leptin nonresponders to have larger evoked EPSCs, post hoc analysis revealed no significant difference in basal NMDAR currents (122.8 ± 26.3 vs 186.6 ± 84.1 pA; t(11) = 1.914, p = 0.082, Student's t test), basal AMPAR currents (310 ± 52.5 vs 439.9 ± 50.6 pA; t(11) = 1.763, p = 0.106, Student's t test), or AMPAR/NMDAR ratio (2.60 ± 0.402 vs 2.598 ± 0.437; t(11) = 0.0029, p = 0.996, Student's t test) between leptin-responsive and nonresponsive neurons. From the shape of the discernible monosynaptic EPSCs, it appears that at least some of the monosynaptic EPSC is mediated by NMDARs and that this current is increased by leptin. However, it is difficult to differentiate the effects of leptin on NMDARs specifically at monosynaptic versus polysynaptic synapses, as polysynaptic inputs are inherently variable and difficult to measure in isolation.

Leptin increases ST-evoked NMDAR currents in NTS LepR neurons. A, Example voltage-clamp trace taken from a LepR+ neuron before (gray) and after (black) 100 nm leptin. Neurons were recorded in the absence of magnesium, and the average NMDAR current was measured at the plateau of the sustained current during a 50 Hz stimulus train. B, Fold change in NMDAR current postleptin comparing responsive and nonresponsive neurons. C, Mean (±SEM) fold change in NMDAR current over time between leptin-responsive and nonresponsive neurons. D, Mean (±SEM) fold change in AMPAR current (EPSC1 peak) over time between leptin-responsive and nonresponsive neurons. †p < 0.05, Student's t test; *p < 0.05, ANOVA.
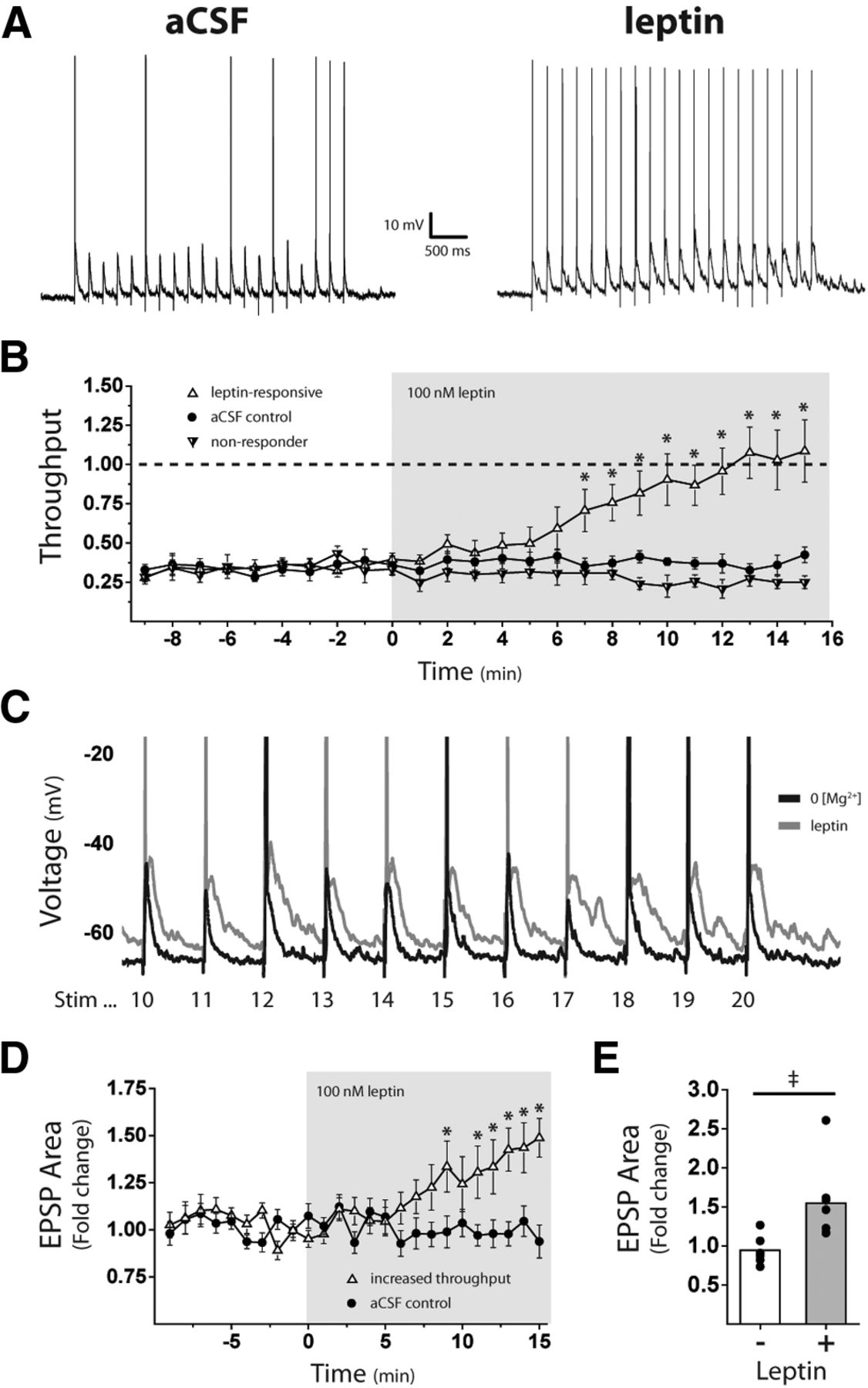
Leptin increases ST-NTS synaptic throughput to LepR neurons
Our results suggest that leptin could increase the sensitivity of NTS LepR neurons to ST input by potentiating postsynaptic NMDAR currents. To determine whether leptin exposure results in increased throughput at the vagal afferent–LepR neuron synapse, we performed CC recordings in the absence of external Mg2+ and measured leptin-induced changes in evoked Aps and EPSPs. NTS LepR neurons were allowed several minutes to acclimate to CC conditions and reach a stable resting membrane potential before ST stimulation (membrane potential = −64.7 ± 0.97 mV; n = 16). The ST was stimulated 20 times at 5 Hz, and the stimulus intensity was slowly raised until an intensity was reached that consistently produced 6–8 evoked APs of a possible 20 in each cell, or a throughput of ∼0.3 (Fig. 4A, left). This stimulus train was then repeated at the same intensity every 30 s for a 10 min control period followed by 15 min of 100 nm leptin. A majority of leptin-treated neurons (7 of 10) had a significant increase in synaptic throughput compared with aCSF controls (n = 6; t(11) = 9.911, p < 0.001, two-way ANOVA and post hoc Bonferroni test; Fig. 4B), with later ST stimulations eliciting one or more APs. In the other three leptin-treated cells, synaptic throughput was not significantly different from aCSF controls (t(7) = 2.04, p = 0.128, Bonferroni t test; Fig. 4B).

Leptin increases ST-NTS synaptic throughput to LepR neurons. A, Example current-clamp traces taken from the same LepR neuron before and after 100 nm leptin. The solitary tract was stimulated 20 times at 5 Hz at a shock intensity that produced 6–8 evoked action potentials of a possible 20 under control conditions (throughput, ∼0.3, where throughput = average number of action potentials per stimulation). B, Mean (±SEM) throughput over time comparing aCSF, leptin-responsive, and nonresponsive neurons. C, Overlaid current-clamp traces from the same LepR neuron before (black) and after (gray) 100 nm leptin. y-Axis scaled up to illustrate the difference in EPSP area and amplitude after leptin. D, Mean (±SEM) fold change in total EPSP area over time between aCSF controls and leptin-responsive neurons. E, Mean EPSP area and distribution in aCSF control and leptin-treated neurons. ‡p < 0.05, Mann–Whitney test; *p < 0.05, ANOVA.
We also found that leptin increased EPSP area during the stimulus train. EPSP area was calculated for each stimulus train by averaging 100 ms of baseline membrane potential and measuring the area under the curve for the entire 4 s stimulus train. In the same leptin-treated cells that had shown a significant increase in synaptic throughput (7 of 10 neurons), EPSP area was also visibly larger than before leptin treatment (Fig. 4A,C). Plotting the fold change in EPSP area over time, those same seven leptin-treated cells showed a significant increase in EPSP area compared with aCSF controls (F(1,24) = 19.35, p < 0.001, two-way ANOVA; Fig. 4D). When averaged over the last 3 min, these leptin-treated cells (n = 7) showed a 1.5-fold increase in EPSP area, while aCSF controls (n = 6) were comparable to baseline (1.55 ± 0.21- vs 0.95 ± 0.08-fold increase; p = 0.008, Mann–Whitney; Fig. 4E).
Postsynaptic NMDARs are required for leptin-induced increases in synaptic throughput
Our data show that leptin increases postsynaptic NMDAR currents and ST–NTS synaptic throughput in the majority of LepR neurons, so we next determined whether NMDARs are required for the effects of leptin on synaptic throughput. First, we confirmed that NMDARs can contribute to synaptic throughput by performing CC recordings under different levels of magnesium block (0, 0.6, and 1.2 mm external Mg2+). Similar to our previous experiments, ST-shock intensity was slowly raised from 0 mA until NTS LepR neurons consistently fired 6–8 APs of a possible 20 for a 10 min control period. Bath application of 10 μm DCP significantly reduced synaptic throughput in 0 and 0.6 mm Mg2+ conditions, but not in 1.2 mm Mg2+ (F(2,24) = 26.32, p < 0.001, ANOVA; Fig. 5A). Following wash throughput remained significantly lower in 0 mm Mg2+, while throughput eventually returned to baseline levels in the 0.6 mm Mg2+ condition. These results show that NMDARs can be recruited during low-frequency stimulation of the ST (5 Hz) and contribute to synaptic throughput at Mg2+ concentrations <1.2 mm.

Postsynaptic NMDARs are required for leptin-induced increases in synaptic throughput. A, Effect of 10 μm DCP on the mean (±SEM) throughput over time from current-clamp recordings in 1.2 mm [Mg2+], 0.6 mm [Mg2+], and 0 [Mg2+]. B, Example current-clamp traces taken from the same LepR neuron in 0 [Mg2+] before and after 10 μm DCP. C, Mean (±SEM) throughput over time comparing aCSF, DCP, and DCP/leptin treatment. D, Overlaid current-clamp traces from the same LepR neuron before (black) and after (gray) 10 μm DCP in 0 [Mg2+]. y-Axis scaled up to illustrate the difference in EPSP area and amplitude after NMDAR antagonism. E, Mean (±SEM) fold change in total EPSP area over time among aCSF, DCP, and DCP/leptin treatments. F, Mean (±SEM) throughput over time comparing aCSF (empty triangles) and leptin-treated neurons (black triangles) with postsynaptic NMDARs selectively blocked. Neurons were recorded in 0 [Mg2+] with MK-801 included in the recording pipette (iMK801).
We next tested whether open NMDARs are required for the effects of leptin on synaptic throughput. To maximize our ability to study the NMDAR contribution, CC recordings were taken in 0 mm Mg2+ and throughput was set to ∼0.3, as previously described. The 10 μm DCP significantly decreased throughput in all neurons treated (n = 16; Fig. 5B,C). Notably, post hoc analysis showed that synaptic throughput in neurons cotreated with DCP and leptin (n = 10) did not differ from the DCP group (t(14) = 0.603, p ∼ 1.00, Bonferroni t test; Fig. 5C). DCP also visibly reduced EPSP area during the stimulus train (Fig. 5D), suggesting that open NMDARs can make significant contributions to LepR neuron membrane depolarization. DCP rapidly reduced EPSP area in both the control (t(7) = 26.68, p < 0.001, Bonferroni t test) and the leptin (t(11) = 27.86, p < 0.001, Bonferroni t test) cotreatment groups, and the EPSP area remained significantly lower than in aCSF controls (n = 3) for the remainder of the recording (Fig. 5E).
NMDARs are expressed by both NTS neurons and vagal afferents. Therefore, it is possible that leptin effects on synaptic throughput might be mediated by presynaptic or postsynaptic NMDARs. To isolate the contribution of postsynaptic NMDARs to leptin-induced changes in throughput, we performed CC recordings in 0 mm Mg2+ and included the NMDAR channel blocker MK-801 (dizocilpine) in the recording pipette solution to selectively block postsynaptic NMDARs from the intracellular side. Before conducting synaptic throughput experiments in current-clamp mode, brief voltage-clamp recordings were made to confirm that 1 mm internal MK-801 (iMK-801) blocked evoked NMDAR currents (data not shown). NTS LepR neurons were able to fire APs in the presence of iMK-801, and throughput was set to ∼0.25 for a 10 min control period followed by 15 min of 100 nm leptin. Synaptic throughput remained fairly constant and did not differ between leptin-treated neurons (n = 9) and iMK-801 controls (n = 8; F(1,24) = 0.859, p = 0.355, ANOVA; Fig. 5F). These data are consistent with our hypothesis that leptin increases postsynaptic NMDAR function in LepR neurons and that open NMDARs are required for the acute effects of leptin on synaptic throughput.
NMDAR antagonism blocks the effect of intra-DVC leptin injections on overnight food intake in rats
We used mice to determine how leptin potentiates transmission across the vagus–NTS synapse as we needed to identify neurons that express the LepR. However, we have previously shown that rat NTS neurons also express postsynaptic NMDARs and that they are also required to maintain fidelity of transmission across the synapse (Zhao et al., 2015), suggesting that the mechanism we describe likely occurs in both mice and rats. There is an extensive literature showing that, in rats, NTS NMDARs participate in meal termination (satiation) and in the reduction of food intake by GI peptide satiation signals such as CCK (Gillespie et al., 2005; Campos et al., 2012). To determine whether NTS NMDARs also contribute to reduction of food intake by intra-NTS leptin, we measured overnight food intake in 10 male rats following nanoinjections of saline, DCP, leptin, and a DCP/leptin cocktail directly into the DVC, which includes the NTS. There was a main effect of injection treatment on overnight food intake (F(3) = 4.821, p = 0.0064, one-way repeated-measures ANOVA), and post hoc analysis indicated that intra-DVC injections of leptin (200 ng) reduced overnight food intake compared with saline (17.0 ± 0.51 vs 20.6 ± 0.66 g; t(18) = 4.31, p < 0.001, Dunnett's test; Fig. 6), similar to what has been reported previously (Grill et al., 2002). Injection of the NMDAR antagonist DCP (20 ng) alone did not alter overnight intake compared with saline (18.9 ± 0.57 vs 20.6 ± 0.66 g; t(18) = 1.94, p = 0.0524, Dunnett's test; Fig. 6). However, coinjection of DCP with leptin prevented leptin-induced reductions in food intake (18.9 ± 0.88 vs 20.6 ± 0.66 g; t(18) = 1.54, p = 0.1236, Dunnett's test; Fig. 6).

NMDAR antagonism attenuates the reduction of overnight food intake following intra-DVC leptin injections rats. Male rats (n = 10) with guide cannulas implanted into the DVC had ad libitum access to water and standard pelleted rodent chow. Overnight food intake was measured for each animal after intracranial injections of saline, 20 ng DCP, 200 ng leptin, and a DCP/leptin cocktail, with each injection spaced ≥48 h apart. Overnight food intake is reported as the mean ± SEM. *p < 0.05 one-way repeated-measures ANOVA with post hoc Dunnett's multiple comparison.
Discussion
Leptin receptors in the NTS are important for normal control of food intake as disrupted NTS leptin signaling causes hyperphagia and reduced sensitivity to gastrointestinal satiation signals (Scott et al., 2011; Kanoski et al., 2012). However, the role of LepR neurons in NTS circuitry and the cellular mechanism by which leptin reduces food intake were not well understood. Hindbrain NMDARs are critical for the control of food intake by vagus-mediated satiation signals (Burns and Ritter, 1998; Treece et al., 1998; Covasa et al., 2000), but their requirement for the actions of leptin had not been investigated. Here we report six novel findings that show how LepR neurons are activated by vagal inputs and demonstrate a role for NMDARs in mediating the actions of leptin, as follows: (1) LepR neurons receive monosynaptic inputs from the ST that summate with polysynaptic inputs; (2) LepR neurons display larger NMDAR-mediated currents than non-LepR neurons; (3) leptin increases NMDAR-mediated currents in LepR neurons; (4) leptin increases ST-NTS synaptic throughput; (5) postsynaptic NMDARs are required for increase in throughput; and (6) NMDARs are required for NTS leptin inhibition of food intake.
LepR-expressing neurons receive convergent monosynaptic and polysynaptic inputs from sensory afferents
We found that NTS LepR neurons receive one or two direct glutamatergic synaptic inputs from ST fibers, as well as an average of three polysynaptic connections through other neurons activated by ST stimulation. This degree of convergence is greater than that seen in other populations of NTS neurons, including catecholamine and GABA NTS neurons (Appleyard et al., 2007; Bailey et al., 2008), suggesting that LepR neurons integrate information from a wider number of sources than other NTS neurons. While the polysynaptic EPSCs were generally smaller than monosynaptic EPSCs, they can summate to produce large compound EPSCs that are often longer lasting and so prolong the exposure of LepR neurons to synaptically released glutamate. Some NTS neurons also receive nonvagal inputs (Browning and Travagli, 2014), and future studies are needed to identity the physiological significance of the different vagal and nonvagal glutamatergic inputs to NTS LepR neurons.
Leptin increases NMDAR currents
We found that LepR neurons express larger NMDAR-mediated synaptic currents than most non-LepR NTS neurons, suggesting that NMDARs are important for how these neurons process information. Vagal afferents have a high probability of releasing glutamate following stimulation, resulting in pronounced frequency-dependent depression of AMPAR-mediated EPSCs (Bailey et al., 2006b; Peters et al., 2010). We previously demonstrated that NTS NMDARs are necessary to sustain the fidelity of synaptic transmission during periods of high-frequency stimulation when glutamate release is depressed (Zhao et al., 2015). Given that the vagus fires at high frequencies (>10 Hz) during a meal (Schwartz et al., 1991), NMDAR currents could provide a mechanism by which NTS LepR neurons maintain the ability to fire action potentials in response to vagal satiation signals. Interestingly, leptin has also been shown to inhibit some NTS neurons (Williams and Smith, 2006); however, we did not see any hyperpolarizing effects in LepR neurons. As Williams and Smith (2006) recorded in coronal slices, while we recorded from horizontal slices, it is possible that we are recording from different areas of the NTS. Furthermore, we recorded from LepR-expressing neurons, while the neuronal phenotype of the neurons Williams and Smith (2006) recorded from is not known. Our findings are consistent with those of the study by Hisadome et al. (2010) who showed leptin increased the firing rate of preproglucagon (PPG) neurons, at least some of which are thought to be LepR-expressing neurons (Garfield et al., 2012). The excitatory effects are also concordant with leptin activating immediate early genes in NTS neurons, suggesting that leptin has an excitatory effect in at least some NTS neurons (Elias et al., 2000; Ellacott et al., 2006; Huo et al., 2007; Ruiter et al., 2010). However, it would be interesting to determine which NTS neurons are excited or inhibited by leptin and whether these effects are altered by different conditions.
Leptin has been shown to increase NMDAR-dependent synaptic plasticity in the hippocampus (Shanley et al., 2001). The relatively high expression of synaptic NMDARs in NTS LepR neurons suggests that synaptic inputs to these neurons, similar to those in the hippocampus, could undergo some form of plasticity in response to leptin exposure. Furthermore, the high density of synaptic NMDARs and the number of glutamatergic inputs that LepR neurons receive suggest that LepR neurons could act as coincidence detectors, integrating various vagal and nonvagal signals to control NTS output and affect food intake.
Postsynaptic NMDAR currents are required for leptin-induced increases in synaptic throughput
Leptin receptors and NMDARs are expressed in both NTS neurons and synaptic terminals of vagal afferents (Elmquist et al., 1998; Aicher et al., 1999; Buyse et al., 2001). Furthermore, leptin has been shown to affect food intake through both presynaptic receptors (de Lartigue et al., 2014) and postsynaptic receptors (Kanoski et al., 2012). Our results suggest that the activation of postsynaptic NMDARs is required for acute leptin-induced enhancement of synaptic throughput. First, we found that leptin increases NMDAR currents in the majority of LepR neurons while AMPAR currents are not acutely affected by leptin. If leptin acted primarily at vagal afferent terminals to increase glutamate release, we would expect both AMPAR and NMDAR currents to be increased. Second, we show that postsynaptic NMDARs are required for acute leptin effects on synaptic throughput, as inclusion of a use-dependent NMDAR channel blocker (MK801) in the recording pipette was sufficient to prevent leptin-induced increases in synaptic throughput. Nevertheless, our findings cannot rule out concomitant or long-term contributions of presynaptic LepRs and NMDARs to leptin action in the NTS. Indeed, both presynaptic vagal afferent LepRs (Peters et al., 2005; de Lartigue et al., 2014) and presynaptic NMDARs (Gillespie et al., 2005; Campos and Ritter, 2015) have been implicated in control of food intake. Presynaptic and postsynaptic actions of leptin may work in concert to increase vagal firing, stimulate glutamate release, and increase NTS sensitivity to more strongly drive activation of downstream targets that impact food intake. In addition, LepRs are also found in glia in the NTS, and this could be another way that leptin indirectly impacts vagal activation of NTS neurons (Dallaporta et al., 2009).
The molecular mechanisms by which leptin increases NMDAR currents in the NTS remain to be established. Leptin activates many signaling pathways, including PI3 kinase, MAP kinase, and Src family kinases (SFKs), all of which have been reported to increase NMDAR surface expression or function (Nicoll and Malenka, 1999; Salter and Kalia, 2004; Groc and Choquet, 2006). Leptin-induced increases in NMDAR EPSC amplitude and the conversion of short-term potentiation to long-term potentiation in hippocampal neurons are both blocked by inhibitors of PI3 kinase, MAP kinase, and Src tyrosine kinase (Shanley et al., 2001). Furthermore, we recently showed that leptin increases NMDAR surface expression in hippocampal neurons through an SFK-dependent mechanism (Bland et al., 2020). It is therefore possible that leptin activates one or more of these kinases in NTS LepR neurons to increase NMDAR trafficking or channel gating.
Role of LepR-expressing neurons
Activation of LepR neurons for 4 d in the NTS suppresses food intake and decreases body weight (Cheng et al., 2020). In addition, these neurons are thought to integrate the response to leptin with forebrain regions as fourth-ventricle, leptin-conjugated saporin decreases the sensitivity to forebrain leptin inhibition of food intake (Harris, 2020). LepR neurons have been shown to colocalize with PPG and POMC-EGFP neurons in the NTS (Garfield et al., 2012) and leptin increases c-fos expression in PPG/GLP-1 neurons (Elias et al., 2000) and p-STAT3 (phosphorylated signal transducer and activator of transcription 3) in POMC-EGFP neurons (Ellacott et al., 2006). Interestingly, both POMC-EGFP and PPG-YFP neurons are directly activated by vagal afferents (Appleyard et al., 2005; Hisadome et al., 2010), consistent with our findings, and activation of both of these populations of neurons also inhibits food intake (Zhan et al., 2013; Holt et al., 2019). Leptin also increases the firing rate of GLP-1 neurons (Hisadome et al., 2010) and potentiation of the NMDAR current could represent one mechanism by which that occurs. Furthermore, GLP1 neurons form local circuits in the NTS, including making connections with each other (Card et al., 2018), and as GLP1 neurons are also glutamatergic they could contribute to some of the polysynaptic circuits onto LepR neurons. However, Cheng et al. (2020) recently suggested that LepR neurons suppress food intake independently of GLP1 in mice (Cheng et al., 2020). The actions of leptin in the NTS are increased in mice lacking PTP1B, specifically in POMC neurons (De Jonghe et al., 2012), supporting a role for this population mediating some of the actions of leptin. Alternatively, other neurotransmitters could mediate the effects of LepR neurons, although LepRs do not colocalize with CART (cocaine- and amphetamine-regulated transcript), BDNF, NPY, nesfatin, catecholamines, GABA, PRP (prolactin-releasing peptide), or NOS (nitric oxide synthase; Garfield et al., 2012). Interestingly, LepR neurons have been shown to express CCK mRNA (Garfield et al., 2012), and the activation of NTS CCK neurons inhibits food intake (Roman et al., 2017); although a more recent study did not see colocalization when CCK-eGFP reporter mice were used (Cheng et al., 2020).
NTS NMDARs are required for leptin-induced inhibition of food intake
NMDARs contribute to inhibition of food intake by vagal-mediated satiation signals, including meal ingestion and CCK (Jahng and Houpt, 2001; Gillespie et al., 2005; Campos et al., 2012). Here we show that they are also required for leptin inhibition of food intake in male rats. Interestingly, leptin increases the efficacy of subthreshold intraluminal nutrients (Emond et al., 2001), cholecystokinin (Emond et al., 1999), or gastric distension to reduce food intake (Huo et al., 2007). One mechanism by which leptin could enhance responsiveness to vagal-mediated GI satiation signals is through potentiation of NMDAR-mediated currents, and consequently the likelihood of vagal stimuli producing action potentials. Conversely, decreased leptin signaling could translate to smaller NMDAR-mediated currents and a reduced sensitivity to ST stimulation. It is interesting that leptin levels exhibit circadian fluctuations (Schoeller et al., 1997), and circulating concentrations are reduced following a fast (Hardie et al., 1996). Thus, leptin could modulate NTS LepR neuron sensitivity and output to produce adaptive changes in food intake related to metabolic state, as has been proposed in the hypothalamus (Friedman and Halaas, 1998).
In summary, our data demonstrate that leptin enhances postsynaptic NMDAR function and increases synaptic throughput in NTS LepR neurons. LepR neurons have extensive glutamatergic inputs and large NMDAR-mediated currents; thus, changes to NMDAR activity can have pronounced effects on LepR neuron firing and synaptic processing. Antagonism of NTS NMDARs blocks the effects of intra-NTS leptin injections on overnight food intake, consistent with the hypothesis that leptin-induced effects on NMDAR currents contributes to control of food intake by the NTS. Our results describe a novel mechanism for leptin-induced changes in NTS sensitivity and further emphasize the importance of hindbrain NMDARs in vagus–NTS synaptic transmission.
Footnotes
The authors declare no competing financial interests.
This work was funded by National Institutes of Health Grants R01DK083452 (to S.M.A.), R01DK052849 (to R.C.R. and S.M.A.), and R01HD092369 (to G.A.W.). We thank Dr. Martin Myers (University of Michigan) for kindly providing the LepR-Cre mice. We also thank Crystal Dillon for help genotyping the mice. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- Correspondence should be addressed to Suzanne M. Appleyard at s.appleyard{at}wsu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}