


Abstract
Muscimol is a psychoactive isoxazole derived from the mushroom Amanita muscaria and a potent orthosteric agonist of the GABAA receptor. The binding of [3H]muscimol has been used to evaluate the distribution of GABAA receptors in the brain, and studies of modulation of [3H]muscimol binding by allosteric GABAergic modulators such as barbiturates and steroid anesthetics have provided insight into the modes of action of these drugs on the GABAA receptor. It has, however, not been feasible to directly apply interaction parameters derived from functional studies to describe the binding of muscimol to the receptor. Here, we employed the Monod-Wyman-Changeux concerted transition model to analyze muscimol binding isotherms. We show that the binding isotherms from recombinant α1β3 GABAA receptors can be qualitatively predicted using electrophysiological data pertaining to properties of receptor activation and desensitization in the presence of muscimol. The model predicts enhancement of [3H]muscimol binding in the presence of the steroids allopregnanolone and pregnenolone sulfate, although the steroids interact with distinct sites and either enhance (allopregnanolone) or reduce (pregnenolone sulfate) receptor function. We infer that the concerted transition model can be used to link radioligand binding and electrophysiological data.
Significance Statement The study employs a three-state resting-active-desensitized model to link radioligand binding and electrophysiological data. We show that the binding isotherms can be qualitatively predicted using parameters estimated in electrophysiological experiments and that the model accurately predicts the enhancement of [3H]muscimol binding in the presence of the potentiating steroid allopregnanolone and the inhibitory steroid pregnenolone sulfate.
Introduction
Muscimol [(5-aminomethyl)-isoxazol-3-ol] is a GABA-mimetic substance found in the mushroom Amanita muscaria. It is a highly potent orthosteric agonist of the GABAA receptor interacting with the receptor via the transmitter binding site (Beaumont et al., 1978; Deng et al., 1986; Smith and Olsen, 1994; Jones et al., 1998). The binding of radiolabeled muscimol ([3H]muscimol) was initially used to characterize the distribution of GABA binding sites in the brain and other tissues (Beaumont et al., 1978; Krause et al., 1980). Subsequent studies probing modulation of [3H]muscimol binding in the presence of various GABAergic drugs indicated a correlation between the ability of a drug to potentiate currents and enhance [3H]muscimol binding to the receptor (Supavilai et al., 1982; Quast and Brenner, 1983; Harrison and Simmonds, 1984; Peters et al., 1988). Studies of modulation of [3H]muscimol binding in the presence of drug combinations have provided mechanistic insight into how the drugs act on the receptor. Specifically, work done using combinations of potentiating steroids and barbiturates indicated that the two classes of ligands interact with distinct sites on the GABAA receptor (Kirkness and Turner, 1988; Peters et al., 1988). Chang et al. (2002) demonstrated that desensitization after prolonged exposure to GABA enhances the binding of [3H]muscimol and proposed that functional desensitization can outlast agonist binding, i.e., receptors can occupy a vacant, desensitized state, thereby providing support for a cyclical model of activation.
A central difficulty in relating receptor activation by agonists to biochemical studies of agonist binding arises because of the existence of agonist-induced conformational changes that alter the affinity of the receptor to agonist. We set out to determine if we could surmount this difficulty by employing the Monod-Wyman-Changeux concerted transition model (Monod et al., 1965) to describe both activation by and binding of muscimol to the human α1β3 GABAA receptor. The model has been used extensively in analysis of electrophysiological data of activation and modulation of the GABAA receptor by orthosteric and allosteric agents (Chang and Weiss, 1999; Rüsch et al., 2004; Shin et al., 2018). In the present work, we extended the model to include a high-affinity desensitized state and derived equations to describe the occupancy function.
To test the validity of the model, we estimated functional parameters of the α1β3 GABAA receptor using electrophysiology, which were then used to predict [3H]muscimol binding isotherms using the derived equations. We show that there is a qualitative agreement between the predicted and observed isotherms. Furthermore, the model explains the observation that both the potentiating steroid allopregnanolone [3α5αP; 1-[(3R,5S,8R,9S,10S,13S,14S,17S)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]ethenone] and the inhibitory steroid pregnenolone sulfate [PS; [(3S,8S,9S,10R,13S,14S,17S)-17-acetyl-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl] hydrogen sulfate], enhance muscimol binding.
Methods
Receptor Expression and Electrophysiological Recordings.
The human α1β3 GABAA receptor was expressed in oocytes from Xenopus laevis (African clawed frog). Oocytes were purchased from Xenoocyte (Dexter, MI) as quarter ovaries. Oocytes were digested in a 2% w/v (mg/ml) collagenase A solution in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES; pH 7.4) with 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C with shaking at 250 rpm for 30–40 minutes. Oocytes were rinsed in ND96 and stored in ND96 with supplements (2.5 mM Na pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μg/ml gentamycin) at 15°C for at least 4 hours before injection.
The oocytes were injected with a total of 12 ng of cRNA per oocyte. The ratio of cRNAs for the α1 and β3 subunits was 5:1 to minimize the expression of β3 homomeric receptors. After injection, the oocytes were incubated in ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5 mM HEPES; pH 7.4) with supplements (2.5 mM Na pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin and 50 μg/ml gentamycin) at 15°C for 2 days prior to conducting electrophysiological recordings.
The electrophysiological recordings were conducted at room temperature using the standard two-electrode voltage clamp technique. The oocytes were clamped at −60 mV. Bath and drug solutions were gravity-applied from glass syringes with glass Luer slips via Teflon tubing to the recording chamber (RC-1Z; Warner Instruments, Hamden, CT) at 5–8 ml/min flow rate.
The current responses were amplified with an OC-725C amplifier (Warner Instruments), digitized with a Digidata 1200 series digitizer (Molecular Devices), and stored using pClamp (Molecular Devices). Analysis of the current traces was done with Clampfit (Molecular Devices).
Analysis of Electrophysiological Data.
The electrophysiological experiments were aimed at determining the properties of 1) receptor activation and desensitization by muscimol, 2) potentiation of muscimol-activated receptors by the steroid 3α5αP, and 3) inhibition of muscimol-activated receptors by the steroid PS.
Activation by muscimol was measured by exposing oocytes to 0.03–30 µM muscimol and measuring the peak response. The drug applications lasted 10–70 seconds. The raw peak amplitudes of the current traces in the presence of muscimol were converted to probability of being in the active state (PA) through normalization to the peak response to saturating (100 µM or 1 mM) GABA + 50 µM propofol, which was considered to have a peak PA indistinguishable from 1. The level of constitutive activity (PA,const) in the absence of any applied agonist (0.00023 of the maximal response; see below) was considered negligible and therefore not included in this calculation.
The current responses in units of PA were analyzed in the framework of the two-state concerted transition model (Forman, 2012; Steinbach and Akk, 2019). The muscimol concentration-response data pooled from five cells were fitted with the state function for peak currents: (1)where [muscimol] denotes the concentration of muscimol, KR,muscimol is the equilibrium dissociation constant for muscimol in the resting receptor, and cmuscimol is the ratio of the equilibrium dissociation constant for muscimol in the active receptor to KR,muscimol. Nmuscimol is the number of binding sites for muscimol, which was held at 2. The parameter L is the ratio of resting to active receptors (R/A in Fig. 1) and expresses the level of background activity. L was calculated from the level of constitutive activity in the absence of applied agonists (PA,const) as (1 − PA,const)/PA,const. PA,const was determined by comparing the effect of 200 µM picrotoxin on holding current with the peak response to saturating (1 mM) GABA plus 50 µM propofol (PA ∼1). This analysis yielded the values of KR,muscimol and cmuscimol.
(1)where [muscimol] denotes the concentration of muscimol, KR,muscimol is the equilibrium dissociation constant for muscimol in the resting receptor, and cmuscimol is the ratio of the equilibrium dissociation constant for muscimol in the active receptor to KR,muscimol. Nmuscimol is the number of binding sites for muscimol, which was held at 2. The parameter L is the ratio of resting to active receptors (R/A in Fig. 1) and expresses the level of background activity. L was calculated from the level of constitutive activity in the absence of applied agonists (PA,const) as (1 − PA,const)/PA,const. PA,const was determined by comparing the effect of 200 µM picrotoxin on holding current with the peak response to saturating (1 mM) GABA plus 50 µM propofol (PA ∼1). This analysis yielded the values of KR,muscimol and cmuscimol.
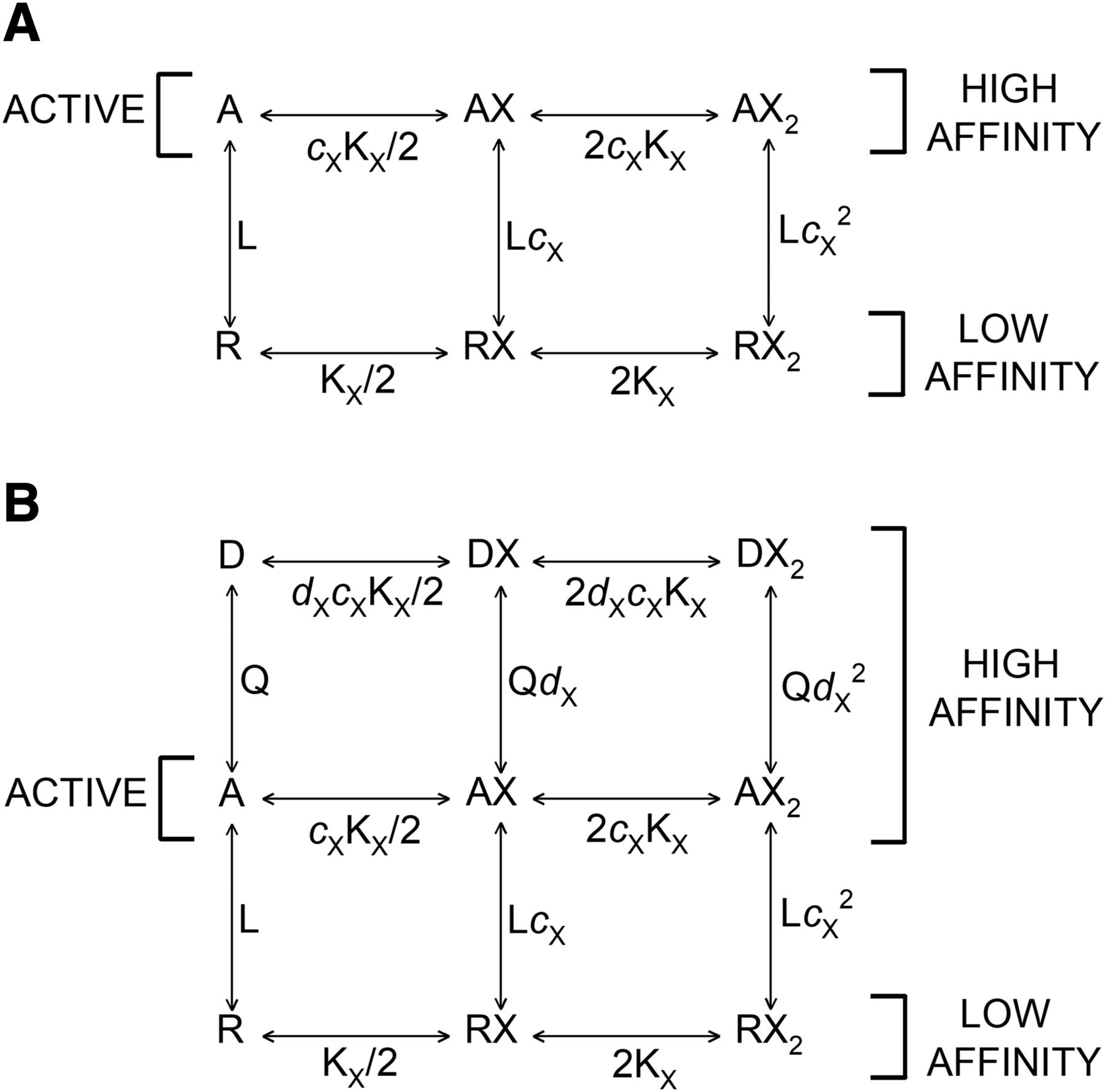
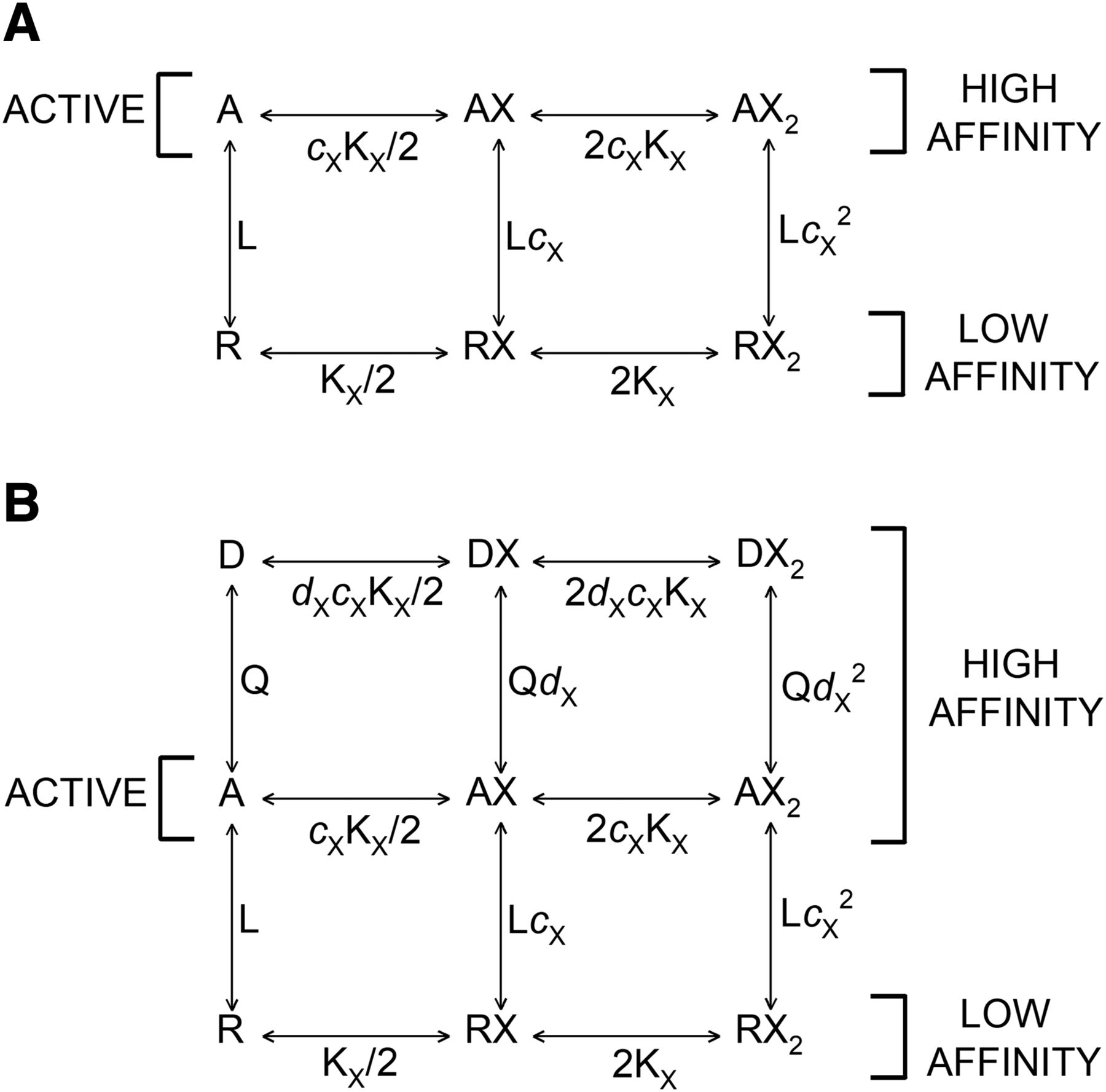
Kinetic schemes. (A) The kinetic scheme for a two-state concerted transition model. R corresponds to the resting state and A to the active state. L (R/A) is the ratio of the resting, low affinity form to the active, high affinity form in the absence of bound agonist. KX is the dissociation constant for the low affinity form, and cX is the ratio of the dissociation constant for the active state to that for the resting state. (B) The kinetic scheme for a three-state concerted transition model. R corresponds to the resting state, A to the active, and D to the desensitized state. Q (A/D) is the ratio of the active to desensitized state in the absence of bound agonist, and dX is the ratio of the dissociation constant for the desensitized state to that for the active state.
The properties of desensitization in the presence of muscimol were determined by exposing the oocytes to long (3–4 minutes) applications of saturating (30–100 µM) muscimol. The value of Q (A/D in Fig. 1B) was estimated using the following equation: (2)where PA,s.s. and PA,peak are the open probabilities of steady-state and peak responses to saturating muscimol, respectively. dmuscimol is the ratio of the equilibrium dissociation constant for muscimol in the desensitized receptor to cmuscimolKR,muscimol. We have assumed that dmuscimol = 1, i.e., that the receptor has indistinguishable affinities to muscimol in the active and desensitized states.
(2)where PA,s.s. and PA,peak are the open probabilities of steady-state and peak responses to saturating muscimol, respectively. dmuscimol is the ratio of the equilibrium dissociation constant for muscimol in the desensitized receptor to cmuscimolKR,muscimol. We have assumed that dmuscimol = 1, i.e., that the receptor has indistinguishable affinities to muscimol in the active and desensitized states.
Potentiation by the steroid 3α5αP was studied by measuring the effect of 0.01–3 µM 3α5αP on the peak response to 0.1 µM muscimol (PA ∼0.09). The affinity and efficacy parameters for allopregnanolone were determined using eq. 3: (3)where L*, a modified value of L, is calculated from the peak PA of the response to 0.1 µM muscimol in the absence of steroid as (1 − PA,0.1 µM muscimol)/PA,0.1 µM muscimol. KR,3α5αP is the equilibrium dissociation constant of the closed receptor for 3α5αP, and c3α5αP is the ratio of the open receptor equilibrium dissociation constant to KR,3α5αP. N3α5αP¸ the number of binding sites for the steroid, was held at 2.
(3)where L*, a modified value of L, is calculated from the peak PA of the response to 0.1 µM muscimol in the absence of steroid as (1 − PA,0.1 µM muscimol)/PA,0.1 µM muscimol. KR,3α5αP is the equilibrium dissociation constant of the closed receptor for 3α5αP, and c3α5αP is the ratio of the open receptor equilibrium dissociation constant to KR,3α5αP. N3α5αP¸ the number of binding sites for the steroid, was held at 2.
Inhibition by the steroid PS was studied by measuring the effect of 0.01–20 µM PS on the steady-state response to 30 µM muscimol. The inhibition concentration-response curve was fitted using eq. 4: (4)where KR,PS is the equilibrium dissociation constant of the resting receptor to the steroid, cPS is the ratio of the steroid equilibrium dissociation constant of the active receptor to KR,PS, and dPS is the ratio of the steroid equilibrium dissociation constant of the desensitized receptor to cPSKR,PS. The term LΓmuscimol describes the level of activity elicited by muscimol. Its value, held at 0.72, was calculated using the experimentally determined PA of the peak response to 30 µM muscimol as (1/PA,peak − 1). Other terms are as described above.
(4)where KR,PS is the equilibrium dissociation constant of the resting receptor to the steroid, cPS is the ratio of the steroid equilibrium dissociation constant of the active receptor to KR,PS, and dPS is the ratio of the steroid equilibrium dissociation constant of the desensitized receptor to cPSKR,PS. The term LΓmuscimol describes the level of activity elicited by muscimol. Its value, held at 0.72, was calculated using the experimentally determined PA of the peak response to 30 µM muscimol as (1/PA,peak − 1). Other terms are as described above.
Expression of Recombinant GABAA Receptors, Cell Culture, and Membrane Protein Preparation.
Construct design and cell culture were performed as described previously (Chen et al., 2019). Briefly, the human α1 and β3 subunits were subcloned into pcDNA3 for molecular manipulations and cRNA synthesis. Using QuikChange mutagenesis (Agilent Technologies, Santa Clara, CA), an 8xHis-FLAG tag was added to the N terminus of the mature α1 subunit. The α1 and β3 subunits were then transferred into the pcDNA4/TO and pcDNA5/TO vectors (Thermo Fisher Scientific), respectively, for tetracycline inducible expression. The tetracycline inducible cell line T-REx-HEK293 (Thermo Fisher Scientific) was cultured under the following conditions: cells were maintained in DMEM/F-12 50/50 medium containing tetracycline-free 10% fetal bovine serum (Takara, Mountain View, CA), penicillin (100 U/ml), streptomycin (100 g/ml), and blasticidin (2 μg/ml) at 37°C in a humidified atmosphere containing 5% CO2. Stably transfected cells were cultured as above with the addition of hygromycin (50 μg/ml) and zeocin (20 μg/ml). A stable cell line was generated by transfecting T-REx-HEK293 cells with human α1-8xHis-FLAG pcDNA4/TO and human β3 pcDNA5/TO, in a 150 mm culture dish, using the Effectene transfection reagent (Qiagen, Germantown, MD). Stably transfected cells selected with hygromycin (50 µg/mol) and zeocin (20 µg/ml) were plated into dishes. After reaching 50% confluency, GABAA receptors were expressed by inducing cells with 1 μg/ml of doxycycline with the addition of 5 mM sodium butyrate. HEK cells, after induction, were grown to 100% confluency, harvested, and washed with PBS (pH 7.5) plus protease inhibitors (Sigma-Aldrich, St. Louis, MO) two times. The cells were collected by centrifugation at 1000g at 4°C for 5 minutes and homogenized with a glass mortar and a Teflon pestle for 10 strokes on ice. The pellet was collected after centrifugation at 20,000g at 4°C for 45 minutes and resuspended in a buffer containing 10 mM potassium phosphate and 100 mM potassium chloride (pH 7.5). The protein concentration was determined with micro-BCA protein assay and adjusted to 2.5 mg/ml for storage at −80°C.
Radioligand Binding Assays.
The [3H]muscimol binding assays were performed using a previously described method (Chen et al., 2019). To perform [3H]muscimol binding isotherms, HEK cell membrane proteins (50 μg/ml final concentration) were incubated with 0.3 nM–1 μM [3H]muscimol (30 Ci/mmol; PerkinElmer, Boston, MA), neurosteroid, and binding buffer (10 mM potassium phosphate, 100 mM potassium chloride, pH 7.5) in a total volume of 1 ml. Assay tubes were incubated for 1 hour on ice in the dark. The specific activity was reduced to 2 Ci/mmol by dilution with nonradioactive muscimol to limit the maximum amount of radioactivity in an assay tube to about 106 cpm or lower (Bylund and Toews, 1993). Nonspecific binding was determined by binding in the presence of 1 mM GABA. The membranes were collected on Whatman/GF-B glass filter papers using a vacuum manifold. Radioactivity bound to the filters was measured by liquid scintillation spectrometry using Bio-Safe II (Research Products International, Mount Prospect, IL).
Drugs in Electrophysiological Recordings.
The stock solution of muscimol was made at 20 mM in ND96 and stored at 4°C. The stock solution of GABA was made in ND96 at 500 mM and stored in aliquots at −20°C. The steroid 3α5αP was dissolved in DMSO at 10 mM and stored at room temperature. The steroid PS was dissolved in DMSO at 50 mM and stored at 4°C. Final dilutions were made on the day of experiment.
Results
Drug Binding in the Monod-Wyman-Changeux Two-State Concerted Transition Model.
To introduce the topic and the form of the equations, we will start by discussing the binding of a drug to a protein that has two states of differing affinity for the drug. The two critical aspects of this model are that 1) all sites of a given class have identical binding properties and 2) the receptor as a whole undergoes a concerted state transition in which all sites change properties simultaneously. That is, there are no receptors with a mixed population of sites. There is no interaction between sites; all effects of ligand binding result from changes in affinity when the receptor changes state. We will provide some examples of the behavior of the predicted occupancy and state functions in the section “Predictions of Binding Curves from Functional Data.”
The original paper (Monod et al., 1965) considers a two-state model (Fig. 1A), in which a protein can adopt two states that have different affinities for a ligand, X. They defined an “occupancy function” for the binding of a drug to a single class of site: (5)
(5) (6)where YX is the fraction of the binding sites occupied, NX is the number of binding sites for X, L is R/A, KX = KR,X, cX = KA,X/KR,X, and αX = [X]/KX.
(6)where YX is the fraction of the binding sites occupied, NX is the number of binding sites for X, L is R/A, KX = KR,X, cX = KA,X/KR,X, and αX = [X]/KX.
The equation for the fraction of receptors in the active form (“state function”) is (7)
(7)
At a saturating concentration of ligand X,  .
.
The occupancy function for X in the presence of drug Y that does not bind to the same sites as X is (8)
(8) (9)where
(9)where and KY = KR,Y, cY = KA,Y/KR,Y and αY = [Y]/KY. The presence of a second drug that binds to distinct sites from X essentially modifies the value of L. Note that when Y is an allosteric inhibitor (cY > 1), increasing concentrations of Y reduce binding of X, and when cY < 1, increasing concentrations enhance binding of X. When cY = 1 (Y binds equally well to the two states), there is no effect of Y (ΓY = 1). The state function in the presence of X and Y is
and KY = KR,Y, cY = KA,Y/KR,Y and αY = [Y]/KY. The presence of a second drug that binds to distinct sites from X essentially modifies the value of L. Note that when Y is an allosteric inhibitor (cY > 1), increasing concentrations of Y reduce binding of X, and when cY < 1, increasing concentrations enhance binding of X. When cY = 1 (Y binds equally well to the two states), there is no effect of Y (ΓY = 1). The state function in the presence of X and Y is (10)
(10)
Drug Binding in the Three-State Resting-Active-Desensitized Model.
A two-state system is not applicable to equilibrium binding to most transmitter-gated channels that undergo a transition to an inactive, high affinity state upon prolonged exposure to agonist (“desensitization”). The resting-active-desensitized (RAD) model extends the original Monod-Wyman-Changeux model by adding an inactive state beyond the active state, as shown in Figure 1B. The presence of this additional state requires an extension of the two-state model treated so far. We have already described and used the “resting-active-desensitized” model in analyzing the steady-state responses of GABAA receptors (Germann et al., 2019a,b; Pierce et al., 2019). We now set out to determine whether it is able to predict steady-state binding data using parameters derived from functional studies.
The RAD model adds a term for binding to desensitized receptors to numerator and denominator in the occupancy function for a drug binding to a single class of sites. For the case of a single drug X, (11)where Q = A/D (ratio of receptors in the active state relative to desensitized state when no drug is bound), dX = KD,X/KA,X, and other terms are as defined previously. The state function is
(11)where Q = A/D (ratio of receptors in the active state relative to desensitized state when no drug is bound), dX = KD,X/KA,X, and other terms are as defined previously. The state function is (12)
(12)
At a saturating concentration of X, 
If the active and desensitized states have the same affinity (dX = 1), (13)
(13)
In this case,  and
and 
The presence of a constant concentration of drug Y (that binds to different sites than X) modifies the L and Q terms: (14)where
(14)where and
and
The state function is (15)
(15)
Despite their simplicity, particularly in the lack of various short-lived states, the resting-active and RAD models have been quite successful in predicting peak and steady-state activity, respectively, of the GABAA receptor in the presence of orthosteric and allosteric agonists and their combinations (Chang and Weiss, 1999; Rüsch et al., 2004; Shin et al., 2018; Germann et al., 2019a,b). A discussion of possible reasons why the simple models could account for the behavior of a channel that is as kinetically complex as the GABAA receptor has been provided previously (Steinbach and Akk, 2019). We note that the parameters estimated in these analyses are not directly applicable to the kinetics of synaptic responses because the resting-active and RAD models deal with equilibrium constants rather than rate constants.
Predictions of Binding Curves from Functional Data.
The analysis of peak and steady-state responses to agonists allows us to derive estimates for all of the parameters in the occupancy functions for the RAD model, so it is possible to predict the normalized binding curves for a ligand and the effects of other ligands on the binding curves. Some examples for predicted activation and binding relationships are shown in Figure 2. Since the concentration dependence of activation and binding may not be intuitively obvious from the equations, we will start with the relationships for a single drug.

Binding and activation simulations. For all of these examples, NX = 2 and KR,X = 100 U. (A) Binding (left) and activation (right) curves are shown for drug X. The predicted curves are shown for a case in which desensitization is minimal (Q = 1000) to illustrate the effects of changes in the relative affinity of the resting and active states. An agonist has a value of cX < 1 and increases the prevalence of the high affinity active state. An allosteric antagonist has a value of cX > 1 and decreases the probability the receptor will be active. The plotted relationships are shown for a receptor that has a relatively high constitutive activity (L = 10, probability of being active in the absence of any agonist is PA,const = 0.091). As shown, a stronger agonist (lower c) activates strongly and has a relatively high apparent affinity (the fitted EC50, binding is 3.31 units when cX = 0.01), whereas the allosteric inhibitor (cX = 3) reduces constitutive activity and has an EC50, binding of 104 units. The equilibrium dissociation constant of the resting state is 100 U, whereas that of the active state is KA,X = cX × KR,X and ranges from 1 to 300 units in the illustrated calculations. (B) In this case, X is an agonist (cX = 0.01) while its affinity for the desensitized state is altered. The ratio of active to desensitized state in the absence of any drug is 1 (Q = 1) and L = 10. When dX > 1, X has a higher affinity for the active state than the desensitized state, and the presence of X increases the prevalence of the active state, whereas when dX < 1, it stabilizes the desensitized state. As shown, when dX = 10, activation is strongly potentiated and binding is dominated by the affinity of the active state (fitted EC50, binding is 3.34 U; compare with the case cX = 0.01 in part A). As dX decreases below 1, the activation is reduced below the constitutive level by increasing [X], but the binding is further left-shifted from that of the resting receptor, and the EC50 (fitted EC50,binding = 0.35 U) approaches that of the desensitized receptor (KD,X = 100 × 0.01 × 0.1 = 0.1 U). (C) The effects of allosteric modulators that bind to different sites than X are shown as a function of [X] for an NAM that reduces function and a PAM that enhances function. For this panel the receptor has a relatively low constitutive activity (L = 100; PA,const = 0.01; Q = 1), and NX = 2, cX = 0.01, dX = 1 and KR,X = 100 U. Both the PAM and NAM have two binding sites on a receptor. In the absence of either modulator X is relatively potent (EC50,activation = 6.6 U and EC50,binding = 7.1) and efficacious (PA,max = 0.498). Both the PAM and NAM were modeled as if they were present at their respective dissociation constants for the resting receptor. The PAM was modeled as a relatively weak agonist (cPAM = 0.1) with identical affinities for the active and desensitized receptors (dPAM = 1). As shown, the basal activity in the absence of X but the presence of the PAM increased from 0.01 to 0.19, the maximal response to X increased very slightly (PA,max = 0.500) and EC50,activation shifted to = 0.9 U, whereas EC50,binding shifted to 1.6 U. The NAM had equal affinities for the resting and active receptor (cNAM = 1) but a higher affinity for desensitized receptors (dNAM = 0.1). The basal activity in the absence of X but the presence of the NAM decreased from 0.01 to 0.008, whereas the maximal response to X was severely reduced (PA,max = 0.032). However, the EC50s for X for both activation (EC50,activation = 1.29 U) and binding (EC50,binding = 2.05) were left shifted to a comparable extent as for the PAM.
There are three states of the receptor: resting (R), active (A), and desensitized (D). The parameters L (PR/PA) and Q (PA/PD) reflect the energy differences between the states. When L is large, the resting state is energetically much favored in the absence of bound ligand, resulting in low baseline activity. For the wild-type heteromeric GABAA receptor, L is ∼1000–10,000, and basal activity is low. If Q is large, then the active state is energetically favored relative to the desensitized state and there is relatively little desensitization. For the wild-type GABAA receptor, Q is 0.05–1; that is, desensitization is usually between 50% and 95% at steady state.
The relative affinities of drug X are expressed by the ligand-specific parameters cX and dX, where cX is the ratio of the dissociation constant for X in the active state to that in the resting state (cX = KA,X/KR,X) and dX is the analogous ratio for the active and desensitized states (dX = KD,X/KA,X). Note that since dissociation constants are used, cX is less than 1 when the affinity is higher for the active state, and similarly dX is less than 1 when the affinity is higher for the desensitized state.
When cX is <1, the presence of X will increase the prevalence of the active form; that is, X is an agonist. Activation will be enhanced as the concentration of X increases, and binding will be dominated by binding to the active, high affinity form. When cX = 1, there is no preference for either state. Consequently, the presence of X has no effect of activity, and binding is determined by the dissociation constant for the resting, lower affinity form. When cX > 1, then the presence of X favors the resting state, and so as the concentration of X and binding increase, activity decreases. Again, binding is determined by the affinity of the resting form.
This is illustrated in Figure 2A for a receptor that has a relatively low energy difference between the R and A states (L = 10) so that both increases and decreases of activity can be seen. To remove the effect of desensitization, Q is set to 1000 (the A state is 1000-fold more prevalent than the D state). As seen in the figure, when cX = 0.01, activity and binding occur at relatively low [X], with EC50s reflecting the higher affinity of the active state (KA,X = cX × KR,X). When cX = 1, the increase in [X] results in no activation. The binding curve is shifted to the right, and the EC50 reflects the affinity of the resting state. When cX > 1, the resting activity is reduced by X, and the binding curve is slightly shifted to the right of that when cX = 1.
The contribution of the desensitized state is illustrated in Figure 2B. Again, only a single drug (X) is present. X is an agonist with higher affinity for the active state than resting (cX = 0.01). To maintain an appreciable constitutive level of activity, L = 10. The ratio of the active to the desensitized state in the absence of any ligand is 1 (Q = 1). When the affinity of X is higher for the active than the desensitized state (dX = 10), activation is quite strong, and the EC50 is close to the dissociation constant for the active state (KA,X = KR,X × cX = 100 × 0.01 for this example). The EC50 for binding is similar. As dX approaches 1, the steady-state response is reduced due to the increased prevalence of the desensitized state, and the EC50 begins to move toward the dissociation constant of the desensitized state, as does the EC50 for binding. When dX < 1, then the presence of X favors the desensitized state, so the steady-state response is more strongly reduced and the EC50 is shifted further toward the affinity of the desensitized state. Finally, when dX is much less than 1, X is a more efficacious inhibitor, and the EC50 for activation approaches the affinity of the desensitized state (KD,X = KR,X × cX × dX = 100 × 0.01 × 0.1 for this case when dX = 0.1).
Note the contrast between the two mechanisms for functional inhibition in the presence of the single drug, X. If cX > 1, the presence of X reduces the constitutive activity of the receptor because X favors the low affinity resting state, and the concentration dependence of inhibition reflects the affinity of the low affinity state (Fig. 2A). In contrast, when cX < 1 but dX < < 1, X inhibits function by enhancing desensitization, and the concentration dependence reflects the high affinity of the desensitized state (Fig. 2B).
Figure 2C illustrates the consequences of the presence of a positive allosteric modulator (PAM) and a negative allosteric modulator (NAM) on the binding of an agonist, X (KR,X = 100, cX = 0.01). Both PAM and NAM bind to different sites than does X. The PAM has a higher affinity for the active than the resting receptor (cPAM = 0.1) but equal affinities for the active and desensitized receptor (dPAM = 1). The NAM, in contrast, has equal affinities for the resting and active receptor (cNAM = 1) but a higher affinity for desensitized than active receptors (dNAM = 0.1). As seen in Figure 2C, the two allosteric modulators have very different effects on the concentration-response relationship for the functional effects of X but essentially identical effects on the binding of X. They both enhance the prevalence of a high affinity state of the receptor, but the PAM increases the prevalence of the active and the desensitized states, whereas the NAM preferentially increases the desensitized state.
Activation and Desensitization Properties of the Human α1β3 GABAA Receptor.
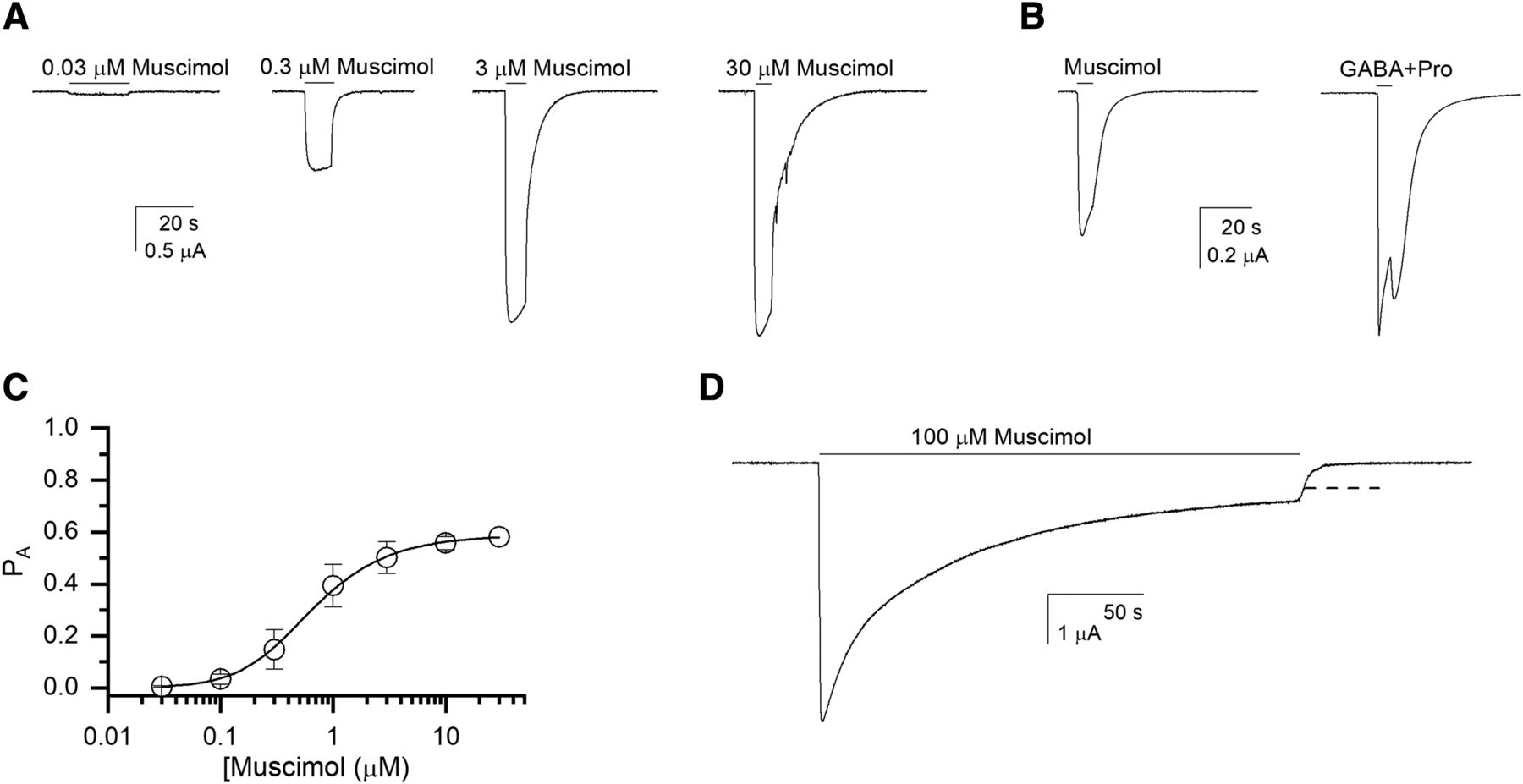
Receptor activation by muscimol was determined by exposing oocytes expressing α1β3 receptors to 0.03–30 µM muscimol. Fitting the concentration-response relationships for the peak currents with the Hill equation gave an EC50 of 0.65 ± 0.22 µM (mean ± S.D.; n = 9 cells) and a Hill coefficient of 1.77 ± 0.71.
The peak responses were analyzed in terms of the two-state Monod-Wyman-Changeux model. This approach is based on the idea that the agonist induces essentially no desensitization at the time of the peak response. Constitutive activity (PA,const) was measured by comparing the effect of 200 µM picrotoxin on holding current with the peak response to 1 mM GABA + 50 µM propofol (Eaton et al., 2016). The estimated PA,const was 0.00023 ± 0.00012 (n = 7 cells), giving a value of 6130 for L. Fitting the muscimol concentration versus PA,peak data to eq. 1 yielded a KR,muscimol of 0.54 ± 0.03 µM (best-fit parameter ± S.D. of the fit) and a cmuscimol of 0.0107 ± 0.0001. The number of binding sites for muscimol was held at 2. Sample currents and the concentration-response curve for muscimol are shown in Figure 3, A–C.

Activation of the α1β3 receptor. (A) Sample current responses to applications of 0.03, 0.3, 3, and 30 µM muscimol. All traces are from the same cell. (B) Comparison of responses to 100 µM muscimol and 1 mM GABA + 50 µM propofol (Popen ∼1). Both traces are from the same cell. (C) Muscimol concentration-response relationship. The data points show means ± S.D. from nine cells. The curve shows a fit to eq. 1, giving a KR,muscimol of 0.54 µM and a cmuscimol of 0.011. The number of binding sites for muscimol was held at 2. (D) A sample current trace in the presence of a 4-minute application of 100 µM muscimol. The dashed line shows the fitted steady-state current level.
Long exposure to high concentrations of muscimol desensitizes the receptor. The extent of desensitization is described by the parameter Q (= A/D in Fig. 1B). We used eq. 2 to estimate the value of the parameter Q as 0.057 ± 0.020 (n = 5). A sample current trace is shown in Figure 3D. Note that we have assumed that dmuscimol = 1.
A similar estimate for Q was obtained from the analysis of responses to saturating concentrations of GABA (Q = 0.099 ± 0.033; n = 7 cells), consistent with the idea that the value of Q is a property of the receptor. This is in agreement with findings in previous studies of α1β2γ2L receptors, which yielded similar values for Q after activation by GABA and taurine (QGABA = 0.29 and Qtaurine = 0.34; Germann et al., 2019b) and GABA and β-alanine (QGABA = 0.27 and Qβ-Alanine = 0.24; Germann et al., 2019a).
Modulation of GABAA Receptor Currents by the Steroids 3α5αP and PS.
The steroid 3α5αP is a positive allosteric modulator of the GABAA receptor (Belelli and Gee, 1989; Puia et al., 1990; Li et al., 2007). In the framework of the concerted transition model, its ability to potentiate results from the energy that its binding contributes to stabilizing the active state, as shown by the decrease in the effective value of L (Germann et al., 2019a; Shin et al., 2019). Receptor modulation by 3α5αP was evaluated by coapplying 0.01–3 µM 3α5αP with 0.1 µM muscimol (PA,muscimol ∼0.09). Each cell was additionally tested with 100 µM GABA + 50 µM propofol to obtain a reference response with PA,peak of ∼1. In nine cells, the EC50 for 3α5αP was 0.13 ± 0.06 µM and the Hill coefficient 1.31 ± 0.18. The steroid concentration–PA data were fitted with eq. 3, yielding a KR,3α5αP of 0.19 ± 0.02 µM and a c3α5αP of 0.132 ± 0.005. The number of binding sites for 3α5αP was constrained to 2 (given that the Hill coefficient was greater than 1) and L* to 10.1. Sample current traces and the 3α5αP concentration-response relationship are given in Figure 4A. As shown in the figure, the parameter values give a good description of the data.
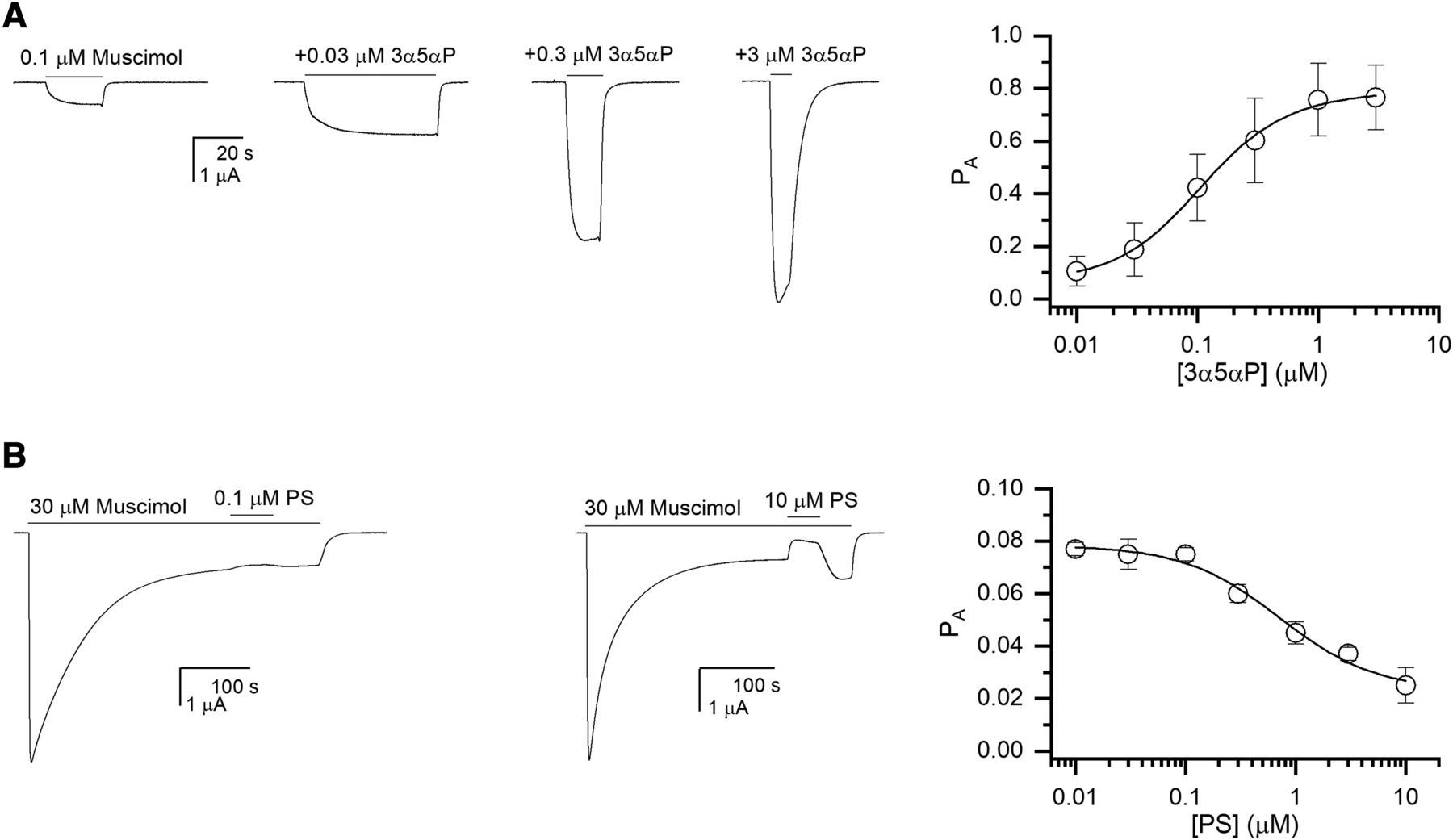
Modulation of the α1β3 receptor currents by 3α5αP and PS. (A) Sample current responses to 0.1 µM muscimol (PA ∼0.09) and to muscimol coapplied with 3α5αP. The graph shows the 3α5αP concentration-response relationship. The data points show means ± S.D. from nine cells. The curve shows a fit to eq. 3, giving a KR,3α5αP of 0.19 µM and a c3α5αP of 0.132. The number of binding sites for 3α5αP was held at 2. (B) Sample current responses showing the effect of PS on steady-state current elicited by 30 µM muscimol. The graph shows the PS concentration-response relationship. The data points show means ± S.D. from five to six cells per concentration. The curve shows a fit to eq. 4, giving a KR,PS of 0.98 µM and a dPS of 0.278. The number of binding sites for PS was held at 1.
The steroid PS is a negative allosteric modulator of the GABAA receptor that acts by promoting desensitization (Majewska et al., 1988; Akk et al., 2001; Germann et al., 2019b). Receptor modulation by PS was determined by coapplying 0.01–20 µM PS during the steady-state response to a saturating concentration of muscimol. Exposure to PS led to a decrease in the steady-state current level (Fig. 4B). Due to extended recording times and drug exposures, each cell was tested with a single concentration of PS. Analysis of the effect of PS using the Hill equation yielded an IC50 of 1.4 ± 0.8 µM and a Hill coefficient of −0.58 ± 0.09 (five to six cells per concentration of PS). Since PS has no discernable effect on holding current or on the peak response to GABA (Eisenman et al., 2003; Akk et al., 2007; Germann et al., 2019b), it was assumed that it has equal affinities for resting and active receptors (cPS = 1). Analysis of PS modulation of currents using eq. 4 gave a KR,PS (or KA,PS) of 0.98 ± 0.28 µM and a dPS of 0.278 ± 0.086. The number of binding sites for PS was held at 1. Again, the parameters provide a good description of the data (Fig. 4B).
Observed and Predicted Steady-State Muscimol Binding Isotherms.
Steady-state binding of muscimol was determined using membranes from HEK 293 cells that stably expressed GABAA α1 and β3 subunits. Binding was performed at 1 hour on ice, so the overall distribution of the receptors among conformational states was likely at steady state. Binding isotherms were conducted three times for muscimol alone, for muscimol plus 10 µM 3α5αP, and for muscimol plus 10 µM PS.
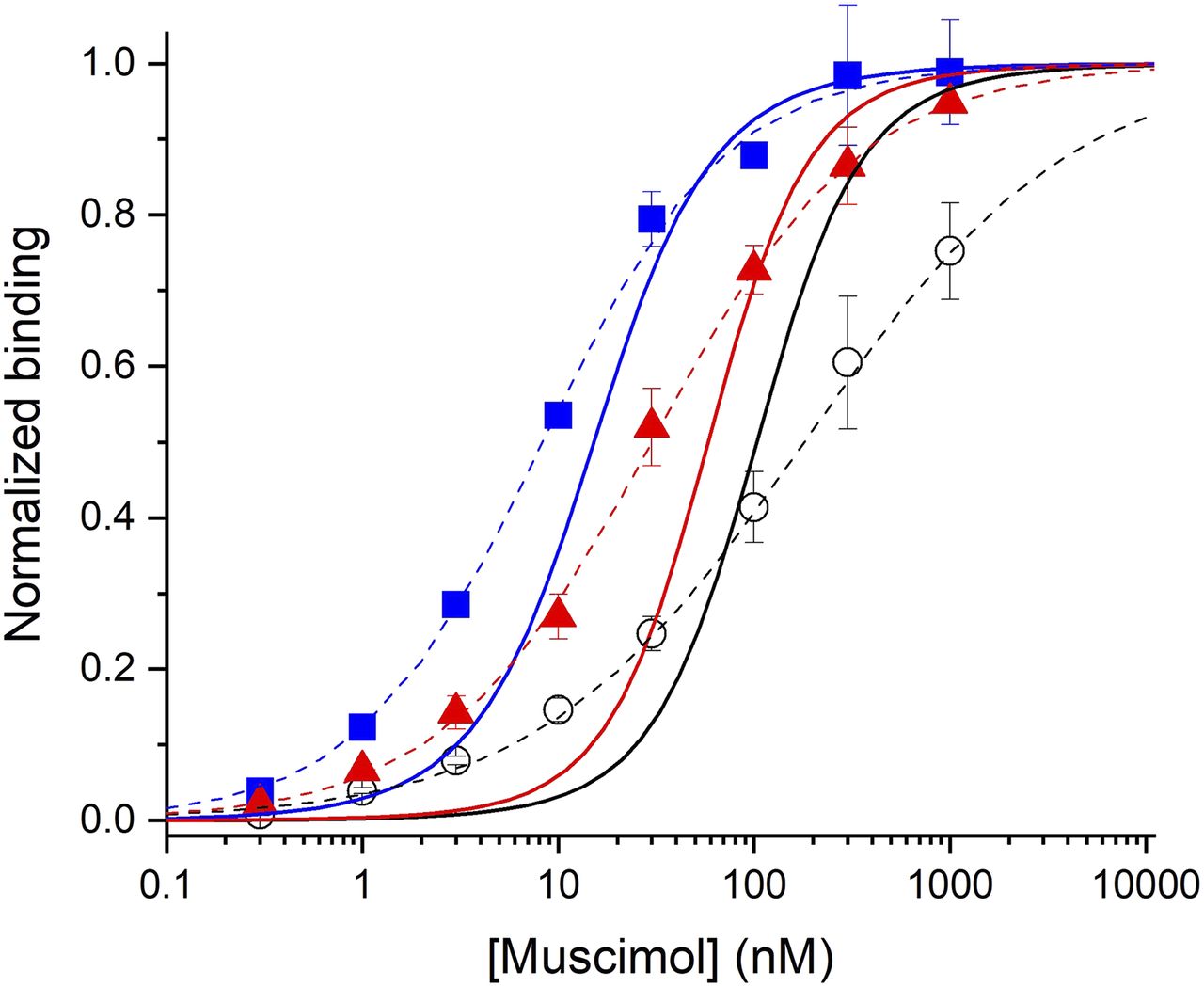
[3H]muscimol binds to α1β3 GABAA receptors with an EC50 of 180 ± 83 nM and a Hill coefficient of 0.64 ± 0.06 (Fig. 5). When the binding experiments were conducted in the presence of 10 µM 3α5αP or 10 µM PS, the binding curves were shifted to lower concentrations of muscimol. In the presence of 3α5αP, the EC50 was 8.3 ± 0.6 nM and the Hill coefficient 0.93 ± 0.07. In the presence of PS, the EC50 was 30 ± 6 nM and the Hill coefficient 0.82 ± 0.02.

Observed and predicted [3H]muscimol binding isotherms. The data points show means ± S.D. from three experiments each for control, i.e., in the absence of any modulators (black circles) and in the presence of 10 µM 3α5αP (blue squares) or 10 µM PS (red triangles). The dashed lines show the predictions of fit Hill equations (control EC50 = 180 ± 83 nM, nHill = 0.64 ± 0.06; +3α5αP EC50 = 8.3 ± 0.6 nM, nHill = 0.93 ± 0.07; +PS EC50 = 30 ± 6 nM, nHill = 0.82 ± 0.02). The data have been normalized to the fitted maximal binding for each separate experiment. The solid lines show the predicted binding curves from the physiologic data (control EC50 = 106 nM, nHill = 1.56; +3α5αP EC50 = 16 nM, nHill = 1.38; +PS EC50 = 60 nM, nHill = 1.61).
The simulations of [3H]muscimol binding were done using eq. 11 employing the activation and desensitization parameters determined earlier in electrophysiological experiments (L = 6130, Q = 0.057, KR,muscimol = 0.54 µM, cmuscimol = 0.0107, dmuscimol = 1, and Nmuscimol = 2). The simulated curve is shown in Figure 5. Fitting the predicted curve with the Hill equation gave an EC50 of 106 nM and a Hill coefficient of 1.56.
Simulations of modulation of [3H]muscimol binding by 3α5αP and PS were conducted using eq. 14 and the properties of the steroids as estimated using analysis of functional responses (K3α5αP = 0.19 µM, c3α5αP = 0.132, d3α5αP = 1, N3α5αP = 2, and KR,PS = 0.98 µM, cPS = 1, dPS = 0.278, NPS = 1). The simulated curves are shown in Figure 5. Fitting the simulated curves with the Hill equation gave an EC50 of 16 nM and a Hill coefficient of 1.38 for binding in the presence of 3α5αP, and an EC50 of 60 nM and a Hill coefficient of 1.61 for binding in the presence of PS.
Discussion
Radioligand binding to the GABAA receptor has been employed to describe receptor expression and distribution in various brain regions and other tissues (Beaumont et al., 1978; Krause et al., 1980; Chandra et al., 2010; Benkherouf et al., 2019). For example, evaluation of [3H]muscimol binding has revealed a loss of GABAA receptors in brain regions involved in face processing in people with autism, whereas increased [3H]muscimol binding was found in prefrontal cortex of people with schizophrenia (Oblak et al., 2011; Verdurand et al., 2013). The binding of [3H]muscimol is generally increased when measured in the presence of allosteric potentiators of the GABAA receptor and decreased in the presence of allosteric inhibitors (Supavilai et al., 1982; Quast and Brenner, 1983; Harrison and Simmonds, 1984; Peters et al., 1988). One notable exception is the inhibitory steroid PS that, at least at some doses, has been shown to enhance [3H]muscimol binding to native GABAA receptors (Majewska et al., 1985).
Ligand binding is governed by its affinity to the target. When the target occupies a single state, the EC50 of the binding saturation curve equals the equilibrium dissociation constant of the ligand. When the target can occupy multiple states, the total binding reflects both the fraction of receptors in various states and the affinities of the ligand to the individual states. In the case of [3H]muscimol binding to the GABAA receptor, the ligand itself is an activator having different affinities to the resting and active or desensitized states, and it is able to alter the distribution of the states, i.e., drive receptors from resting to active and desensitized states. Allosterically acting ligands can influence the distribution of the receptor population among the states by binding preferentially to particular states. Our calculations predict that both positive and negative allosteric modulators can enhance binding of an orthosteric ligand if the allosteric agents bind preferentially to a high affinity state of the receptor for the orthosteric ligand.
Binding isotherms for [3H]muscimol confirm this prediction, as both 3α5αP (a PAM) and PS (an NAM) shift the isotherm to lower concentrations of muscimol. The predicted binding curves have EC50 values in the same sequence as the observed: the observed EC50 values for control:+10 µM PS:+10 µM 3α5αP are 180:30:8 nM, whereas the predicted EC50 values are 106:60:16 nM. The predicted isotherms are consistently steeper, with Hill coefficients about 1.5–2-fold larger than the observed isotherms that have Hill coefficients less than 1. This difference suggests that there might be several populations of receptors observed in the binding experiments, possibly reflecting receptors in different compartments of the cell (internal vs. surface), at different stages of maturation, or with different posttranslational modifications. When the predicted and observed isotherms are compared, the observed isotherms appear to contain a higher affinity population of sites than the predicted isotherm rather than a lower affinity component, which suggests that the lower slope is not the result of a population of resting (or other low affinity) receptors. We note that the muscimol binding isotherm for α1β3 GABAA receptors transiently expressed in HEK cells is steeper than observed with the stably expressed receptors, perhaps because of differences in the cell biology of the receptors (Hill coefficient 1.2; Y. Sugasawa et al., preprint, DOI: https://doi.org/10.1101/2020.04.27.063404). It is important to note that there were no free parameters in the predicted isotherms (Fig. 5), i.e., all parameters were derived from the functional studies.
There is some disagreement in the literature about whether muscimol binding isotherms show evidence for multiple components (e.g., “high affinity” and “low affinity” classes of receptor). In the case of receptors isolated from brain tissue it is perhaps not surprising that the multiplicity of subunit compositions might generate a broad isotherm. Even in the case of recombinant receptors there is some disagreement, with reports of a single (Baur and Sigel, 2003; Dostalova et al., 2014) or multiple components (Newell et al., 2000). The RAD model predicts single component binding curves, although the Hill coefficient of the occupancy relationship may be less than the postulated number of binding sites.
In Figure 6, we show the muscimol concentration dependence of the distribution of receptors in the resting, active, and desensitized states and the predicted binding of [3H]muscimol to each state. At low concentrations (up to ∼0.01 µM), muscimol binding is dominated by binding to the resting state. Even though the affinity of muscimol to this state is low, the vast majority of receptors occupy the resting state at low agonist concentration. At higher concentrations, binding to the desensitized state becomes prevalent. At the highest [3H]muscimol concentration tested (1 µM), the model predicts that 8% of the receptors are in the resting state, 5% in the active state, and 87% in the desensitized state, and 5% of the bound muscimol is bound to sites on resting receptors, 5% to sites on active receptors, and 86% to sites on desensitized receptors (Fig. 6A). Note that the high prevalence of the desensitized state is the result of the low value for Q, implying that the majority of receptors in the high affinity (active or desensitized) states will be in the desensitized state.
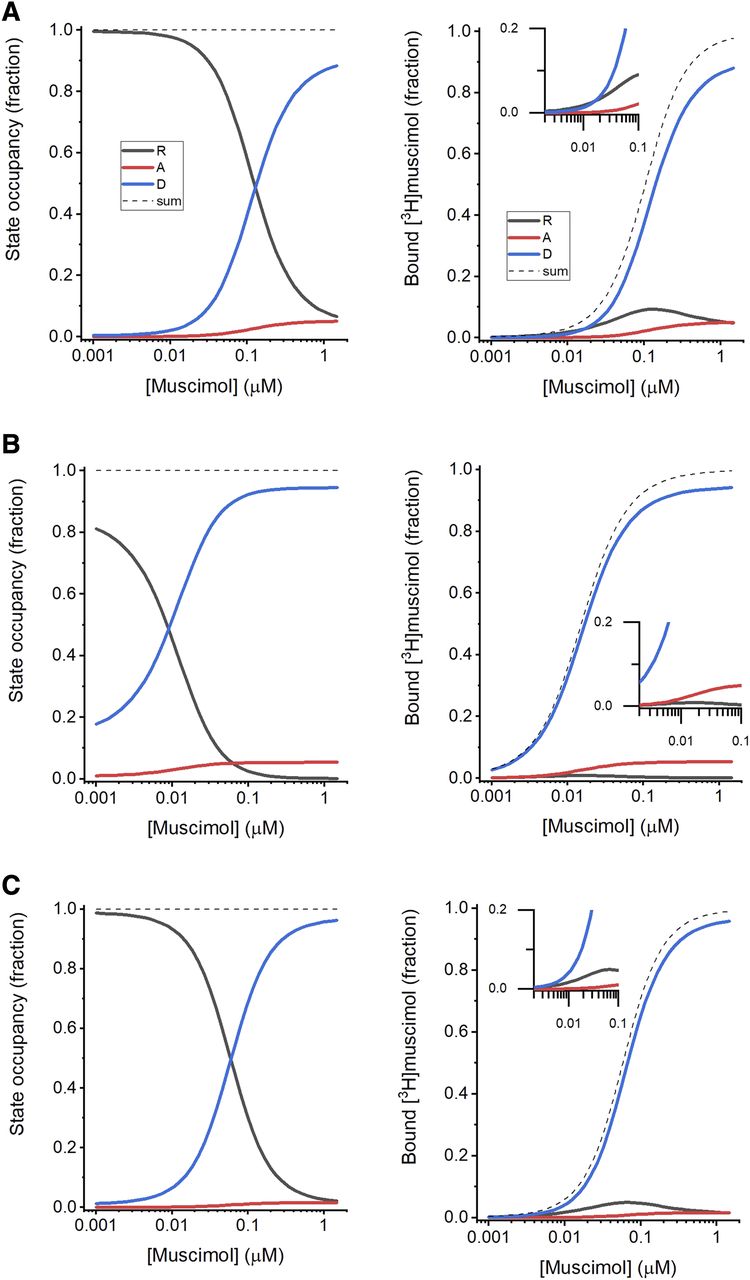
State occupancy and ligand binding. (A) The left panel shows occupancies of the resting (black), active (red), and desensitized (blue) states as a function of muscimol concentration. The occupancies were calculated using the receptor properties (L = 6130, Q = 0.057) and muscimol activation parameters estimated in electrophysiological experiments (KR,muscimol = 0.54 µM, cmuscimol = 0.0107, dmuscimol = 1, Nmuscimol = 2). The dashed black line shows the sum (1) of the fractional occupancies of the individual states. The right panel shows the fractions of the sites that are occupied by muscimol on receptors in the resting (black), active (red), and desensitized (blue) states that have bound [3H]muscimol. The dashed black line shows total fractional occupancies for all states. The inset shows the predictions at low muscimol concentrations in greater detail. (B) The left panel shows state occupancies as a function of muscimol concentration in the presence of 10 µM 3α5αP. The right panel shows the fractions of receptors in various states that have bound [3H]muscimol. The effect of 3α5αP was determined using its properties estimated in electrophysiological recordings (KR,3α5αP = 0.19 µM, c3α5αP = 0.132, d3α5αP = 1, N3α5αP = 2). (C) The left panel shows state occupancies as a function of muscimol concentration in the presence of 10 µM PS. The right panel shows the fractions of receptors in various states that have bound [3H]muscimol. The effect of PS was determined using its properties estimated in electrophysiological recordings (KR,PS = 0.98 µM, cPS = 1, dPS = 0.278, NPS = 1).
When 1 µM muscimol is combined with 10 µM 3α5αP, the fraction of receptors in the resting states decreases to 0.2%. The fractions of receptors in the active and desensitized states are slightly increased (to 5.4% and 95%, respectively). Note that the active to desensitized ratio (i.e., Q) is not altered in the presence of 3α5αP. In the presence of 10 µM 3α5αP and 1 µM [3H]muscimol, more than 99% of total sites will be occupied, with 0.1% bound to sites on resting receptors, 5.3% on active receptors, and 94% on desensitized (Fig. 6B).
Exposure to 1 µM muscimol in the presence of 10 µM PS increases the fraction of receptors in the desensitized state from 87% to ∼96%. Only 2.7% of receptors are in the resting and 1.6% in the active state when exposed to muscimol + PS. A total of 99% of sites are predicted to have bound [3H]muscimol, 1.7% in the resting, 1.6% in the active, and 95% in the desensitized state (Fig. 6C).
Exposure to either 3α5αP or PS shifts the [3H]muscimol binding curve to lower concentrations. The underlying mechanisms in the RAD model are, however, different. The potentiating steroid 3α5αP enhances [3H]muscimol binding by drawing more receptors from the resting to active state (through a c < 1), thereby reducing the fraction of receptors in the low affinity resting state. The inhibitory steroid PS shifts receptors from active to desensitized state (through a d < 1), which leads to reduced occupancy of the low affinity resting state to maintain the resting to active ratio.
In summary, we used a cyclic concerted transition model that incorporates a high affinity desensitized state to examine activation and desensitization of α1β3 GABAA receptors by the orthosteric agonist muscimol, and to derive parameters for binding to the resting, active, and desensitized states. We then examined the ability of PAM and NAM neurosteroids to modulate responses and derived parameters for the binding of the neurosteroids to the different states. The parameters obtained from functional studies allowed us to predict the normalized binding isotherms for muscimol in the absence or presence of the allosteric neurosteroids, with no free parameters. There is a close qualitative agreement between the observations and the predictions that is perhaps surprising given the very different natures of the preparations (live cells vs. membrane fragments) and the measurements (membrane currents vs. binding). There are, however, some quantitative differences, most notably in the steepness of the concentration dependencies predicted and observed. Still, the overall agreement provides strong support for the overall validity of the RAD model in studies of the GABAA receptor.
Authorship Contributions
Participated in research design: Akk, Sugasawa, Evers, Steinbach.
Conducted experiments: Germann, Sugasawa, Pierce.
Performed data analysis: Akk, Germann, Sugasawa, Pierce, Steinbach.
Wrote or contributed to the writing of the manuscript: Akk, Evers, Steinbach.
Footnotes
- Received May 11, 2020.
- Accepted July 17, 2020.
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grants GM108580 and GM108799] and funds from the Taylor Family Institute for Innovative Psychiatric Research.
Primary laboratory of origin: Department of Anesthesiology, Washington University School of Medicine, St. Louis, MO (G.A.).
Abbreviations
- 3α5αP
- 1-[(3R,5S,8R,9S,10S,13S,14S,17S)-3-hydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl]ethanone (allopregnanolone)
- NAM
- negative allosteric modulator
- PAM
- positive allosteric modulator
- PS
- [(3S,8S,9S,10R,13S,14S,17S)-17-acetyl-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-yl] hydrogen sulfate (pregnenolone sulfate)
- RAD
- resting-active-desensitized
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}