
Abstract
The SLC2 genes code for a family of GLUT proteins that are part of the major facilitator superfamily (MFS) of membrane transporters. Crystal structures have recently revealed how the unique protein fold of these proteins enables the catalysis of transport. The proteins have 12 transmembrane spans built from a replicated trimer substructure. This enables 4 trimer substructures to move relative to each other, and thereby alternately opening and closing a cleft to either the internal or the external side of the membrane. The physiological substrate for the GLUTs is usually a hexose but substrates for GLUTs can include urate, dehydro-ascorbate and myo-inositol. The GLUT proteins have varied physiological functions that are related to their principal substrates, the cell type in which the GLUTs are expressed and the extent to which the proteins are associated with subcellular compartments. Some of the GLUT proteins translocate between subcellular compartments and this facilitates the control of their function over long- and short-time scales. The control of GLUT function is necessary for a regulated supply of metabolites (mainly glucose) to tissues. Pathophysiological abnormalities in GLUT proteins are responsible for, or associated with, clinical problems including type 2 diabetes and cancer and a range of tissue disorders, related to tissue-specific GLUT protein profiles. The availability of GLUT crystal structures has facilitated the search for inhibitors and substrates and that are specific for each GLUT and that can be used therapeutically. Recent studies are starting to unravel the drug targetable properties of each of the GLUT proteins.
Similar content being viewed by others
Introduction
The SLC (SoLute Carrier) gene family has been classified into 65 sub-families with identities within the sub-family that differ more than 20–25% from other SLCs [38, 89]. The SLC2 sub-families of 14 related genes are thus distinct from the closest relatives (which are the SLC19 sub-family) and lie within the major facilitator superfamily (MFS) group which codes for proteins whose function is to facilitate membrane transport of substrates [89]. The 14 SLC2 genes code for proteins that are further sub-divided into phylogenetically distinct classes 1-3 of GLUT (glucose transport) proteins [106].
The proteins of the SLC2 family have 12 transmembrane spans (TMs 1-12) with intracellular N- and C-termini. Signature motifs in the GLUTs include highly conserved salt bridging motifs RXGRR between cytoplasmic loop of TM2 and TM3, with a repeated of the salt bridge sequence between the cytoplasmic loop between TM8 and TM9. These motifs are present in all proteins of the MFS superfamily and are associated with catalytic conformational change that occurs in mediated transport. The proteins are thought to have evolved from 4 inverted trimer repeats as TM1-3 has some sequence similarity to an inverted TM4-6 and TM7-9 has some sequence similarity to an inverted TM10-12. Such inverted repeats are thought to emerge from gene duplication and fusion [75, 167]. There is also structural pseudo-symmetry in which TM1-6 (the N-terminal half) is mirrored by TM7-12 (the C-terminal half) with the 2 half proteins separated by a large cytoplasmic loop between TM6 and TM7 [161]. There are also sequence motifs that are highly conserved and unique to the GLUT family and these regions may play a role in recognition of hexose-like substrates [11].
All the GLUT proteins were originally presumed to catalyse hexose transport into and out of cells. This is clearly the case for the class 1 GLUT proteins. However, class 2 and class 3 GLUT proteins do not necessarily have a primary function in catalysing glucose transport. In many cases, they can be shown to catalyse glucose uptake in experimental systems. However, some of these proteins (particularly the class 3 group) have alternative substrates and therefore alternative functions within their physiological settings, and at an appropriate endogenous cellular location.
Much of the experimental work on the GLUT proteins has focussed on the mammalian glucose transporters that are most abundant. These GLUTs (GLUTs 1-4) catalyse (facilitate) passive movement of glucose down concentration gradients. These gradients are usually from the blood system to the cell interior, but in the case of the liver, these gradients can be from the cell to the systemic blood stream. Once transport down a concentration gradient has been achieved, then net flux into or out of the cell is zero, but transport of glucose continues by a process of equilibrium exchange. Glucose influx and efflux through the protein continue but these exchange fluxes are equal [67]. The GLUT family of transport proteins thereby cooperatively function to provide a supply of glucose in the direction needed for cell metabolic processes while maintaining a remarkably constant blood glucose level (5 mM after fasting in humans) [84, 154]. These glucose transporters are often rate limiting for subsequent metabolism [82] and thus provide a point at which metabolic flux can be controlled.
Class 1: GLUTs 1, 2, 3, 4 and 14
Class1 GLUTs have an N-linked asparagine with varying chain length glycan polymers that often give a broad spread of the protein when detected by Western blotting after resolution on SDS-PAGE gels. The topography of the GLUTs, particularly GLUT1, has been explored by a wide range of scanning mutagenesis experiments [152] that have revealed regions of the protein that are essential for transport.
GLUT1
The GLUT1 transporter is present in high amounts in human erythrocytes and because of the relative ease of working with these cells, it has been most studied from the structure related to function perspective [49, 152, 154]. The large amount of the protein (approximately 5% of the membrane protein) was an important factor in its purification. A peptide sequence was obtained and used to identify a cDNA clone and ultimately the DNA sequence [152]. GLUT1 is still the only endogenous GLUT that has been purified to homogeneity and which can be identified as a Coomassie-staining protein on an SDS-PAGE gel. Methods for purification of recombinant GLUT1 have gradually been improved over several decades and ultimately GLUT1 was crystallised in an inward facing conformation by the Yan group [56].
GLUT1 has a Km for glucose influx (around 2 mM) which is significantly lower than blood glucose levels [16, 112, 187]. The kinetics of glucose and galactose uptake have been extensively studied and reveal that below physiological temperatures, the Km and Vmax for efflux can up to 10-fold higher than for influx. In addition, the kinetic parameters for exchange transport (Km and Vmax) are both approximately 10-fold higher than for net influx at low temperatures. These data suggest that influx (but not efflux or exchange transport) can be rate limited by the availability of binding sites at the exofacial surface of the erythrocyte. The physiological importance of this property is unclear and indeed, the asymmetry of binding site availability is much less evident at physiological temperatures [27, 219]. However, the property may allow rapid glucose efflux from the erythrocyte when these cells reach conditions of relatively low plasma glucose concentrations. The transport kinetic properties associated with GLUT1 in erythrocytes may be specific for this cell type and the asymmetry has not been extensively studied in other cell types. GLUT1 has very low affinity for the ketoses fructose and sorbose with affinity constants in the molar range [14].
GLUT1 is very abundant at the endothelial cells of blood-brain barrier and this supply of glucose to the brain must always be efficient, even at the expense of depriving other tissues of glucose. In this location, it provides a rate limiting barrier for glucose reaching the brain [215]. Consistent with this hypothesis, GLUT1 levels at this barrier are increased when blood glucose concentrations are low and in clinical hypoglycaemia [191]. GLUT1 is also present at high levels in the endometrial stromal cells of the placenta and is essential for viability and survival of the foetus [76, 149].
GLUT1 is expressed in varying degrees in most human cells, often along with other class 1 glucose transporters, and its availability at the plasma membrane can be regulated by membrane protein translocation in tissues where glucose uptake is hormonally controlled [92]. Several stress stimuli, including osmotic shock, are known to increase GLUT1 levels and possibly GLUT1 catalytic activity [12, 15]. GLUT1 can bind ATP and other nucleotides and these interactions may have a regulatory role [42]. GLUT1 knockout is embryonically lethal [221] but GLUT1 haplo-insufficiencies (due to inherited GLUT1 mutations) lead to range of metabolic imbalances and symptoms that include epileptic seizures [36, 162]. The study of these naturally occurring mutations has aided the mapping of important regions and residues in GLUT1 that are necessary for transport [49, 95, 153].
GLUT2
GLUT 2 is present at high levels in the liver, pancreatic β cells, the intestine and other cells of the endoderm lineage [210]. By contrast with GLUT1, it has relatively high affinity for both glucose and fructose with a Km of 20 mM for glucose and Km of 67 mM for fructose [43, 84]
Glucose and fructose enter the portal circulation via GLUT2-mediated transport, primarily at the basolateral border of intestinal epithelial cells. However, GLUT2 can also be translocated to the brush border of intestinal cells depending upon the dietary sugar load [117]. In the liver, GLUT2 avidly removes fructose from the portal vein so that only low levels of fructose are present in the systemic circulation [163]. GLUT2 on the basolateral borders of kidney epithelial cells is required for glucose reabsorption and GLUT2 knockout mice have elevated glucosuria [87]. GLUT2 has a Km value of influx of approximately 20 mM which is higher than fasting blood glucose reflecting the functions of this protein in supplying glucose to the systemic circulation.
In the mouse [211] but not human [144] pancreas, GLUT2 is a key component (along with glucokinase and Kir6.2/SUR potassium channels) in glucose-stimulated insulin and glucagon secretion. Glucose metabolism in the pancreas is mainly rate limited by glucokinase [211]. Transport by GLUT2 is also associated with extra-pancreatic glucose sensing in neurones, and in blood vessels of the portal and systemic circulations. In the some brain regions, including the hypothalamus, GLUT2 and hexokinase may co-ordinately control aspects of feeding and energy expenditure [211]. Consistent with this, GLUT2 knockout mice have abnormal feeding behaviour [87] and mutations in the human SLC2A2 gene are associated with a preference for sugar feeding and type 2 diabetes [69]. Human mutations result in the Fanconi-Bickel syndrome, originally described as a glycogen storage disorder and associated with a range of metabolic traits including glucosuria. A large range of rare inherited mutations in the SLC2A2 gene occur. The range of phenotypic changes associated with different GLUT2 mutations include those due to a loss of GLUT2-mediated glucose transport and GLUT2 targeting to the plasma membrane [68]
GLUT3 and GLUT14
GLUT3 is mainly present in the brain [139]. The Km of 1–2 mM is low in relation to blood glucose and the transport catalysis rate is very fast in cerebellar granule neurones [138]. These kinetic factors allow the neurones to avidly remove and metabolise glucose that has passed through the blood-brain barrier endothelial cells [192]. Some brain cancers, such as those arising in glioblastoma cells, could be specifically targeted by therapeutic reagents because of their dependence on the high rates of GLUT3-mediated glucose uptake [45].
GLUT3 is important for mouse embryo development and (together with GLUT1) with the supply of glucose to the foetus [77, 192]. GLUT3 is upregulated in response to hypoxia [2], and this upregulation is mediated by the transcription factor HIF, possibly in response to a long non-coding RNA [128]. Depletion of cellular ATP levels also leads to upregulation of GLUT3 by a slowing of degradation [118].
GLUT14 is a GLUT3 variant that has also been found in the genome as a duplicon of GLUT3 [226]. It is essentially uncharacterised in terms of substrate specificity, which is assumed to be similar to that of GLUT3. The function and tissue distribution are uncharacterised, although there is some disease association, specifically in inflammatory bowel disease [3].
Heterozygous knockout of GLUT3 in mice leads to abnormal brain development and some behavioural abnormalities that resemble those of autistic spectrum disorders. These GLUT3 partial knockout phenotypes indicate an important role in many aspects of neuronal function [34, 182, 233].
GLUT4
GLUT4 is present mainly in the insulin-sensitive tissues of adipose, heart and skeletal muscle. Its Km for glucose substrate is close to the fasting blood glucose level and this is unchanged by insulin action, which instead leads to a massive increase in the Vmax for transport, which can be over 10-fold in some tissues [209]. The transport system is kinetically symmetric with equal Km and Vmax for uptake, efflux and exchange transport [208].
There are three main ways in which GLUT4 can be regulated: post-translocation modification including glycosylation, possible ubiquitinoylation, phosphorylation and palmitoylation; changes in turnover including transcription and degradation; membrane protein translocation and compartmentalisation. Each of these processes would be expected to have a different degree of temporal control, ranging from rapid control of translocation to slower control of protein turnover, but it is often difficult to distinguish these separate processes. For the GLUT4 transporter, the extents of post-translational modification by ubiquitinylation [127], phosphorylation [176] and palmitoylation [64, 171] may be quite low and are not easily quantified. However, if GLUT4 traffics through low abundance but critical subcellular compartments in which these modifications occur, then these steps may be quite influential in determining the storage and fate of GLUT4 vesicles. Further studies may reveal the extent to which these modifications are important for long-term regulation of GLUT4 and control of its turnover.
The insulin-stimulated increase in the Vmax for transport in insulin-target tissues occurs rapidly and is considered to be almost entirely due to the rapid translocation of GLUT4 to the cell surface from an intracellular reservoir compartment, but often it is also associated with a smaller fold translocation of GLUT1 [92], and possibly other GLUTs [145].
GLUT translocation was originally discovered in adipose cells in 1980 [50, 202]. This was followed by the cloning of the GLUT4 isoform [20, 101]. Since then, many new techniques for studying the subcellular locations of GLUT4 have been developed and important aspects of the link between GLUT4 traffic and insulin signalling have been revealed (reviewed in [120] and summarised in Fig. 1) Early insulin signalling from the insulin receptor tyrosine kinase activation to the downstream serine kinase Akt is now well understood [206]. Activated Akt leads to phosphorylation and inactivation of several GAP proteins that control metabolism. These include the Rab GAPs TBC1D1 and TBC1D4 which maintain their Rab substrates in the GDP form. Rab-GAP inactivation therefore leads to increases in the loading of Rab proteins with GTP. One of the main targets of TBC1D1 and TBC1D4 is Rab10, but there are possibly other Rab proteins that are involved in the control of GLUT4 traffic that operate in parallel with Rab10 or downstream of it [123, 201]. A cascade of linked Rab GAPs and Rabs is also likely [172]. TBC1D1 and TBC1D4 are also targets of AMPK and there may be some convergence with signalling from insulin activation of Akt [71, 103]. However, it has yet to be demonstrated that GLUT4 exocytosis occurs in response to AMPK signalling to the Rab GAPS. The activities of the Rab GAPs are further discussed by Al Hasani and Chadt in this Special Issue of Pflügers Archives.

Compartmentalisation and retention of GLUT4 in insulin-target tissues. In basal cells, most of the cellular GLUT4 is sorted into intracellular compartments (EE: early endosomes; SE sorting endosomes; TGN: trans-Golgi network; Golgi: Golgi stacks; ERGIC; endoplasmic reticulum- Golgi intermediate compartment; ER endoplasmic reticulum; lysosomes: lysosomes including multi-vesicular body (MVB) pre-lysosomal compartments; GSV/IRV GLUT4 storage vesicles/insulin-responsive vesicles. Several processing and sorting steps (blue text) are involved in maintained intracellular GLUT4. These include EE retrieval of GLUT4 from the plasma membrane via CHC17 clathrin-coated vesicles. The SE compartments segregate GLUT4 either for recycling or for lysosomal degradation via MBV compartments that lead to lysosomes (this appears to be dependent on ubiquitinoylation of GLUT4 or GLUT4 associated proteins). In the absence of ubiquitinoylation, or following deubiquitinylation by USP25, vesicle recoating with clathrin (CHC22 in humans) occurs in a process involving sortilin and GGA2 and with retrograde transfer to the TGN and GSV/IRV. GLUT4 is also trafficked from ER to ERGIC. It is retained in ERGIC but is available for release (possibly directly to GSV/IRV). The candidate proteins for this further processing and sorting are clathrin-coating CHC22 in humans, the Golgi tether p115, the insulin-responsive amino-peptidase IRAP, TUG and the enzyme tankarase, UBC9 (reviewed in detail in ref [26, 120]). Insulin signalling through tyrosine phosphorylation leads to activation of SNARE proteins (VAMP2 on GLUT4 vesicles and the SNAP23, Syntaxin4, Munc18c complex at the plasma membrane). These activations facilitate fusion of GLUT4 vesicles with the plasma membrane. Insulin signalling through serine kinase cascades leads to phosphorylation and inactivation of the Rab-GAP activities of TBC1D1 and TBC1D4. This is associated with GTP loading of the Rabs on GLUT4 vesicles (mainly Rab10-GTP) and these GTP-Rabs direct the recruitment of the vesicles to the plasma membrane, ref [120] for review. This figure is adapted from the GLUT4 traffic map described in [32] and kindly supplied by the Brodsky group https://www.ucl.ac.uk/research/domains/food-metabolism-and-society/events/glut4-traffic-map-workshop
GLUT4 is compartmentalised in multiple subcellular compartments in insulin-target cells [28, 32, 93, 193] and much effort has been involved in determining how and when the insulin-stimulated redistribution of GLUT4 between the plasma membrane, endosomes, pre-lysosomal multivesicular body (MBV) compartments, trans-Golgi stacks and ER-Golgi intermediate compartment (ERGIC) compartments occurs (Fig. 1) (reviewed in [25, 120]). An intracellular targeting sequences, FQQI at the N-terminus and dileucine and an acidic cluster motif at the C-terminus [18, 23, 142, 188] determine the intracellular distribution and are necessary for GLUT4 sequestration to a specialised reservoir compartment in unstimulated cells.
Kinetic studies on GLUT4 traffic have revealed a major effect of insulin is to increase the rate of exocytosis of GLUT4 from the intracellular reservoir compartment to the plasma membrane in adipose cells [180, 232] and to the sarcolemma and T-tubules in muscle [72, 113]. This change in the rate of exocytosis rate quantitatively and temporally accounts for increase in cell surface GLUT4. Several insulin-regulated steps may be involved in stimulation of GLUT4 exocytosis. These include release of GLUT4 from intracellular retention [155] and insulin-stimulated fusion of GLUT4 vesicles at the plasma membrane [9, 122]. The fusion step may be controlled directly at the plasma membrane as insulin-activated plasma membrane alone can activate fusion in a cell free system in which vesicles and cytoplasm are maintained in a basal state [122]. In addition to steps downstream of Akt activation [9], insulin-activated receptor tyrosine kinase stimulates vesicle fusion proteins of the SNARE complex through changes in syntaxin4 and Munc18c interaction [6, 104, 119, 168].
Intracellular retention of GLUT4 in a specialised reservoir compartment is essential for maintaining a low rate of glucose transport in the basal state. Retention and GLUT4 sorting mechanisms vary depending on the cell type and cell line. However, collectively the retention mechanisms appear to involve a series of interacting, adapter and tethering proteins including TUG, sortilin, golgi associated, gamma adaptin ear containing, ARF-binding protein 2 (GGA2), p115 and insulin-responsive aminopeptidase (IRAP), the clathrin heavy chain (CHC) components CHC17 and CHC22 [26, 32, 177]. The enzyme tankarase, UBC9 (ubiquitinylation) and USP25 (deubiquitnylation) also appear to be involved in post-translational control of GLUT4 traffic and sorting [124, 127, 177, 227]. Figure 1 summarises the compartments in which GLUT4 is localised, together with the GLUT4-vesicle-interacting proteins which control GLUT4 retention and sorting. These processes ultimately lead to the generation of the GLUT4 storage compartment and insulin-responsive vesicles.
In rat adipose cells, a large proportion to the total internal GLUT4 resides in specialised vesicles of approximately 60 nm that contain very few additional components other than IRAP and VAMP2 [109, 110, 169], and it is these vesicles that are rapidly recruited to the plasma membrane in response to insulin signalling. The fusion of these vesicles with the plasma membrane occurs at a rate that is consistent with this step limiting the whole GLUT4 exocytosis process [9, 122]. It would be useful to obtain rates for the insulin-stimulated release of GLUT4 from retention compartments and restraining steps, such as TUG proteolytic cleavage and changes in ubiquitylation of GLUT4 vesicle-associated proteins. This would allow determination of whether these release steps occur at rates that are comparable with the insulin-stimulated GLUT4-vesicle fusion rate.
Endocytosis of GLUT4 occurs through both clathrin-coated [70, 72, 197] and non-clathrin vesicles [22]. Although most of the insulin-stimulated translocation of GLUT4 occurs through a stimulation of exocytosis, hypoxia and cell-energy depletion (through mitochondrial electron transport inhibition) lead mainly to reductions in GLUT4 endocytosis. This is evident from experiments in which metformin, hypoxia, oligomycin and DNP treatments have been studied [4, 165, 229, 230]. Exercise and insulin action impinge on GLUT4 traffic through different mechanisms [71, 173]. These effects may be partly related to changes in the AMP/ATP level and activation of AMPK [4, 113, 121, 125]. Stimulation of the levels of cell surface GLUT4 through inhibition of endocytosis is necessarily much slower than that mediated by stimulation of exocytosis [93, 113]. Many of the steps involved in synthesis, degradation and post-translational modification of GLUT4 might also be expected to be slow processes. However, GLUT4 endocytosis at the plasma membrane, and steps that lead to GLUT4 degradation through pre-lysosomal compartments, may be drug targets for treatment of type 2 diabetes as they may bypass defects in insulin signalling to GLUT4 exocytosis. Long-term regulation and dysregulation of these processes may occur. Exercise and training lead to an upregulation of GLUT4 in skeletal muscle [90], while in severe type 2 diabetes GLUT4, and the ubiquitin conjugating enzyme UBC9 are both lost in both adipose tissue and muscle [108], but the importance of these correlations is unclear.
Homozygous knockout of GLUT4 in mice is not lethal, while heterozygous GLUT4 knockout mice develop diabetic histo-pathologies but are lean [114, 147]. Transgenic mice in which GLUT4 is over-expressed in muscle have improved glucose disposal [218].
Class 2: GLUTs 5, 7, 9 and 11
One of the distinguishing features of class 2 GLUTs is the occurrence of the exofacial glycation site on the arginine in exofacial loop 5 (the linker between TM9 and TM10), with no glycation site on loop1 as in class 1 and class 3 GLUTs. There seems to be preference for fructose rather than glucose as substrate. There are alternative non-hexose substrates for some class 2 transporters.
GLUT5
GLUT5 was cloned and sequenced from the small intestine [116]. Levels of GLUT5 are regulated in response to changes in the levels of fructose in the intestine [47, 100]. GLUT5 is a fructose transporter with higher affinity for fructose than glucose [29, 207] and is abundant in the small intestine, kidney and sperm with lower levels in fat and skeletal muscle [62].
In adipose and muscle, GLUT5-mediated fructose transport is responsive to insulin stimulation [98]. GLUT5 is upregulated in the skeletal muscle and intestine in type 2 diabetes patients and reversed by treatment with anti-diabetes drugs [198]. Recently there has been a major advance in the study of GLUT5 with the determination of the 3D crystal structures of GLUT5 by the Drew group [158].
GLUT7
GLUT7 was found at high levels in the small intestine, colon, testis and prostate [131]. The GLUT7 substrate remains obscure and controversial. Manolescu et al. [140, 141] studied GLUT7 in Xenopus oocytes and reported that GLUT7 was a high affinity and low capacity (Vmax) fructose transporter. However, more recent studies have shed some doubt on whether fructose is a substrate for GLUT7. Ebert et al. [65, 66] have shown that in comparison with GLUT5, GLUT7 has negligible fructose transport in mammalian NIH-3T3 fibroblasts. Differences between oocyte and mammalian cell expression data may be related to a leak pathway and assay conditions that lead to trapping of fructose by a low Km fructokinase. The comparison between GLUT5 and GLUT7 is therefore useful and it has been observed that GLUT7 domains introduced into GLUT5 generate functionally inactive chimeras [65].
GLUT9
GLUT9 was identified by the Moley group [164]. It has two splice variants and is present at highest levels in the liver and kidney. The splice variations have different tissue distributions and subcellular localisations. GLUT9 is important in early development and the pre-implanted embryo [7]. GLUT9 was originally proposed to transport fructose based on studies in Xenopus oocytes [141]. Later studies, also working with the Xenopus expression system, found insignificant levels of fructose transport and instead reported that GLUT9 was a urate transporter [5, 40, 194]. Knockout of GLUT9 in mice led to hyperuricemia, hyperuricosuria and metabolic syndrome [54], consistent with a major function for this transporter in urate transport. Although urate seems to be the most important substrate, exchange of urate for glucose and fructose in the kidney has been reported suggesting that urate loss may be usefully accompanied by sugar reabsorption [37].
GLUT11
Cloning and characterisation of GLUT11 revealed that it transports both glucose and fructose [59, 179]. Its sequence has highest similarity to GLUT5, and this suggests the possibility that fructose may be the preferred substrate. GLUT11 is absent in mice and so it is likely that GLUT11 function is non-essential. It is expressed mainly in the skeletal muscle and heart. In the human skeletal muscle, GLUT11 has a fibre-type specific distribution and is abundant in slow-twitch but not fast twitch fibres [78]. Three different length transcripts occur. All of these transcripts can transport fructose but they each have somewhat different tissue distributions [181].
Class 3: GLUTs 6, 8, 10, 12 and GLUT13 (HTMI)
This class of protein has the typical 12 TM spans. The asparagine N-linked glycation to asparagine occurs in loop 1. This class of GLUTs has intracellular targeting sequences, typically dileucine motifs, within the N-terminal domains. The presence of these sequences suggests at least some intracellular targeting and compartmentalisation that may be related to function.
GLUT6
GLUT6 was identified and cloned by the Joost group [58], but initially named GLUT9. The preferred substrate for GLUT6 is unclear but it has low affinity for glucose and fructose [107]. It is present mainly in the spleen, lymphocytes and brain. GLUT6 is upregulated in inflammatory cells [35, 115, 137] and endothelial cells [212]. GLUT6 has an N-terminal dileucine group that is associated with intracellular retention [132]. It may be localised primarily to lysosomal membranes [137]. GLUT6 knockout has minimal effects on glucose metabolic physiology in mice [31].
GLUT8
GLUT8 was identified by the Joost and Mueckler groups [33, 60]. GLUT8 contains a lysosomal targeting motif and localises to late endosomes and lysosomes [8, 184, 225]. It transports glucose and fructose and is upregulated (along with GLUT12) on high-fat diets [53].
GLUT8 has recently been demonstrated to transport the disaccharide trehalose, and it has homology to the trehalose transport Tret1 in drosophila [143]. It appears to be present at low levels in many tissues but is high in testis [60], spermatozoa [83] and in lactating mammary gland alveolar cells [217]. In the latter cell type, it plays a role in lactose synthesis and accumulation [217]. It is important in the blastocyst and in these cells, it translocates to the plasma membrane in response to insulin. In the early stages of development, it may functionally act instead of GLUT4 (which is absent) [33].
As trehalose and lactose are GLUT8 substrates, it would be of interest to explore the range of disaccharides that GLUT8 transports and further explore whether its subcellular translocation is responsive to hormonal stimulation. GLUT8 knockout mice have a mild phenotype with evidence for reduced sperm motility and some abnormal neuronal and locomotor activities [183].
GLUT10
GLUT10 is somewhat larger than most GLUTs with 541 amino acids. It occurs at high levels in the liver and endocrine pancreas [51, 146]. It is also present in vascular smooth muscle cells where it has mainly an intracellular distribution [166]. Human GLUT10 mutations are associated with tortuosity and stenosis of large arteries leading to a propensity for aneurysms [24, 46]. In artery cells, there is intracellular targeting of GLUT10 to mitochondria, where it functions in redox control through the accumulation of dehydro-ascorbate [24]. Consistent with a role of GLUT10 in compartmentalisation of ascorbate within arterial cells, mice with a GLUT10 G128E mutation exhibit increased levels of reactive oxygen species, fragmented mitochondria and cross-linking of proteins at the endoplasmic reticulum [203].
GLUT12
GLUT12 is another large protein of 596 amino acids. It is mainly present in insulin-responsive, adipose, heart and skeletal muscle cells and mammary gland alveolar cells [136, 199]. In unstimulated cells, it localises to intracellular vesicle associated with the Golgi and at the plasma membrane [1, 74] in a manner that is dependent on its dileucine targeting motif. It is recruited to the plasma membrane of its target tissues in response to insulin stimulation [199]. It is important in foetal development, particularly of the heart, while GLUT12 deficiency is associated with heart failure [105]. GLUT12 expression is upregulated by androgens in the prostate and levels are increased in prostate cancers [10, 39, 224].
GLUT13
GLUT13 is a very large protein of 618 amino acids and has clear specificity for myo-inositol in a manner that is facilitated by protons. It is therefore more commonly named HMIT or H(+)-myo-inositol transporter [213]. It is mainly localised to neuronal tissues [57]. Its activity can be regulated by changes in cell membrane potential but its coupling to proton movement may not be obligatory.
Crystal structure of GLUTs
In 2014, Yan and colleagues described the crystal structure of GLUT1 with bound to nonyl-β-D-glucoside [56]. This structure has in an inward-facing conformation. The same GLUT1 structural conformation was crystallised with the GLUT inhibitor cytochalasin B bound [111]. This was followed in 2015 by the reporting of a crystal structure of GLUT3 in outward-facing conformations. Structures with GLUT3 bound to glucose or to the disaccharide maltose were obtained [55].
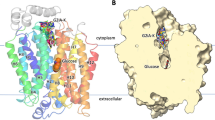
The structure of GLUT1 with nonyl-β-D-glucoside bound to the central substrate site revealed a cleft which is open to the inside solution of the cell, with the cleft to the outside clearly occluded and inaccessible (Fig. 2 OPI-S). By contrast, the structure of GLUT3 with exofacially bound maltose has a cleft only open to the outside solution. The leading glucose moiety occupies a deep central region of the cleft while the trailing glucose moiety of the disaccharide (the non-reducing end) lies nearer the surface of the cleft but is still well within the cleft (Fig. 2 OPO-S). In a separate crystal structure with bound glucose in GLUT3 (pdb:4zw9), the sugar occupies the same central location and orientation as that which occurs in the maltose and nonyl-β-D-glucoside structures. In each case, the C4/C6-OH of the glucose trails behind the leading C1-OH. The central position of the bound glucose moiety remains essentially the same in all three structures while the protein changes shape around the ligand in the OPO-S to OPI-S transition.

Crystal structures of GLUT proteins define the structural basis for alternate exposure of binding site clefts to either outside or inside solutions. Discrete conformations for the GLUTs are identified as OPO (open-outside without substrate; rat GLUT5, pdb:4ybq), OPO-S (open-outside with substrate; human GLUT3 with maltose, pdb:4zwc), OPI-S (open-inside with substrate; human GLUT1 with nonyl-glucoside, 4pyp) and OPI (open-inside without substrate; bovine GLUT5, pdb:4yb9). The structural fold has four inverted trimer repeats with TM1-3 and TM7-9 showing inverted repeat similarity to TM4-6 and TM10-12, respectively. The first TM helix of each of the four trimers is shown as cartoon representation: TM1 (blue); TM4 (green); TM7 (yellow); TM10 (orange), while the rest of the protein is shown as transparent ribbon. The upper half of TM7 (TM7b) and the lower half of TM10 (TM10b) are particularly important for occluding the binding site from the external and internal solutions, respectively. The occlusion OPO to OPI is associated with hydrophobic residues in TM7b moving closer to TM1 while hydrophobic residues in TM10b move away from TM4. The reverse occurs in the OPI to OPO conformational changes. TM7b hydrophobic residues 291, 292, 293 and 294 and TM10b residues 386 and 387 in GLUT1 (with equivalent residues in GLUT3 and GLUT5) are shown with space filling to illustrate this occlusion. In addition, salt bridges between residues at the cytosolic ends of the TMs are formed to bunch these TM ends, and the C-terminal ICH5 region, closer together in the OPO conformations. The location of the substrate glucose moiety that is revealed from both the GLUT3 structures with maltose (OPO-S, and GLUT1 with nonyl-glucose (OPI-S) is the same as in the GLUT3 structure with glucose (not shown, pdb:4zw9). In all cases, the glucose in the central site is polarised with C1-O projecting toward the internal solution with C4-O trailing. The structures shown are constructed using the VMD software and using the pdb files reported and described by the Yan group ([55, 56] for GLUT1, 3) and the Drew group ([158] for GLUT5)
Crystal structures of GLUT5 have been obtained by the Drew group [158]. GLUT5 has been crystallised (without the substrate fructose present) but in both outward open (rat GLUT5, OPO) and inward open (bovine GLUT5, OPI) conformations. All 4 structures, OPO; open-outside: OPI; open-outside with substrate bound: OPO-S; open-inside with substrate bound: OPI-S, show the same basic protein structural folds. Having all 4 structures strongly supports the alternating access hypothesis for transport in which there are cyclic transitions between the 4 conformations OPO→ OPO-S→ OPI-S→ OPI and back to OPO. The mechanism of alternate binding-cleft opening based on descriptions of the GLUT1, GLUT3 and GLUT5 structures [55, 56, 158, 200] and homologous bacterial glucose transporters [200] are remarkably consistent. There is a pseudo 2-fold axis of symmetry such that the locations of helices in the C- and N-terminal halves are superimposable. In addition, within each half of the proteins (6 helices), there is trimer symmetry such that trimer TM1,2,3 has inverted repeat sequence similarities to trimer TM4,5,6 while trimer TM7,8,9 has inverted repeat sequence similarities to trimer TM10,11,12. This enables 4 trimer substructures to move relative to each other. The OPO to OPI transitions are partly due to rigid-body rotation and tilt of the helices in the four trimer sections of the protein. The N-terminal half may act as a platform for the conformational changes and hydrogen bonding interactions with substrate that occur in the C-terminal half [55, 158].
The outward-facing conformations are stabilised by salt bridge formation between conserved glutamates and arginines of endofacial loop sequences RXGRR that occurs between TM3-TM4 and TM10-TM11. It is these stabilising motifs that are typical of the whole of the SLC family of transporters extending beyond the SLC2 family of glucose transporters. Previous mutagenesis studies highlighted the importance of these interactions in stabilising the outward-facing conformation. Mutation of conserved glutamates and arginines leads to a locking of GLUT4 into the inward-facing conformation that could not bind exofacial ligand Bio-LC-ATB-BMPA but could bind the endofacial ligand cytochalasin B [186]. Furthermore, a GLUT1 mutation in conserved glutamate 329 (E329Q) facilitated crystallisation of GLUT1 in the inward-facing formation [56]. In the inward-facing conformations, salt bridges between these helical segments are lost and instead there is salt bridging to the cytoplasmic loop domains between TM6 and TM7 (ICH1-4) and the C-terminus ICH5. The C-terminal segment ICH5 moves to open an ICH network of interactions, and this may be associated with more access to the binding site from the internal side (Fig. 2).
In addition to the rigid body movement, the crystallographic studies on GLUT1, GLUT3,GLUT5 and the bacterial homologue XylE now highlight the importance of localised movement of helical segments and side chains within the C-terminal half of the protein, and particularly with movement of the top half of TM7 (TM7b) and its inverted repeat of TM10 (TM10b) [55, 158]. These half-helices occur at hinge points that contain multiple glycine residues [204]. Thus, TM7 and TM10 both have broken helical shapes so that when they move, the substrate-binding cavity opens to either the outside or the inside. The TM7B sequence G286INAVFYYST295 is highly important in sugar binding and transport as revealed both in mutagenesis studies [61, 96, 160, 220]. Furthermore, a threonine 295 mutation to methionine has been found to be a human GLUT1 haploinsufficiency disease [222]. In particular, the sequence FYYST is of interest as mutations in these residues produce a stable outward-facing conformation that binds the exofacial reagent Bio-LC-ATB-BMPA but not the endofacial reagent cytochalasin B and very low glucose transport activity. Therefore, it appears that the entire TM7B sequence may have a role in opening and closing access (occluding) at the exofacial side of the GLUT transporters. Comparisons of crystal structures of GLUT1, GLUT3 and GLUT5, and illustrated in Fig. 2, reveal that movements of hydrophobic side chain groups of tyrosine, tryptophan and phenylalanine residues are involved in shutting off the glucose cleft from the external solution (involving TM7b residues Phe291, Tyr292 and Tyr293 in GLUT1 and the equivalent Tyr, Tyr, Tyr in GLUT5). Mutagenesis studies highlighted the importance of these hydrophobic regions in TM7b [61, 96, 150, 160, 220]. The same conformational change occurs both with and without substrate (Fig. 2). Residues in TM7b close against residues in TM1 to occlude the outside site while residues in TM10b move away from TM4 to open the inside site. When the reverse occurs, with TM7b moving away from TM1 and TM10b moving toward TM4, then the outside site opens and the internal site closes (Fig. 2).
The availability of crystal structures for the GLUTs is a tremendous advance and will pave the way for more studies that reveal the dynamics of the transport catalysis process and provide information that will be useful in the design of GLUT-specific inhibitors. The crystallography data strongly support the alternating access model for transport that was proposed for the phosphate exchanger by Mitchell [148] and for the Na/K ATPase by Jardetzky [102]. Experimental evidence for the applicability of this hypothesis to GLUT proteins was first obtained from analogue probes specific for either OPO-S or OPI-S conformations [14, 94] (section: GLUT specificity for substrates and inhibitors).
Inferred transport catalysis mechanism
The alternative access mechanism is distinct from a mechanism in which there is free movement of glucose through an ungated membrane pore or channel. The distinguishing characteristics of glucose transport that must be accounted for mechanistically are the related phenomena of substrate exchange and counterflow [196]. Counterflow can best be described by considering two substrates (although it is often measured by following the fluxes of radioactive tracers of a single substrate). When substrate A in the external solution is exchanged with substrate B in the internal solution, then it can be temporarily driven up its concentration gradient (so that A-inside > A-outside) while B is moving down its concentration gradient. Experimentally, counterflow has proven to be very useful in assays as the transport signal from A is enhanced [55]. Counterflow stops only when both A and B are at equilibrium concentrations. A simple pore model cannot account for this phenomenon. B moving down its concentration gradient through a simple pore might be expected to inhibit the flux of A, but this is not observed. The GLUTs are not trans-inhibited by substrate on the opposing side of the membrane [81]. For GLUT1 in erythrocytes, trans-acceleration is observed such that B increases the initial rate of transport of A. The alternate access model accounts for exchange and counterflow as the single central site is not exposed to both external and internal solutions at the same time, and therefore A and B do not compete. It can also account for accelerated exchange. If the conformational change OPI-S→ OPO-S is faster than OPI→ OPO (Fig. 2), then exchange flux can be faster than net influx which would be rate limited only by OPI→ OPO. The GLUT1 crystallography data indicate a very restrictive and narrow choke point within the transporter (around Trp288), Fig. 2 in OPI-S. This provides strong evidence that sugars A and B, initially from opposite sides of the membrane, would not be able to move past each other at this point. In addition, sugar analogue studies (section: GLUT specificity for substrates and inhibitors) suggest that the sugar is tightly polarised in its orientation within this central region. A comparative molecular dynamic study on the plant SWEET and bacterial semi-SWEET glucose transporters has suggested a sliding movement of glucose (with restricted orientation) in SWEET transporters while semi-SWEET transporters have a tumbling movement (with a less restricted orientation of glucose). The sliding movement was associated with lower free energy and faster transport, possibly associated with less steric clashes with the protein during the transport [41].
More complex multiple-occupancy pore models for glucose transport have been proposed in which sugars A and B move past each other within the protein and do not impede each other’s progress [48]. Currently, it is not clear whether the central polarisation of substrate in the GLUT crystal structures is maintained in the wide clefts leading to the central binding site, but less restricted orientation within the wider regions of the cleft is likely. The crystallography data indicate that the cavities leading to the binding site are certainly large enough at the exofacial site to accommodate the trailing glucose of maltose and bis-hexoses (see Fig. 2 OPO-S and Fig. 3 e and f). Cis-allostery in which multiple occupancy by substrate and/or inhibitor occurs at the exofacial site of GLUT1 has been demonstrated [159]. However, cis-allostery is consistent with the alternating access mechanism [133]. Trans-allostery (with the possibility that substrates and inhibitors from opposite sides of the membrane bind simultaneously and bypass one another within the protein) is a much more controversial issue [134, 154, 156, 157]. Trans-allostery would be consistent with the alternating access mechanism if the simultaneous trans-occupancy occurred within a dimer of two alternating-access monomers that were conformationally linked. One monomer could be in the OPO-S while the other was in the OPI-S conformation [52, 134]. Although much of the literature suggests that GLUTs behaves as monomers in most situations [154], it is possible that when GLUT1 levels are very high within cells (such as human erythrocytes), then dimerization and associated allosteric-like effects are more likely.

Substrates and inhibitors reveal differing specificity requirements for the GLUTs and may aid the therapeutic targeting of individual GLUTs that are implicated in disease. In a substrate and inhibitor, interactions with the GLUTs are associated with H-bonding (involving either electron donating or withdrawing groups) that can be examined using analogues that are H-bond accepting only (fluorine substitution for –OH). Spatial limitations to binding can be explored using O-alkyl groups. In b, β-methyl-D-glucoside has very low affinity for the outside site of GLUT1 suggesting a close approach to C1-O, while a 4-O-propyl group (c) is well tolerated at the outside site. The outside site can accommodate quite bulky substitutions at C4-OH with disaccharides such as maltose (e and Fig. 2 in OPO-S), and bis-glucose propylamine BGPA (f) derivatives being well tolerated. In f, the R group on the phenyl-diazirine photoreactive moiety can be a very large spacer arm with biotin. In contrast to these spatial restraints at the outside site, C1-O substitutions as in β-O-propyl-glucoside (d) or β-O-nonyl-glucoside (Fig. 2 in OPI-S) are well tolerated at the inward-facing site. Both fructofuranose and fructopyranose forms of fructose are transported by GLUTs such as GLUT5. The closed ring forms, including 2-5-anhydro-D-mannitol (h) and the β-methyl-fructofuranosides and β-methyl-fructopyranosides (j and k), are good substrates and inhibitors. Several new derivatives, including fluorescent and photolabeling compounds, based on 2-5-anhydro-D-mannitol have been described. The introduction of an H-bond accepting fluoro group at C3 of 2-5-anhydro-D-mannitol (i) reduces affinity for GLUT5 but increases affinity for GLUT1 suggesting tuning analogues for a specific GLUT is possible. The GLUT family is not restricted to glucose or fructose as substrate. GLUT9 is a transporter for urate (l). Dehydro-ascorbate in solution as a hydrate (m) is transported well by several GLUTs, and particularly GLUT10. GLUT13 transports myo-inositol (n) with good specificity. Now that GLUT structures are available, therapeutic targeting of individual GLUTs is becoming possible. In silico docking-aided screening of compound libraries has led to the identification of Bay 876 (o) as a high affinity inhibitor of GLUT1 (and not other class 1 GLUTs) and MSNBA (p) as a specific inhibitor of GLUT5 with negligible affinity for class 1 GLUTs
GLUT specificity for substrates, inhibitors and drug targets
Defining the interactions of GLUTs with substrates is often useful in attributing the function of the GLUT. Most GLUTs exhibit some level of glucose or hexose transport. However, it is now clear that although they have the same structural folds, which are associated with similar modes of transport catalysis mechanism, often quite subtle differences in amino acid side chains can radically alter substrate preference and interaction. Likewise, subtle differences in substrate analogues can alter which of the GLUTs transports the analogue most efficiently. Considerations of GLUT specificity are therefore ongoing and much needs to be done in terms of defining both the physiological substrates and the related functions of some of the class 2 and class 3 transporters.
GLUT specificity for substrates and inhibitors
The specificity and transport kinetic features of the GLUT proteins for hexose analogues and transport inhibitors have been most extensively studied for class 1 (GLUTs1-4) and for GLUT5. Such studies are important for defining the key features of the substrate that are necessary for efficient transport, for defining differences between each of the GLUT proteins and ultimately for defining routes to the development of high affinity transport inhibitors
Sugar analogues including deoxy sugars and epimers have been useful in defining these substrate specificities in GLUTs1-4 [14, 43, 170]. The hydroxyls at C1, C3, C4 and C6 and the ring oxygen are all important for hexose binding to GLUT1. The C4 hydroxyl seems to be more important for binding to GLUT1 where galactose has 10-fold lower affinity than glucose. By contrast, GLUT4 and GLUT3 have similar affinity for glucose and galactose [43, 170]. The C2 hydroxy seems to be unimportant for transport as 2-deoxy-glucose is transported well by all class 1 GLUTs. The direction of H-bonding from glucose hydroxyls to both GLUT1, GLUT3 and GLUT4 transporters has been examined using fluorine substitution. If the bond is from the protein sidechain to the electronegative O of the –OH, then the electronegative F will substitute. If the H-bond involves this H of the –OH, then it will not. Fluorine substituted D-glucoses indicate H-bonding to the C1-β-O of glucose and to the C3-O, C4-OH, C6-O positions [14], Fig. 3a.
Many GLUT protein residues interact with glucose in the deep binding cleft identified by the GLUT1 and GLUT3 crystallography studies [55, 56]. These include polar residues Gln282, Gln283, Asn288, Asn317 and Glu380 in the C-terminal half of the protein (GLUT1 numbering). Only Gln161 in the N-terminal half of the protein contributes to the H-bonding network. However, these H-bonding interactions are not all essential as single amino acid mutations do not always lead to a large loss of function [88, 96]. This probably occurs in the single point mutation variants because there is sufficient H-bonding to the substrate from other residues that surround the glucose, and/or assist its passage to and from the central binding site.
Glucose has α- and β-glucopyranose anomers in solution (in the ratio 66% β, and 33% α) and vanishing small amounts of glucofuranose forms. Studies on C1-fluoro glucoses (which do not interconvert) suggest that the β anomer binds more strongly than the α isomer [14] and is transported more rapidly by GLUT1 [135]. However, additional experiments in which glucose anomer mixtures are studied [130, 189] suggest that the α anomer is preferred. These experiments are complicated by rapid interconversion of α and β glucose anomers and by hetero exchanges (αβ and βα, exchanges). The latter dominates the transport kinetics and particularly the maximum rate of transport, which is the sum of the maximum exchange rates for α + β, no matter which anomer is being followed [170]. However, the rate of transport at low substrate concentrations (or the ratio of apparent Km over apparent Vmax) is clearly faster for the α anomer [189]. Furthermore, in the crystal structure of GLUT3, both α and β glucose are present but α glucose is more abundant [55].
Studies on alkyl substitutions on the glucose hydroxyls have been informative and have provided experimental evidence for an alternating cleft hypothesis for the conformational changes that alter binding specificity at external and internal sites (section: Crystal structure of GLUTs). C1-substituted β-O-methyl glucose is not an inhibitor of substrate transport when added to the outside of the erythrocyte GLUT1 or adipocyte GLUT4. This suggests that the relatively small methyl group is not tolerated at the exofacial site of GLUT1 and GLUT4 and that there is a close approach between the C1-OH position of glucose and the protein cleft. By contrast, there is good tolerance of bulky substitutions around C4 and C6 at the external site of GLUT1; for example, 4-O-propyl-D-glucose, 6-O-propyl-D-galactose, 4,6-O-ethylidene-D-glucose and maltose bind well from outside the cell. However, these analogues are not transported by the GLUTs suggesting that bulk around C4-O/C6-O prevents a conformational change associated with transport. Hydrophobic substitutions at C6-O lead to increased binding of the inhibitors which is consistent with interaction of the substituted groups with hydrophobic amino acid side chains in the binding cleft [14, 151].
Binding constraints are different at the internal side of the GLUTs. C1-O-alkyl glucose compounds do not enter the cell through GLUT1 or GLUT4. However, a long chain glucoside (propyl-β-D-glucoside) (Fig. 3d) enters through the membrane lipid and is a good inhibitor from the inside site of both these transporters. The studies with alkyl substitutions at either C1-O or C4-O thus reveal a polarised binding of glucose at the centre of the protein with the substrate binding site alternately opening to either the inside or the outside solutions [13] such that these sites accommodated either substitutions at C-4-O in OPO-S or C-1-O in OPI-S. These specificity factors are now evident in the crystal structures with the C4-OH substituted maltose binding well in OPO-S and nonyl-glucoside binding well in OPI-S (Fig. 2).
Bis-hexose analogues with bulky substitutions into the C4-OH position have also been useful for development of photoaffinity labels that cross-link to the exofacial site of the GLUT proteins (Fig. 3f). The bis-hexose structure with hydrophobic substitutions in the trailing C4-OH position has affinities for the GLUTs that are 10-fold higher than the parent compound D-glucose. The bis-mannose and bis-glucose photolabels have been used extensively for tracking the kinetics of GLUT movement to and from the plasma membrane in response to insulin (reviewed in [91]).
Fructose adopts α and β forms of fructopyranose and fructofuranose in solution (αP:βP:αF:βF; < 1%:75%:4%:21%). GLUT5 mainly interacts with β forms since both ring-closed furanose and pyranose derivatives are good inhibitors of transport with Ki values comparable with the Km of D-fructose (Fig. 3 j and k). C2-β-O-methyl D-frucofuranoside and C2-β-O-methyl-D-fructopyranoside both have approx. 5-fold higher affinity than the corresponding C2-α-O-methyl compounds [207]. 2,5-Anhydro-D-mannitol (2-deoxy-D-fructofuranose, Fig. 3h) is a particularly useful GLUT5 analogue that retains a furanose ring shape, as it cannot ring open and therefore cannot form anomers. Fructose transporter photolabels, based on the 2,5-anhydro-D-mannitol core structure, interact well with GLUT5 [228]. Fluorescent GLUT5 probes based on the 2,5 anhydro-mannitol ring have also recently been designed [17, 126]. The presence of a hydrogen bond donor at the C-3 position of 2,5-anhydro-D-mannitol derivatives is essential for effective binding to GLUT5 and fructose transport into tumour cells. Interestingly, replacement of the C3-OH with a fluorine group (Fig. 3i), that can function only as a hydrogen bond acceptor, resulted in selective recognition by GLUT1 rather than GLUT5 [126].
The crystal structure of GLUT5 has been obtained without bound substrate but inferences have been drawn concerning binding interactions with D-fructose. An important feature of GLUT5 structure that is distinct from the structure of class 1 GLUTs occurs in the binding cleft and at residue Trp388 (GLUT1 numbering). This tryptophan is absent in GLUT5 which instead has alanine at the equivalent position. This difference makes the GLUT5-binding cavity less sterically restrictive (Fig. 2 OPO and OPI). The lack of Trp388 also accounts for the lack of inhibition of fructose transport by cytochalasin B, a powerful inhibitor of the class 1 GLUTs which is known to interact with Trp388 [158]. A replacement of the GLUT5 Ala for Trp leads to reduced fructose transport and some gain of glucose transport [79]. GLUT5 and GLUT7 have many sequence similarities. However, GLUT5 is a much better transporter for fructose than GLUT7. Nomura et al. [158] identified that this difference could be accounted for by the absence of Gln166 (GLUT5) which is Glu in GLUT7. They found that a GLUT5 Q166E mutation led to much reduced fructose binding but improved glucose binding. Fructose transport activity of this mutant has also been reported to be impaired, but evidence for improved glucose transport was not obtained [65].
DHA is transported by many GLUTs but detailed comparison of the relative transport rates through each of the proteins in the 3 classes of GLUTs is lacking. However, it has been reported that GLUT1, 3 and 4 are DHA transporters but GLUT5 and GLUT2 are not [174, 216]. Whether this affinity for DHA is related to the ability to transport glucose, not fructose remains to be investigated. The function of DHA transport may lie in maintaining redox levels in cells (for example in human erythrocytes with high GLUT1 levels) or in subcellular compartments [178]. GLUT10 deficiency is associated with blood vessel damage [24, 203], while a deficiency in GLUT14 transport of DHA is associated with inflammatory bowel disease [3]. A more complete study of all the GLUTs that transport DHA is necessary before ascorbate is considered the main substrate for any of the GLUT proteins. In addition, it is necessary to establish whether circulating glucose levels competitively compete with DHA uptake. Other proteins of the SLC family including the ion-coupled transporters seem to be primarily associated with ascorbate transport [30]. Regardless of the unknown physiological significance of DHA transport by the GLUT proteins, it is of interest to discover whether the planar structure of this molecule (Fig. 3m) allows less steric constraint to transport than that which occurs during the transport of glucose and fructose. The transport of urate must in some way be determined by its pharmacophore of neighbouring steric and electronic features (Fig. 3l) but relating these features to the structure of hexoses that share the same transport protein is challenging. However, myo-inositol (Fig 3n) is structurally similar to glucose, and the extent of GLUT13 evolutionary adaptation of ancestral SLC2 genes for specialised transport of this polyol would probably have been minimal.
GLUT proteins as potential drug targets
With the availability of the new crystallographic data on the GLUT family of proteins, it is likely that new reagents and substrate analogues can be developed that are specific for individual GLUTs. It has long been clear that pharmaceutical control of glucose transport is desirable, particularly in the cancer biology field [190, 195, 205]. However, as glucose uptake is so essential to all cells of the body, a generalised knockdown of GLUT activity is unlikely to be useful. In addition, preventing glucose entering cells may in some cases be desirable but could potentially lead to hyperglycaemia which is clearly undesirable. There are now increasing numbers of chemical biology studies in the literature that use chemical library screening to identify GLUT inhibitors [205]. These are often based on cell screens used in conjunction with in silico screens that involve the docking of potential inhibitors on computer models of known crystal structures of the GLUTs
A GLUT1-specific inhibitor might be useful as this isoform is known to be upregulated in many cancers. GLUT1-mediated glucose transport is associated with the reliance of cancer cells on glycolytic metabolism, the Warburg effect [2, 223]. One of the many typical screens has been described by Siebeneicher et al. [190]. This group initially screened 3 million compounds and then took the lead compound and further chemically modified it to end up with the compound Bay 876 that has low nM affinity for GLUT1, but much less affinity for the other GLUTs tested. The Siebeneicher group refined their GLUT inhibitors by obtaining a crystal structure of GLUT1 with bound cytochalasin B. A range of highly modified peptide-like compounds (Glut-i1 and Glut-i2) that bound to the same inward-facing site as cytochalasin B were studied in detailed crystal structures with bound ligands [111]. Of course, there is no information yet on the range of off-target interactions that these screening-derived compounds might have. However, there seems to be real potential for GLUT-specific targeting and possibly GLUT-inhibitor-based therapies.
GLUT5 is known to be upregulated in cancer of some tissues [126, 205, 231] and fructose (derived from all the sucrose we consume) probably does more harm than good in the body as it can contribute toward non-alcoholic fatty liver disease [63]. In addition, fructose is more rapidly converted to advanced glycation end products than glucose, but the significance of this fructation is debated [86]. As described above, fructopyranose and fructofuranose analogues that are specific for fructose transporters have been synthesised. The potential for targeting the unique specificity of GLUT5 is therefore good [80, 126, 195], particularly now that crystal structures of GLUT5 are available [158]. Detailed modelling is required to design fructose analogues that are transported only by GLUT5 and not by GLUT2 (which has significant affinity for fructose [21, 44, 80]). The compound MSNBA (Fig 3p) was obtained from library screening and in silico docking and modelling [80]. It is a specific inhibitor for GLUT5 with negligible affinity for class 1 GLUTs. Importantly it has no affinity for GLUT2. Docking studies have suggested that the H-bond acceptor nitro moiety binds strongly to protonated His387, which is unique to GLUT5 while other GLUTs have a Phe group at the equivalent position.
GLUT4-mediated glucose transport into the major tissues of fat, muscle and heart is of vital importance and reductions in signalling to this process lead to insulin resistance. Transgenic mouse models of diabetes in which GLUT4 expression is increased in muscle have improvements in whole body glucose homeostasis [218]. Furthermore maintenance of normal blood glucose levels through insulin therapy is recognised as being important in any hospitalised patients undergoing intensive critical care [214]. Clearly insulin resistance in mammals is undesirable and inhibition of this major protein responsible for glucose disposal would potentially lead to transient hyperglycaemia. By contrast, in lower organisms, one of the consequences of reducing insulin signalling to glucose transport and metabolism is to extend lifespan, suggesting that there may be some potential health benefits of limiting excess glucose transport and metabolism [19, 73, 97, 129, 175, 185]. Some of these beneficial effects are driven by reductions in whole organism calorie intake. Of course, comparisons with simple organism lifespan will over-simplify of the role of glucose transport in human physiology. However, specific targeting of GLUT4 could be explored in animals with a view to it providing better management and rebalancing of glucose homeostasis in conditions where cellular uptake of glucose is excessive.
Conclusions and perspectives
In recent years, our understanding of GLUT proteins has been greatly enhanced by phylogeny and by the availability of crystal structures of GLUTs in a range of conformations. The crystal structures support the central tenant of the alternating access mode of transport. While this model is consistent with most available data on the kinetics of glucose transport, there remains evidence for a slightly more complex model with the possibility of additional (perhaps rare) states of the protein in which allosteric interactions occur. Recent papers provide a useful discussion of these additional stochastic processes in alternative access modes of membrane transport [85, 99].
The GLUTs1-4, GLUT5 and GLUT13 are well-characterised transporters with often quite clear and separate functions related to their kinetic, specificity and cellular distributions. This group of 6 transporters is already the subject of approaches that aim to generate tools and reagents that can specifically target the individual isoforms, with the goal of generating therapeutics. However, the other 8 GLUTs that make up the family are relatively uncharacterised and importantly, the substrate specificity is in many cases unknown (or in the case of GLUT7 uncertain). Crystal structures of these GLUTs are currently not available but in silico modelling of these GLUT structures based on the GLUT1, 3 and 5 structures will now be possible and may lead to some inferences regarding their likely substrates. Further characterisation of the substrate specificities and clarification of the subcellular distributions of these 8 isoforms will aid in attributing function.
A quantitative approach to defining absolute levels of protein abundance in different tissues is still disappointingly lacking for many GLUTs, and often mRNA levels alone are reported. Using some of the newer protein detection techniques, such as fluorescently tagged protein constructs, it will also be important to establish the extent of regulation of the more recently discovered GLUTs, particularly those of the class 2 and 3. Following methods utilised to study class 1 GLUTs, it would be of interest to define whether there is hormonal and energy-level control of translocation between subcellular compartments. In addition, examination of longer-term changes—including post-translocation palmitoylation, ubiquitinoylation and phosphorylation of the GLUTs and associated proteins, and changes in turnover (transcription and degradation)—will reveal whether these GLUTs are necessary for organism viability and health. In future, with wider human genome sequencing, inherited mutations in these transporters may emerge. Such studies will aid in ascribing function to the class 2 and 3 GLUTs, and thereby increase understanding of any related disease syndromes.
Data availability
Not applicable.
Abbreviations
- GGAP2:
-
Golgi associated, gamma adaptin ear containing, ARF-binding protein 2
- VAMP:
-
Vesicle-associated membrane protein
- IRAP:
-
Insulin-responsive aminopeptidase
- UBC9:
-
Polyubiquitin-C precursor 9
- USP25:
-
Ubiquitin carboxyl-terminal hydrolase 25
- TM:
-
Transmembrane segment
- p115:
-
General vesicular transport factor p115
- MSNBA:
-
N-[4-(methylsulfonyl)-2-nitrophenyl]-1,3-benzodioxol-5-amine
- TBC:
-
Tre-2/Bub2/Cdc16
- CHC:
-
Clathrin heavy chain
- ERGIC:
-
ER-Golgi intermediate compartment
- TGN:
-
Trans-Golgi network
- MVB:
-
Multivesicular body
References
Aerni-Flessner LB, Otu MC, Moley KH (2011) The amino acids upstream of NH(2)-terminal dileucine motif play a role in regulating the intracellular sorting of the Class III transporters GLUT8 and GLUT12. Mol Membr Biol 28:30–41. https://doi.org/10.3109/09687688.2010.508196
Airley RE, Mobasheri A (2007) Hypoxic regulation of glucose transport, anaerobic metabolism and angiogenesis in cancer: novel pathways and targets for anticancer therapeutics. Chemotherapy 53:233–256
Amir Shaghaghi M, Zhouyao H, Tu H, El-Gabalawy H, Crow GH, Levine M, Bernstein CN et al (2017) The SLC2A14 gene, encoding the novel glucose/dehydroascorbate transporter GLUT14, is associated with inflammatory bowel disease. Am J Clin Nutr 106:1508–1513. https://doi.org/10.3945/ajcn.116.147603
Antonescu CN, Diaz M, Femia G, Planas JV, Klip A (2008) Clathrin and non-clathrin mediated Glut4 endocytosis in myocytes: effect of mitochondrial uncoupling. Traffic 9:1173–1190
Anzai N, Ichida K, Jutabha P, Kimura T, Babu E, Jin CJ, Srivastava S et al (2008) Plasma urate level is directly regulated by a voltage-driven urate efflux transporter URATv1 (SLC2A9) in humans. J Biol Chem 283:26834–26838. https://doi.org/10.1074/jbc.C800156200
Aran V, Bryant NJ, Gould GW (2011) Tyrosine phosphorylation of Munc18c on residue 521 abrogates binding to Syntaxin 4. BMCBiochem 12:19
Augustin R, Carayannopoulos MO, Dowd LO, Phay JE, Moley JF, Moley KH (2004) Identification and characterization of human glucose transporter-like protein-9 (GLUT9): alternative splicing alters trafficking. J Biol Chem 279:16229–16236. https://doi.org/10.1074/jbc.M312226200
Augustin R, Riley J, Moley KH (2005) GLUT8 Contains a [DE]XXXL[LI] Sorting motif and localizes to a late endosomal/lysosomal compartment. Traffic 6:1196–1212
Bai L, Wang Y, Fan J, Chen Y, Ji W, Qu A, Xu P et al (2007) Dissecting multiple steps of GLUT4 trafficking and identifying the sites of insulin action. Cell Metab 5:47–57. https://doi.org/10.1016/j.cmet.2006.11.013
Bakht MK, Lovnicki JM, Tubman J, Stringer KF, Chiaramonte J, Reynolds MR, Derecichei I et al (2019) Differential expression of glucose transporters and hexokinases in prostate cancer with a neuroendocrine gene signature: a mechanistic perspective for FDG imaging of PSMA-suppressed tumors. J Nucl Med. https://doi.org/10.2967/jnumed.119.231068
Baldwin SA (1993) Mammalian passive glucose transporters: members of an ubiquitous family of active and passive transport proteins. Biochim Biophys Acta 1154:17–49
Barnes K, Ingram JC, Porras OH, Barros LF, Hudson ER, Fryer LG, Foufelle F et al (2002) Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP-activated protein kinase (AMPK). J Cell Sci 115:2433–2442
Barnett JE, Holman GD, Munday KA (1973) An explanation of the asymmetric binding of sugars to the human erythrocyte sugar-transport systems. Biochem J 135:539–541
Barnett JEG, Holman GD, Munday KA (1973) Structural requirements for binding to the sugar transport system of the human erythrocyte. Biochem J 131:211–221
Barros LF, Barnes K, Ingram JC, Castro J, Porras OH, Baldwin SA (2001) Hyperosmotic shock induces both activation and translocation of glucose transporters in mammalian cells. Arch Eur J Physiol 442:614–621
Basketter DA, Widdas WF (1978) Asymmetry of the hexose transfer system in human erythrocytes. Comparison of the effects of cytochalasin B, phloretin and maltose as competitive inhibitors. JPhysiol 278:389–401
Begoyan VV, Weselinski LJ, Xia S, Fedie J, Kannan S, Ferrier A, Rao S et al (2018) Multicolor GLUT5-permeable fluorescent probes for fructose transport analysis. Chem Commun (Camb) 54:3855–3858. https://doi.org/10.1039/c7cc09809j
Bernhardt U, Carlotti F, Hoeben RC, Joost HG, Al Hasani H (2009) A dual role of the N-terminal FQQI motif in GLUT4 trafficking. Biol Chem 390:883–892
Biddinger SB, Kahn CR (2006) From mice to men: insights into the insulin resistance syndromes. Annu Rev Physiol 68:123–158. https://doi.org/10.1146/annurev.physiol.68.040104.124723
Birnbaum MJ (1989) Identification of a novel gene encoding an insulin-responsive glucose transporter protein. Cell 57:305–315
Blakemore SJ, Aledo JC, James J, Campbell FC, Lucocq JM, Hundal HS (1995) The GLUT5 hexose transporter is also localized to the basolateral membrane of the human jejunum. Biochem J 309(Pt 1):7–12
Blot V, McGraw TE (2006) GLUT4 is internalized by a cholesterol-dependent nystatin-sensitive mechanism inhibited by insulin. EMBO J 25:5648–5658
Blot V, McGraw TE (2008) Molecular mechanisms controlling GLUT4 intracellular retention. Mol Biol Cell
Boel A, Veszelyi K, Nemeth CE, Beyens A, Willaert A, Coucke P, Callewaert B et al (2019) Arterial tortuosity syndrome: an ascorbate compartmentalization disorder? Antioxid Redox Signal. https://doi.org/10.1089/ars.2019.7843
Bogan JS (2012) Regulation of glucose transporter translocation in health and diabetes. Annu Rev Biochem 81:507–532. https://doi.org/10.1146/annurev-biochem-060109-094246
Bogan JS, Kandror KV (2010) Biogenesis and regulation of insulin-responsive vesicles containing GLUT4. Curr Opin Cell Biol 22:506–512
Brahm J (1983) Kinetics of glucose transport in human erythrocytes. J Physiol 339:339–354. https://doi.org/10.1113/jphysiol.1983.sp014720
Bryant NJ, Govers R, James DE (2002) Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol 3:267–277
Burant CF, Takeda J, Brotlaroche E, Bell GI, Davidson NO (1992) Fructose transporter in human spermatozoa and small-intestine is Glut5. J Biol Chem 267:14523–14526
Burzle M, Hediger MA (2012) Functional and physiological role of vitamin C transporters. Curr Top Membr 70:357–375. https://doi.org/10.1016/B978-0-12-394316-3.00011-9
Byrne FL, Olzomer EM, Brink R, Hoehn KL (2018) Knockout of glucose transporter GLUT6 has minimal effects on whole body metabolic physiology in mice. Am J Physiol Endocrinol Metab 315:E286–E293. https://doi.org/10.1152/ajpendo.00082.2018
Camus SM, Camus MD, Figueras-Novoa C, Boncompain G, Sadacca LA, Esk C, Bigot A et al (2019) CHC22 clathrin mediates traffic from early secretory compartments for human GLUT4 pathway biogenesis. J Cell Biol 219. https://doi.org/10.1083/jcb.201812135
Carayannopoulos MO, Chi MM, Cui Y, Pingsterhaus JM, McKnight RA, Mueckler M, Devaskar SU et al (2000) GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc Natl Acad Sci U S A 97:7313–7318. https://doi.org/10.1073/pnas.97.13.7313
Carayannopoulos MO, Xiong F, Jensen P, Rios-Galdamez Y, Huang H, Lin S, Devaskar SU (2014) GLUT3 gene expression is critical for embryonic growth, brain development and survival. Mol Genet Metab 111:477–483. https://doi.org/10.1016/j.ymgme.2014.01.013
Caruana BT, Byrne FL, Knights AJ, Quinlan KGR, Hoehn KL (2019) Characterization of glucose transporter 6 in lipopolysaccharide-induced bone marrow-derived macrophage function. J Immunol 202:1826–1832. https://doi.org/10.4049/jimmunol.1801063
Castellotti B, Ragona F, Freri E, Solazzi R, Ciardullo S, Tricomi G, Venerando A et al (2019) Screening of SLC2A1 in a large cohort of patients suspected for Glut1 deficiency syndrome: identification of novel variants and associated phenotypes. J Neurol 266:1439–1448. https://doi.org/10.1007/s00415-019-09280-6
Caulfield MJ, Munroe PB, O’Neill D, Witkowska K, Charchar FJ, Doblado M, Evans S et al (2008) SLC2A9 is a high-capacity urate transporter in humans. PLoS Med 5:e197. https://doi.org/10.1371/journal.pmed.0050197
Cesar-Razquin A, Snijder B, Frappier-Brinton T, Isserlin R, Gyimesi G, Bai X, Reithmeier RA et al (2015) A call for systematic research on solute carriers. Cell 162:478–487. https://doi.org/10.1016/j.cell.2015.07.022
Chandler JD, Williams ED, Slavin JL, Best JD, Rogers S (2003) Expression and localization of GLUT1 and GLUT12 in prostate carcinoma. Cancer 97:2035–2042. https://doi.org/10.1002/cncr.11293
Cheeseman C (2008) GLUT7: a new intestinal facilitated hexose transporter. Am J Physiol Endocrinol Metab 295:E238–E241. https://doi.org/10.1152/ajpendo.90394.2008
Cheng KJ, Selvam B, Chen LQ, Shukla D (2019) Distinct substrate transport mechanism identified in homologous sugar transporters. J Phys Chem B 123:8411–8418. https://doi.org/10.1021/acs.jpcb.9b08257
Cloherty EK, Levine KB, Graybill C, Carruthers A (2002) Cooperative nucleotide binding to the human erythrocyte sugar transporter. Biochemistry 41:12639–12651
Colville CA, Seatter MJ, Jess TJ, Gould GW, Thomas HM (1993) Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: substrate specificities and effects of transport inhibitors. Biochem J 290(Pt 3):701–706. https://doi.org/10.1042/bj2900701
Corpe CP, Basaleh MM, Affleck J, Gould G, Jess TJ, Kellett GL (1996) The regulation of GLUT5 and GLUT2 activity in the adaptation of intestinal brush-border fructose transport in diabetes. Pflug Archiv Eur J Phys 432:192–201
Cosset E, Ilmjarv S, Dutoit V, Elliott K, von Schalscha T, Camargo MF, Reiss A et al (2017) Glut3 addiction is a druggable vulnerability for a molecularly defined subpopulation of glioblastoma. Cancer Cell 32(856-868):e855. https://doi.org/10.1016/j.ccell.2017.10.016
Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J, Fox JE et al (2006) Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat Genet 38:452–457. https://doi.org/10.1038/ng1764
Cui XL, Jiang L, Ferraris RP (2003) Regulation of rat intestinal GLUT2 mRNA abundance by luminal and systemic factors. Biochim Biophys Acta 1612:178–185. https://doi.org/10.1016/s0005-2736(03)00129-9
Cunningham P, Afzal-Ahmed I, Naftalin RJ (2006) Docking studies show that D-glucose and quercetin slide through the transporter GLUT1. J Biol Chem 281:5797–5803. https://doi.org/10.1074/jbc.M509422200
Cura AJ, Carruthers A (2012) Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Compr Physiol 2:863–914. https://doi.org/10.1002/cphy.c110024
Cushman SW, Wardzala LJ (1980) Potential mechanism of insulin action on glucose transport in the isolated rat adipose cell. Apparent translocation of intracellular transport systems to the plasma membrane. J Biol Chem 255:4758–4762
Dawson PA, Mychaleckyj JC, Fossey SC, Mihic SJ, Craddock AL, Bowden DW (2001) Sequence and functional analysis of Glut10: a glucose transporter in the type 2 diabetes-linked region of chromosome 20q12-13.1. Mol Genet Metab 74:186–199
De Zutter JK, Levine KB, Deng D, Carruthers A (2013) Sequence determinants of GLUT1 oligomerization: analysis by homology-scanning mutagenesis. J Biol Chem 288:20734–20744. https://doi.org/10.1074/jbc.M113.469023
DeBosch BJ, Chi M, Moley KH (2012) Glucose transporter 8 (GLUT8) regulates enterocyte fructose transport and global mammalian fructose utilization. Endocrinology 153:4181–4191. https://doi.org/10.1210/en.2012-1541
DeBosch BJ, Kluth O, Fujiwara H, Schurmann A, Moley K (2014) Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun 5:4642. https://doi.org/10.1038/ncomms5642
Deng D, Sun P, Yan C, Ke M, Jiang X, Xiong L, Ren W et al (2015) Molecular basis of ligand recognition and transport by glucose transporters. Nature 526:391–396. https://doi.org/10.1038/nature14655
Deng D, Xu C, Sun P, Wu J, Yan C, Hu M, Yan N (2014) Crystal structure of the human glucose transporter GLUT1. Nature 510:121–125. https://doi.org/10.1038/nature13306
Di Daniel E, Mok MH, Mead E, Mutinelli C, Zambello E, Caberlotto LL, Pell TJ et al (2009) Evaluation of expression and function of the H+/myo-inositol transporter HMIT. BMC Cell Biol 10:54. https://doi.org/10.1186/1471-2121-10-54
Doege H, Bocianski A, Joost HG, Schurmann A (2000) Activity and genomic organization of human glucose transporter 9 (GLUT9), a novel member of the family of sugar-transport facilitators predominantly expressed in brain and leucocytes. Biochem J 350:771–776
Doege H, Bocianski A, Scheepers A, Axer H, Eckel J, Joost HG, Schurmann A (2001) Characterization of human glucose transporter (GLUT) 11 (encoded by SLC2A11), a novel sugar-transport facilitator specifically expressed in heart and skeletal muscle. Biochem J 359:443–449. https://doi.org/10.1042/0264-6021:3590443
Doege H, Schurmann A, Bahrenberg G, Brauers A, Joost HG (2000) GLUT8, a novel member of the sugar transport facilitator family with glucose transport activity. J Biol Chem 275:16275–16280
Doege H, Schurmann A, Ohnimus H, Monser V, Holman GD, Joost HG (1998) Serine-294 and threonine-295 in the exofacial loop domain between helices 7 and 8 of glucose transporters (GLUT) are involved in the conformational alterations during the transport process. Biochem J 329(Pt 2):289–293
Douard V, Ferraris RP (2008) Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab 295:E227–E237. https://doi.org/10.1152/ajpendo.90245.2008
Douard V, Ferraris RP (2013) The role of fructose transporters in diseases linked to excessive fructose intake. J Physiol 591:401–414. https://doi.org/10.1113/jphysiol.2011.215731
Du K, Murakami S, Sun Y, Kilpatrick CL, Luscher B (2017) DHHC7 palmitoylates glucose transporter 4 (Glut4) and regulates Glut4 membrane translocation. J Biol Chem 292:2979–2991. https://doi.org/10.1074/jbc.M116.747139
Ebert K, Ewers M, Bisha I, Sander S, Rasputniac T, Daniel H, Antes I et al (2018) Identification of essential amino acids for glucose transporter 5 (GLUT5)-mediated fructose transport. J Biol Chem 293:2115–2124. https://doi.org/10.1074/jbc.RA117.001442
Ebert K, Ludwig M, Geillinger KE, Schoberth GC, Essenwanger J, Stolz J, Daniel H et al (2017) Reassessment of GLUT7 and GLUT9 as putative fructose and glucose transporters. J Membr Biol 250:171–182. https://doi.org/10.1007/s00232-016-9945-7
Eilam Y, Stein WD (1972) A simple resolution of the kinetic anomaly in the exchange of different sugars across the membrane of the human red blood cell. Biochim Biophys Acta 266:161–173. https://doi.org/10.1016/0005-2736(72)90132-0
Enogieru OJ, Ung PMU, Yee SW, Schlessinger A, Giacomini KM (2019) Functional and structural analysis of rare SLC2A2 variants associated with Fanconi-Bickel syndrome and metabolic traits. Hum Mutat 40:983–995. https://doi.org/10.1002/humu.23758
Eny KM, Wolever TM, Corey PN, El-Sohemy A (2010) Genetic variation in TAS1R2 (Ile191Val) is associated with consumption of sugars in overweight and obese individuals in 2 distinct populations. Am J Clin Nutr 92:1501–1510. https://doi.org/10.3945/ajcn.2010.29836
Esk C, Chen CY, Johannes L, Brodsky FM (2010) The clathrin heavy chain isoform CHC22 functions in a novel endosomal sorting step. J Cell Biol 188:131–144
Espelage L, Al-Hasani H, Chadt A (2019) RabGAPs TBC1D1 and TBC1D4 in skeletal muscle function and exercise. J Mol Endocrinol. https://doi.org/10.1530/JME-19-0143
Fazakerley DJ, Scott SP, Lizunov VA, Cushman SW, Holman GD (2009) A common trafficking route for GLUT4 translocation in cardiomyocytes following insulin, contraction and energy-status signalling. J Cell Sci 122:727–734
Feng Y, Williams BG, Koumanov F, Wolstenholme AJ, Holman GD (2013) FGT-1 is the major glucose transporter in C. elegans and is central to aging pathways. Biochem J 456:219–229. https://doi.org/10.1042/BJ20131101
Flessner LB, Moley KH (2009) Similar [DE]XXXL[LI] motifs differentially target GLUT8 and GLUT12 in Chinese hamster ovary cells. Traffic 10:324–333. https://doi.org/10.1111/j.1600-0854.2008.00866.x
Forrest LR (2015) Structural symmetry in membrane proteins. Annu Rev Biophys 44:311–337. https://doi.org/10.1146/annurev-biophys-051013-023008
Frolova AI, Moley KH (2011) Quantitative analysis of glucose transporter mRNAs in endometrial stromal cells reveals critical role of GLUT1 in uterine receptivity. Endocrinology 152:2123–2128. https://doi.org/10.1210/en.2010-1266
Ganguly A, McKnight RA, Raychaudhuri S, Shin BC, Ma Z, Moley K, Devaskar SU (2007) Glucose transporter isoform-3 mutations cause early pregnancy loss and fetal growth restriction. Am J Physiol Endocrinol Metab 292:E1241–E1255. https://doi.org/10.1152/ajpendo.00344.2006
Gaster M, Handberg A, Schurmann A, Joost HG, Beck-Nielsen H, Schroder HD (2004) GLUT11, but not GLUT8 or GLUT12, is expressed in human skeletal muscle in a fibre type-specific pattern. Pflugers Arch 448:105–113. https://doi.org/10.1007/s00424-003-1219-4
George Thompson AM, Iancu CV, Nguyen TT, Kim D, Choe JY (2015) Inhibition of human GLUT1 and GLUT5 by plant carbohydrate products; insights into transport specificity. Sci Rep 5:12804. https://doi.org/10.1038/srep12804
George Thompson AM, Ursu O, Babkin P, Iancu CV, Whang A, Oprea TI, Choe JY (2016) Discovery of a specific inhibitor of human GLUT5 by virtual screening and in vitro transport evaluation. Sci Rep 6:24240. https://doi.org/10.1038/srep24240
Ginsburg H, Stein WD (1975) Zero-trans and infinite-cis uptake of galactose in human erythrocytes. Biochim Biophys Acta 382:353–368. https://doi.org/10.1016/0005-2736(75)90277-1
Gliemann J, Rees WD, Foley JA (1984) The fate of labelled glucose molecules in the rat adipocyte. Dependence on glucose concentration. Biochim Biophys Acta 804:68–76. https://doi.org/10.1016/0167-4889(84)90100-9
Gomez O, Romero A, Terrado J, Mesonero JE (2006) Differential expression of glucose transporter GLUT8 during mouse spermatogenesis. Reproduction 131:63–70. https://doi.org/10.1530/rep.1.00750
Gould GW, Holman GD (1993) The glucose-transporter family-structure, function and tissue-specific expression. Biochem J 295:329–341
Grabe M, Zuckerman DM, Rosenberg JM (2020) EmrE reminds us to expect the unexpected in membrane transport. J Gen Physiol:152. https://doi.org/10.1085/jgp.201912467
Gugliucci A (2017) Formation of fructose-mediated advanced glycation end products and their roles in metabolic and inflammatory diseases. Adv Nutr 8:54–62. https://doi.org/10.3945/an.116.013912
Guillam MT, Hummler E, Schaerer E, Yeh JI, Birnbaum MJ, Beermann F, Schmidt A et al (1997) Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet 17:327–330
Hashiramoto M, Kadowaki T, Clark AE, Muraoka A, Momomura K, Sakura H, Tobe K et al (1992) Site-directed mutagenesis of glut1 in helix-7 residue-282 results in perturbation of exofacial ligand-binding. J Biol Chem 267:17502–17507
Hoglund PJ, Nordstrom KJ, Schioth HB, Fredriksson R (2011) The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol Biol Evol 28:1531–1541. https://doi.org/10.1093/molbev/msq350
Holloszy JO (2011) Regulation of mitochondrial biogenesis and GLUT4 expression by exercise. Compr Physiol 1:921–940. https://doi.org/10.1002/cphy.c100052
Holman GD (2018) Chemical biology probes of mammalian GLUT structure and function. Biochem J 475:3511–3534. https://doi.org/10.1042/BCJ20170677
Holman GD, Kozka IJ, Clark AE, Flower CJ, Saltis J, Habberfield AD, Simpson IA et al (1990) Cell-surface labeling of glucose transporter isoform GLUT4 by Bis-Mannose photolabel: correlation with stimulation of glucose transport in rat adipose cells by insulin and phorbol ester. J Biol Chem 265:18172–18179
Holman GD, Leggio LL, Cushman SW (1994) Insulin-stimulated GLUT4 glucose transporter recycling-a problem in membrane-protein subcellular trafficking through multiple pools. J Biol Chem 269:17516–17524
Holman GD, Pierce EJ, Rees WD (1981) Spatial requirements for insulin-sensitive sugar transport in rat adipocytes. Biochim Biophys Acta 646:382–388
Hresko RC, Kruse M, Strube M, Mueckler M (1994) Topology of the Glut 1 glucose transporter deduced from glycosylation scanning mutagenesis. J Biol Chem 269:20482–20488
Hruz PW, Mueckler MM (1999) Cysteine-scanning mutagenesis of transmembrane segment 7 of the GLUT1 glucose transporter. J Biol Chem 274:36176–36180
Hsin H, Kenyon C (1999) Signals from the reproductive system regulate the lifespan of C. elegans. Nature 399:362–366. https://doi.org/10.1038/20694
Hundal HS, Ahmed A, Guma A, Mitsumoto Y, Marette A, Rennie MJ, Klip A (1992) Biochemical and immunocytochemical localization of the GLUT5 glucose transporter in human skeletal-muscle. Biochem J 286:339–343
Hussey GA, Thomas NE, Henzler-Wildman KA (2020) Highly coupled transport can be achieved in free-exchange transport models. J Gen Physiol:152. https://doi.org/10.1085/jgp.201912437
Iizuka K (2017) The role of carbohydrate response element binding protein in intestinal and hepatic fructose metabolism. Nutrients 9. https://doi.org/10.3390/nu9020181
James DE, Strube M, Mueckler M (1989) Molecular-cloning and characterization of an insulin-regulatable glucose transporter. Nature 338:83–87
Jardetzky O (1966) Simple allosteric model for membrane pumps. Nature 211:969–970
Jessen N, An D, Lihn AS, Nygren J, Hirshman MF, Thorell A, Goodyear LJ (2011) Exercise increases TBC1D1 phosphorylation in human skeletal muscle. Am J Physiol Endocrinol Metab 301:E164–E171. https://doi.org/10.1152/ajpendo.00042.2011
Jewell JL, Oh E, Ramalingam L, Kalwat MA, Tagliabracci VS, Tackett L, Elmendorf JS et al (2011) Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J Cell Biol 193:185–199
Jimenez-Amilburu V, Jong-Raadsen S, Bakkers J, Spaink HP, Marin-Juez R (2015) GLUT12 deficiency during early development results in heart failure and a diabetic phenotype in zebrafish. J Endocrinol 224:1–15. https://doi.org/10.1530/JOE-14-0539
Joost HG, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H et al (2002) Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol 282:E974–E976
Joost HG, Thorens B (2001) The extended GLUT-family of sugar/polyol transport facilitators: nomenclature, sequence characteristics, and potential function of its novel members. Mol Membr Biol 18:247–256
Kampmann U, Christensen B, Nielsen TS, Pedersen SB, Orskov L, Lund S, Moller N et al (2011) GLUT4 and UBC9 protein expression is reduced in muscle from type 2 diabetic patients with severe insulin resistance. PLoS One 6:e27854. https://doi.org/10.1371/journal.pone.0027854
Kandror KV, Pilch PF (1998) Multiple endosomal recycling pathways in rat adipose cells. Biochem J 331:829–835
Kandror KV, Pilch PF (2011) The sugar is sIRVed: sorting Glut4 and its fellow travelers. Traffic 12:665–671. https://doi.org/10.1111/j.1600-0854.2011.01175.x
Kapoor K, Finer-Moore JS, Pedersen BP, Caboni L, Waight A, Hillig RC, Bringmann P et al (2016) Mechanism of inhibition of human glucose transporter GLUT1 is conserved between cytochalasin B and phenylalanine amides. Proc Natl Acad Sci U S A 113:4711–4716. https://doi.org/10.1073/pnas.1603735113
Karlish SJ, Lieb WR, Ram D, Stein WD (1972) Kinetic parameters of glucose efflux from human red blood cells under zero-trans conditions. Biochim Biophys Acta 255:126–132
Karlsson HK, Chibalin AV, Koistinen HA, Yang J, Koumanov F, Wallberg-Henriksson H, Zierath JR et al (2009) Kinetics of GLUT4 trafficking in rat and human skeletal muscle. Diabetes 58:847–854. https://doi.org/10.2337/db08-1539
Katz EB, Stenbit AE, Hatton K, DePinho R, Charron MJ (1995) Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature 377:151–155. https://doi.org/10.1038/377151a0
Kavanagh Williamson M, Coombes N, Juszczak F, Athanasopoulos M, Khan MB, Eykyn TR, Srenathan U et al (2018) Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV-1. Viruses 10. https://doi.org/10.3390/v10030114
Kayano T, Burant CF, Fukumoto H, Gould GW, Fan YS, Eddy RL, Byers MG et al (1990) Human facilitative glucose transporters - isolation, functional- characterization, and gene localization of cdnas encoding an isoform (GLUT5) expressed in small-intestine, kidney, muscle, and adipose- tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6). J Biol Chem 265:13276–13282
Kellett GL (2001) The facilitated component of intestinal glucose absorption. JPhysiol 531:585–595
Khayat ZA, McCall AL, Klip A (1998) Unique mechanism of GLUT3 glucose transporter regulation by prolonged energy demand: increased protein half-life. Biochem J 333(Pt 3):713–718. https://doi.org/10.1042/bj3330713
Kioumourtzoglou D, Gould GW, Bryant NJ (2018) Proximity ligation assay to study the GLUT4 membrane trafficking machinery. Methods Mol Biol 1713:217–227. https://doi.org/10.1007/978-1-4939-7507-5_16
Klip A, McGraw TE, James DE (2019) Thirty sweet years of GLUT4. J Biol Chem 294:11369–11381. https://doi.org/10.1074/jbc.REV119.008351
Koistinen HA, Galuska D, Chibalin AV, Yang J, Zierath JR, Holman GD, Wallberg-Henriksson H (2003) 5-amino-imidazole carboxamide riboside increases glucose transport and cell-surface GLUT4 content in skeletal muscle from subjects with type 2 diabetes. Diabetes 52:1066–1072
Koumanov F, Jin B, Yang J, Holman GD (2005) Insulin signaling meets vesicle traffic of GLUT4 at a plasma-membrane-activated fusion step. Cell Metab 2:179–189. https://doi.org/10.1016/j.cmet.2005.08.007
Koumanov F, Pereira VJ, Richardson JD, Sargent SL, Fazakerley DJ, Holman GD (2015) Insulin regulates Rab3-Noc2 complex dissociation to promote GLUT4 translocation in rat adipocytes. Diabetologia 58:1877–1886. https://doi.org/10.1007/s00125-015-3627-3
Koumanov F, Pereira VJ, Whitley PR, Holman GD (2012) GLUT4 traffic through an ESCRT-III-dependent sorting compartment in adipocytes. PLoS One 7:e44141. https://doi.org/10.1371/journal.pone.0044141
Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K et al (2006) Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55:2067–2076
Kumar Kondapi VP, Soueidan OM, Cheeseman CI, West FG (2017) Tunable GLUT-hexose binding and transport via modulation of hexose C-3 hydrogen-bonding capabilities. Chemistry 23:8073–8081. https://doi.org/10.1002/chem.201701329
Lamb CA, McCann RK, Stockli J, James DE, Bryant NJ (2010) Insulin-regulated trafficking of GLUT4 requires ubiquitination. Traffic 11:1445–1454
Lauer V, Grampp S, Platt J, Lafleur V, Lombardi O, Choudhry H, Kranz F et al (2019) Hypoxia drives glucose transporter 3 expression through HIF-mediated induction of the long non-coding RNA NICI. J Biol Chem. https://doi.org/10.1074/jbc.RA119.009827
Lee SJ, Murphy CT, Kenyon C (2009) Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab 10:379–391
Leitch JM, Carruthers A (2009) alpha- and beta-monosaccharide transport in human erythrocytes. Am J Phys Cell Phys 296:C151–C161. https://doi.org/10.1152/ajpcell.00359.2008
Li Q, Manolescu A, Ritzel M, Yao S, Slugoski M, Young JD, Chen XZ et al (2004) Cloning and functional characterization of the human GLUT7 isoform SLC2A7 from the small intestine. Am J Physiol Gastrointest Liver Physiol 287:G236–G242. https://doi.org/10.1152/ajpgi.00396.2003
Lisinski I, Schurmann A, Joost HG, Cushman SW, Al Hasani H (2001) Targeting of GLUT6 (formerly GLUT9) and GLUT8 in rat adipose cells. BiochemJ 358:517–522
Lloyd KP, Ojelabi OA, De Zutter JK, Carruthers A (2017) Reconciling contradictory findings: glucose transporter 1 (GLUT1) functions as an oligomer of allosteric, alternating access transporters. J Biol Chem 292:21035–21046. https://doi.org/10.1074/jbc.M117.815589
Lloyd KP, Ojelabi OA, Simon AH, De Zutter JK, Carruthers A (2018) Kinetic basis of cis- and trans-allostery in GLUT1-mediated sugar transport. J Membr Biol 251:131–152. https://doi.org/10.1007/s00232-017-0006-7
London RE, Gabel SA (1995) Fluorine-19 NMR studies of glucosyl fluoride transport in human erythrocytes. Biophys J 69:1814–1818. https://doi.org/10.1016/S0006-3495(95)80051-2
Macheda ML, Williams ED, Best JD, Wlodek ME, Rogers S (2003) Expression and localisation of GLUT1 and GLUT12 glucose transporters in the pregnant and lactating rat mammary gland. Cell Tissue Res 311:91–97. https://doi.org/10.1007/s00441-002-0661-5
Maedera S, Mizuno T, Ishiguro H, Ito T, Soga T, Kusuhara H (2019) GLUT6 is a lysosomal transporter that is regulated by inflammatory stimuli and modulates glycolysis in macrophages. FEBS Lett 593:195–208. https://doi.org/10.1002/1873-3468.13298
Maher F, Davies-Hill TM, Simpson IA (1996) Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem J 315(Pt 3):827–831. https://doi.org/10.1042/bj3150827
Maher F, Vannucci SJ, Simpson IA (1994) Glucose transporter proteins in brain. FASEB J 8:1003–1011. https://doi.org/10.1096/fasebj.8.13.7926364
Manolescu A, Salas-Burgos AM, Fischbarg J, Cheeseman CI (2005) Identification of a hydrophobic residue as a key determinant of fructose transport by the facilitative hexose transporter SLC2A7 (GLUT7). J Biol Chem 280:42978–42983. https://doi.org/10.1074/jbc.M508678200
Manolescu AR, Augustin R, Moley K, Cheeseman C (2007) A highly conserved hydrophobic motif in the exofacial vestibule of fructose transporting SLC2A proteins acts as a critical determinant of their substrate selectivity. Mol Membr Biol 24:455–463. https://doi.org/10.1080/09687680701298143
Marsh BJ, Alm RA, McIntosh SR, James DE (1995) Molecular regulation of GLUT-4 targeting in 3 T3-L1 adipocytes. J Cell Biol 130:1081–1091
Mayer AL, Higgins CB, Heitmeier MR, Kraft TE, Qian X, Crowley JR, Hyrc KL et al (2016) SLC2A8 (GLUT8) is a mammalian trehalose transporter required for trehalose-induced autophagy. Sci Rep 6:38586. https://doi.org/10.1038/srep38586
McCulloch LJ, van de Bunt M, Braun M, Frayn KN, Clark A, Gloyn AL (2011) GLUT2 (SLC2A2) is not the principal glucose transporter in human pancreatic beta cells: implications for understanding genetic association signals at this locus. Mol Genet Metab 104:648–653. https://doi.org/10.1016/j.ymgme.2011.08.026
McMillin SL, Schmidt DL, Kahn BB, Witczak CA (2017) GLUT4 is not necessary for overload-induced glucose uptake or hypertrophic growth in mouse skeletal muscle. Diabetes 66:1491–1500. https://doi.org/10.2337/db16-1075
McVie-Wylie AJ, Lamson DR, Chen YT (2001) Molecular cloning of a novel member of the glut family of transporters, slc2a10 (glut10), localized on chromosome 20q13.1: a candidate gene for niddm susceptibility. Genomics 72:113–117
Minokoshi Y, Kahn CR, Kahn BB (2003) Tissue-specific ablation of the GLUT4 glucose transporter or the insulin receptor challenges assumptions about insulin action and glucose homeostasis. J Biol Chem 278:33609–33612. https://doi.org/10.1074/jbc.R300019200
Mitchell P (1957) A general theory of membrane transport from studies of bacteria. Nature 180:134–136. https://doi.org/10.1038/180134a0
Moley KH (1999) Diabetes and preimplantation events of embryogenesis. Semin Reprod Endocrinol 17:137–151. https://doi.org/10.1055/s-2007-1016221
Mori H, Hashiramoto M, Clark AE, Yang J, Muraoka A, Tamori Y, Kasuga M et al (1994) Substitution of tyrosine-293 of glut1 locks the transporter into an outward facing conformation. J Biol Chem 269:11578–11583
Mori H, Hashiramoto M, Clark AE, Yang J, Muraoka A, Tamori Y, Kasuga M et al (1994) Substitution of tyrosine 293 of GLUT1 locks the transporter into an outward facing conformation. J Biol Chem 269:11578–11583
Mueckler M, Caruso C, Baldwin SA, Panico M, Blench I, Morris HR, Allard WJ et al (1985) Sequence and structure of a human glucose transporter. Science 229:941–945
Mueckler M, Makepeace C (1997) Identification of an amino acid residue that lies between the exofacial vestibule and exofacial substrate-binding site of the Glut1 sugar permeation pathway. J Biol Chem 272:30141–30146
Mueckler M, Thorens B (2013) The SLC2 (GLUT) family of membrane transporters. Mol Asp Med 34:121–138. https://doi.org/10.1016/j.mam.2012.07.001
Muretta JM, Romenskaia I, Mastick CC (2008) Insulin releases Glut4 from static storage compartments into cycling endosomes and increases the rate constant for Glut4 exocytosis. J Bio Chem 283:311–323
Naftalin RJ (2008) Alternating carrier models of asymmetric glucose transport violate the energy conservation laws. Biophys J 95:4300–4314. https://doi.org/10.1529/biophysj.108.136366
Naftalin RJ (2018) A critique of the alternating access transporter model of uniport glucose transport. Biophys Rep 4:287–299. https://doi.org/10.1007/s41048-018-0076-9
Nomura N, Verdon G, Kang HJ, Shimamura T, Nomura Y, Sonoda Y, Hussien SA et al (2015) Structure and mechanism of the mammalian fructose transporter GLUT5. Nature 526:397–401. https://doi.org/10.1038/nature14909
Ojelabi OA, Lloyd KP, De Zutter JK, Carruthers A (2018) Red wine and green tea flavonoids are cis-allosteric activators and competitive inhibitors of glucose transporter 1 (GLUT1)-mediated sugar uptake. J Biol Chem 293:19823–19834. https://doi.org/10.1074/jbc.RA118.002326
Olsowski A, Monden I, Keller K (1998) Cysteine-scanning mutagenesis of flanking regions at the boundary between external loop i or iv and transmembrane segment ii or vii in the glut1 glucose transporter. Biochemistry 37:10738–10745
Pao SS, Paulsen IT, Saier MH Jr (1998) Major facilitator superfamily. Microbiol Mol Biol Rev 62:1–34
Pascual JM, Wang D, Lecumberri B, Yang H, Mao X, Yang R, De Vivo DC (2004) GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol 150:627–633. https://doi.org/10.1530/eje.0.1500627
Patel C, Sugimoto K, Douard V, Shah A, Inui H, Yamanouchi T, Ferraris RP (2015) Effect of dietary fructose on portal and systemic serum fructose levels in rats and in KHK-/- and GLUT5-/- mice. Am J Physiol Gastrointest Liver Physiol 309:G779–G790. https://doi.org/10.1152/ajpgi.00188.2015
Phay JE, Hussain HB, Moley JF (2000) Cloning and expression analysis of a novel member of the facilitative glucose transporter family, SLC2A9 (GLUT9). Genomics 66:217–220
Pryor PR, Liu SC, Clark AE, Yang J, Holman GD, Tosh D (2000) Chronic insulin effects on insulin signalling and GLUT4 endocytosis are reversed by metformin. Biochem J 348(Pt 1):83–91
Pyla R, Poulose N, Jun JY, Segar L (2013) Expression of conventional and novel glucose transporters, GLUT1, -9, -10, and -12, in vascular smooth muscle cells. Am J Phys Cell Phys 304:C574–C589. https://doi.org/10.1152/ajpcell.00275.2012
Quistgaard EM, Low C, Guettou F, Nordlund P (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol 17:123–132. https://doi.org/10.1038/nrm.2015.25
Ramalingam L, Oh E, Yoder SM, Brozinick JT, Kalwat MA, Groffen AJ, Verhage M et al (2012) Doc2b is a key effector of insulin secretion and skeletal muscle insulin sensitivity. Diabetes 61:2424–2432. https://doi.org/10.2337/db11-1525
Ramm G, Slot JW, James DE, Stoorvogel W (2000) Insulin recruits GLUT4 from specialized VAMP2-carrying vesicles as well as from the dynamic endosomal/trans-Golgi network in rat adipocytes. Mol Biol Cell 11:4079–4091
Rees WD, Holman GD (1981) Hydrogen bonding requirements for the insulin sensitive sugar transport system of rat adipocytes. BiochimBiophysActa 642:251–260
Ren W, Sun Y, Du K (2015) Glut4 palmitoylation at Cys223 plays a critical role in Glut4 membrane trafficking. Biochem Biophys Res Commun 460:709–714. https://doi.org/10.1016/j.bbrc.2015.03.094
Rivera-Molina FE, Novick PJ (2009) A Rab GAP cascade defines the boundary between two Rab GTPases on the secretory pathway. Proc Natl Acad Sci U S A 106:14408–14413
Rose AJ, Richter EA (2005) Skeletal muscle glucose uptake during exercise: how is it regulated? Physiology (Bethesda) 20:260–270
Rumsey SC, Kwon O, Xu GW, Burant CF, Simpson I, Levine M (1997) Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J Biol Chem 272:18982–18989
Russell SJ, Kahn CR (2007) Endocrine regulation of ageing. Nat Rev Mol Cell Biol 8:681–691. https://doi.org/10.1038/nrm2234
Sadler JB, Bryant NJ, Gould GW, Welburn CR (2013) Posttranslational modifications of GLUT4 affect its subcellular localization and translocation. Int J Mol Sci 14:9963–9978. https://doi.org/10.3390/ijms14059963
Sadler JBA, Lamb CA, Welburn CR, Adamson IS, Kioumourtzoglou D, Chi NW, Gould GW et al (2019) The deubiquitinating enzyme USP25 binds tankyrase and regulates trafficking of the facilitative glucose transporter GLUT4 in adipocytes. Sci Rep 9:4710. https://doi.org/10.1038/s41598-019-40596-5
Sage JM, Carruthers A (2014) Human erythrocytes transport dehydroascorbic acid and sugars using the same transporter complex. Am J Phys Cell Phys 306:C910–C917. https://doi.org/10.1152/ajpcell.00044.2014
Sasaki T, Minoshima S, Shiohama A, Shintani A, Shimizu A, Asakawa S, Kawasaki K et al (2001) Molecular cloning of a member of the facilitative glucose transporter gene family GLUT11 (SLC2A11) and identification of transcription variants. Biochem Biophys Res Commun 289:1218–1224. https://doi.org/10.1006/bbrc.2001.6101
Satoh S, Nishimura H, Clark AE, Kozka IJ, Vannucci SJ, Simpson IA, Quon MJ et al (1993) Use of bis-Mannose photolabel to elucidate insulin-regulated GLUT4 subcellular trafficking kinetics in rat adipose cells: evidence that exocytosis is a critical site of hormone action. J Biol Chem 268:17820–17829
Scheepers A, Schmidt S, Manolescu A, Cheeseman CI, Bell A, Zahn C, Joost HG et al (2005) Characterization of the human SLC2A11 (GLUT11) gene: alternative promoter usage, function, expression, and subcellular distribution of three isoforms, and lack of mouse orthologue. Mol Membr Biol 22:339–351. https://doi.org/10.1080/09687860500166143
Schmidt S, Hommel A, Gawlik V, Augustin R, Junicke N, Florian S, Richter M et al (2009) Essential role of glucose transporter GLUT3 for post-implantation embryonic development. J Endocrinol 200:23–33. https://doi.org/10.1677/JOE-08-0262
Schmidt S, Joost HG, Schurmann A (2009) GLUT8, the enigmatic intracellular hexose transporter. Am J Physiol Endocrinol Metab 296:E614–E618. https://doi.org/10.1152/ajpendo.91019.2008
Schmidt U, Briese S, Leicht K, Schurmann A, Joost HG, Al-Hasani H (2006) Endocytosis of the glucose transporter GLUT8 is mediated by interaction of a dileucine motif with the beta2-adaptin subunit of the AP-2 adaptor complex. J Cell Sci 119:2321–2331. https://doi.org/10.1242/jcs.02943
Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6:280–293
Schurmann A, Doege H, Ohnimus H, Monser V, Buchs A, Joost HG (1997) Role of conserved arginine and glutamate residues on the cytosolic surface of glucose transporters for transporter function. Biochemistry 36:12897–12902
Sen AK, Widdas WF (1962) Determination of the temperature and pH dependence of glucose transfer across the human erythrocyte membrane measured by glucose exit. J Physiol 160:392–403
Shewan AM, Marsh BJ, Melvin DR, Martin S, Gould GW, James DE (2000) The cytosolic C-terminus of the glucose transporter GLUT4 contains an acidic cluster endosomal targeting motif distal to the dileucine signal. Biochem J 350:99–107
Shishmarev D, Fontenelle CQ, Kuprov I, Linclau B, Kuchel PW (2018) Transmembrane exchange of fluorosugars: characterization of red cell GLUT1 kinetics using (19)F NMR. Biophys J 115:1906–1919. https://doi.org/10.1016/j.bpj.2018.09.030
Siebeneicher H, Cleve A, Rehwinkel H, Neuhaus R, Heisler I, Muller T, Bauser M et al (2016) Identification and optimization of the first highly selective GLUT1 inhibitor BAY-876. ChemMedChem 11:2261–2271. https://doi.org/10.1002/cmdc.201600276
Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F, KoehlerStec EM et al (1999) Blood-brain barrier glucose transporter: effects of hypo- and hyperglycemia revisited. J Neurochem 72:238–247
Simpson IA, Dwyer D, Malide D, Moley KH, Travis A, Vannucci SJ (2008) The facilitative glucose transporter GLUT3: 20 years of distinction. Am J Physiol Endocrinol Metab 295:E242–E253. https://doi.org/10.1152/ajpendo.90388.2008
Slot JW, Geuze HJ, Gigengack S, Lienhard GE, James DE (1991) Immuno-localization of the insulin regulatable glucose transporter in brown adipose-tissue of the rat. J Cell Biol 113:123–135
So A, Thorens B (2010) Uric acid transport and disease. JClinInvest 120:1791–1799
Soueidan OM, Trayner BJ, Grant TN, Henderson JR, Wuest F, West FG, Cheeseman CI (2015) New fluorinated fructose analogs as selective probes of the hexose transporter protein GLUT5. Org Biomol Chem 13:6511–6521. https://doi.org/10.1039/c5ob00314h
Stein W (2012) Transport and diffusion across cell membranes. Elsevier Science
Stenkula KG, Lizunov VA, Cushman SW, Zimmerberg J (2010) Insulin controls the spatial distribution of GLUT4 on the cell surface through regulation of its postfusion dispersal. Cell Metab 12:250–259
Stuart CA, Howell ME, Yin D (2007) Overexpression of GLUT5 in diabetic muscle is reversed by pioglitazone. Diabetes Care 30:925–931. https://doi.org/10.2337/dc06-1788
Stuart CA, Howell ME, Zhang Y, Yin D (2009) Insulin-stimulated translocation of glucose transporter (GLUT) 12 parallels that of GLUT4 in normal muscle. J Clin Endocrinol Metab 94:3535–3542. https://doi.org/10.1210/jc.2009-0162
Sun L, Zeng X, Yan C, Sun X, Gong X, Rao Y, Yan N (2012) Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature 490:361–366. https://doi.org/10.1038/nature11524
Sun Y, Bilan PJ, Liu Z, Klip A (2010) Rab8A and Rab13 are activated by insulin and regulate GLUT4 translocation in muscle cells. Proc Natl Acad Sci U S A 107:19909–19914. https://doi.org/10.1073/pnas.1009523107
Suzuki K, Kono T (1980) Evidence that insulin causes translocation of glucose transport activity to the plasma membrane from an intracellular storage site. Proc Natl Acad Sci U S A 77:2542–2545
Syu YW, Lai HW, Jiang CL, Tsai HY, Lin CC, Lee YC (2018) GLUT10 maintains the integrity of major arteries through regulation of redox homeostasis and mitochondrial function. Hum Mol Genet 27:307–321. https://doi.org/10.1093/hmg/ddx401
Tamori Y, Hashiramoto M, Clark AE, Mori H, Muraoka A, Kadowaki T, Holman GD et al (1994) Substitution at Pro385 of GLUT1 perturbs the glucose transport function by reducing conformational flexibility. J Biol Chem 269:2982–2986
Tanasova M, Begoyan VV, Weselinski LJ (2018) Targeting sugar uptake and metabolism for cancer identification and therapy: an overview. Curr Top Med Chem 18:467–483. https://doi.org/10.2174/1568026618666180523110837
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96
Tatibouet A, Yang J, Morin C, Holman GD (2000) Synthesis and evaluation of fructose analogues as inhibitors of the D-fructose transporter GLUT5. Bioorg Med Chem 8:1825–1833
Taylor LP, Holman GD (1981) Symmetrical kinetic parameters for 3- O -methyl-D-glucose transport in adipocytes in the presence and absence of insulin. Biochim Biophys Acta 642:325–335
Taylor LP, Holman GD (1981) Symmetrical kinetic parameters for 3-O-methyl-D-glucose transport in adipocytes in the presence and in the absence of insulin. Biochim Biophys Acta 642:325–335
Thorens B (2001) GLUT2 in pancreatic and extra-pancreatic gluco-detection. Mol Membr Biol 18:265–273
Thorens B (2001) GLUT2 in pancreatic and extra-pancreatic gluco-detection (review). Mol Membr Biol 18:265–273. https://doi.org/10.1080/09687680110100995
Tumova S, Kerimi A, Porter KE, Williamson G (2016) Transendothelial glucose transport is not restricted by extracellular hyperglycaemia. Vasc Pharmacol 87:219–229. https://doi.org/10.1016/j.vph.2016.11.001
Uldry M, Ibberson M, Horisberger JD, Chatton JY, Riederer BM, Thorens B (2001) Identification of a mammalian H(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J 20:4467–4477
van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D et al (2001) Intensive insulin therapy in critically ill patients. N Engl J Med 345:1359–1367. https://doi.org/10.1056/NEJMoa011300
Vannucci SJ, Clark RR, Koehler-Stec E, Li K, Smith CB, Davies P, Maher F et al (1998) Glucose transporter expression in brain: relationship to cerebral glucose utilization. Dev Neurosci 20:369–379. https://doi.org/10.1159/000017333
Vera JC, Rivas CI, Fischbarg J, Golde DW (1993) Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 364:79–82. https://doi.org/10.1038/364079a0
Villagran M, Munoz M, Inostroza E, Venegas C, Ruminot I, Parra-Valencia E, Maldonado M et al (2019) GLUT1 and GLUT8 support lactose synthesis in Golgi of murine mammary epithelial cells. J Physiol Biochem 75:209–215. https://doi.org/10.1007/s13105-019-00679-3
Wallberg-Henriksson H, Zierath JR (2001) GLUT4: a key player regulating glucose homeostasis? Insights from transgenic and knockout mice (review). Mol Membr Biol 18:205–211. https://doi.org/10.1080/09687680110072131
Walmsley AR (1988) The dynamics of the glucose transporter. Trends Biochem Sci 13:226–231
Wandel S, Schurmann A, Becker W, Summers SA, Shanahan MF, Joost HG (1994) Substitution of conserved tyrosine residues in helix 4 (Y143) and 7 (Y293) affects the activity, but not IAPS-forskolin binding, of the glucose transporter GLUT4. FEBS Lett 348:114–118
Wang D, Pascual JM, Yang H, Engelstad K, Mao X, Cheng J, Yoo J et al (2006) A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet 15:1169–1179. https://doi.org/10.1093/hmg/ddl032
Wang D, Yang H, Shi L, Ma L, Fujii T, Engelstad K, Pascual JM et al (2008) Functional studies of the T295M mutation causing Glut1 deficiency: glucose efflux preferentially affected by T295M. Pediatr Res 64:538–543. https://doi.org/10.1203/PDR.0b013e318184d2b5
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
White MA, Tsouko E, Lin C, Rajapakshe K, Spencer JM, Wilkenfeld SR, Vakili SS et al (2018) GLUT12 promotes prostate cancer cell growth and is regulated by androgens and CaMKK2 signaling. Endocr Relat Cancer 25:453–469. https://doi.org/10.1530/ERC-17-0051
Widmer M, Uldry M, Thorens B (2005) GLUT8 subcellular localization and absence of translocation to the plasma membrane in PC12 cells and hippocampal neurons. Endocrinology 146:4727–4736. https://doi.org/10.1210/en.2005-0668
Wu X, Freeze HH (2002) GLUT14, a duplicon of GLUT3, is specifically expressed in testis as alternative splice forms. Genomics 80:553–557
Xu Y, Rubin BR, Orme CM, Karpikov A, Yu C, Bogan JS, Toomre DK (2011) Dual-mode of insulin action controls GLUT4 vesicle exocytosis. J Cell Biol 193:643–653
Yang J, Dowden J, Tatibouet A, Hatanaka Y, Holman GD (2002) Development of high-affinity ligands and photoaffinity labels for the D-fructose transporter GLUT5. Biochem J 367:533–539. https://doi.org/10.1042/BJ20020843
Yang J, Holman GD (2005) Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J Biol Chem 280:4070–4078. https://doi.org/10.1074/jbc.M410213200
Yang J, Holman GD (2006) Long-term metformin treatment stimulates cardiomyocyte glucose transport through an AMP-activated protein kinase-dependent reduction in GLUT4 endocytosis. Endocrinology 147:2728–2736. https://doi.org/10.1210/en.2005-1433
Zamora-Leon SP, Golde DW, Concha II, Rivas CI, Delgado-Lopez F, Baselga J, Nualart F et al (1996) Expression of the fructose transporter GLUT5 in human breast cancer. Proc Natl Acad Sci U S A 93:1847–1852. https://doi.org/10.1073/pnas.93.5.1847
Zeigerer A, McBrayer MK, McGraw TE (2004) Insulin stimulation of GLUT4 exocytosis, but not Its inhibition of endocytosis, is dependent on RabGAP AS160. Mol Biol Cell 15:4406–4415
Zhao Y, Fung C, Shin D, Shin BC, Thamotharan S, Sankar R, Ehninger D et al (2010) Neuronal glucose transporter isoform 3 deficient mice demonstrate features of autism spectrum disorders. Mol Psychiatry 15:286–299. https://doi.org/10.1038/mp.2009.51
Funding
This work was supported by a grant from the Leverhulme Trust UK, EM-2018-039.
Author information
Authors and Affiliations
Contributions
The author (GDH) is responsible for all aspects of the review and its content.
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
The author hereby gives consent to publish.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the special issue on Glucose Transporters in Health and Disease in Pflügers Archiv—European Journal of Physiology
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Holman, G.D. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflugers Arch - Eur J Physiol 472, 1155–1175 (2020). https://doi.org/10.1007/s00424-020-02411-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-020-02411-3