
Abstract
Maximum likelihood (ML) analysis of nucleotide or amino-acid alignments is widely used to infer evolutionary relationships among species. Computing the likelihood of a phylogenetic tree from such alignments is a complicated task because the evolutionary processes typically vary across sites. A number of studies have shown that partitioning alignments into sub-alignments of sites, where each sub-alignment is analyzed using a different model of evolution (e.g., GTR + I + G), is a sensible strategy. Current partitioning methods group sites into subsets based on the inferred rates of evolution at the sites. However, these do not provide sufficient information to adequately reflect the substitution processes of characters at the sites. Moreover, the site rate-based methods group all invariant sites into one subset, potentially resulting in wrong phylogenetic trees. In this study, we propose a partitioning method, called mPartition, that combines not only the evolutionary rates but also substitution models at sites to partition alignments. Analyses of different partitioning methods on both real and simulated datasets showed that mPartition was better than the other partitioning methods tested. Notably, mPartition overcame the pitfall of grouping all invariant sites into one subset. Using mPartition may lead to increased accuracy of ML-based phylogenetic inference, especially for multiple loci or whole genome datasets.





Similar content being viewed by others

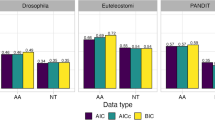
References
Baca SM, Toussaint EFA, Miller KB (2017) Molecular phylogeny of the aquatic beetle family Noteridae (Coleoptera: Adephaga) with an emphasis on data partitioning strategies. Mol Phylogenet Evol 107:282–292
Ballesteros J, Sharma P (2019) A critical appraisal of the placement of Xiphosura (Chelicerata) with account of known sources of phylogenetic error. Syst Biol 68:896–917
Brandley MC, Schmitz A, Reeder TW (2005) Partitioned Bayesian analyses, partition choice, and the phylogenetic relationships of scincid lizards. Syst Biol 54:373–390
Chen MY, Liang D, Zhang P (2015) Selecting question-specific genes to reduce incongruence in phylogenomics: a case study of jawed vertebrate backbone phylogeny. Syst Biol 64:1104–1120
Crotty SM et al (2020) GHOST: recovering historical signal from heterotachously evolved sequence alignments. Syst Biol 69(2):249–264
Cummins CA, McInerney JO (2011) A method for inferring the rate of evolution of homologous characters that can potentially improve phylogenetic inference, resolve deep divergence and correct systematic biases. Syst Biol 60:833–844
Dziak JJ et al (2020) Sensitivity and specificity of information criteria. Brief Bioinform 21(2):553–565
Felsenstein J (2003) Sunderland Inferring Phytogenies. Sinauer Associates, Sunderland
Frandsen PB, Calcott B, Mayer C, Lanfear R (2015) Automatic selection of partitioning schemes for phylogenetic analyses using iterative k-means clustering of site rates. BMC Evol Biol 15:13
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52(5):696–704
Hoang DT et al (2017) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35(2):518–522
Hurvich CM, Tsai C-L (1989) Regression and time series model selection in small samples. Biometrika 76:297–307
Irisarri I et al (2017) Phylotranscriptomic consolidation of the jawed vertebrate timetree. Nat Ecol Evol 1:1370–1378
Jones DT, Taylor WR, Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics 8(3):275–282
Kainer D, Lanfear R (2015) The effects of partitioning on phylogenetic inference. Mol Biol Evol 32:1611–1627
Kalyaanamoorthy S et al (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14:587–589
Katja R, Mappes J, Lauri K, Wahlberg N (2016) Putting Parasemia in its phylogenetic place: a molecular analysis of the subtribe Arctiina (Lepidoptera): molecular phylogeny of Arctiina. Syst Entomol 41:844–853
Kodandaramaiah U et al (2009) Phylogenetics of Coenonymphina (Nymphalidae: Satyrinae) and the problem of rooting rapid radiations. Mol Phylogenet Evol 54:386–394
Kumar S et al (2012) Statistics and truth in phylogenomics. Mol Biol Evol 29:457–472
Lanfear R, Calcott B, Ho S, Guindon S (2012) PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol 29:1695–1701
Lartillot N, Philippe H (2004) A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol 21:1095–1109
Le SQ, Dang CC, Gascuel O (2012) Modeling protein evolution with several amino-acid replacement matrices depending on site rates. Mol Biol Evol 29:2921–2936
Le SQ, Gascuel O (2008) An improved general amino-acid replacement matrix. Mol Biol Evol 25(7):1307–1320
Lemey P, Salemi M, Vandamme AM (2009) The phylogenetic handbook: a practical approach to phylogenetic analysis and hypothesis testing, 2nd edn. Cambridge University Press, Cambridge
Matos-Maraví P et al (2014) Causes of endemic radiation in the Caribbean: Evidence from the historical biogeography and diversification of the butterfly genus Calisto (Nymphalidae: Satyrinae: Satyrini). BMC Evol Biol 14:199
Minh BQ et al (2020) IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37(5):1530–1534
Nylander J, Ronquist F, Huelsenbeck J, Nieves-Aldrey J (2004) Bayesian phylogenetic analysis of combined data. Syst Biol 53:47–67
Pagel M, Meade A (2004) A phylogenetic mixture model for detecting pattern-heterogeneity in gene sequence or character-state data. Syst Biol 53:571–581
Penz C, Devries P, Wahlberg N (2012) Diversification of Morpho butterflies (Lepidoptera, Nymphalidae): a re-evaluation of morphological characters and new insight from DNA sequence data. Syst Entomol 37:670–685
Ran J-H, Shen T-T, Wang M-M, Wang X-Q (2018) Phylogenomics resolves the deep phylogeny of seed plants and indicates partial convergent or homoplastic evolution between gnetales and angiosperms. Proc Royal Soc B. https://doi.org/10.1098/rspb.2018.1012
Robinson DF, Foulds LR (1981) Comparison of phylogenetic trees. Math Biosci 53:131–147
Rodréguez-Ezpeleta N et al (2007) Detecting and overcoming systematic errors in genome-scale phylogenies. Syst Biol 56:389–399
Rota J et al (2018) A simple method for data partitioning based on relative evolutionary rates. PeerJ 6:e5498
Rota J, Wahlberg N (2012) Exploration of data partitioning in an eight-gene data set: phylogeny of metalmark moths (Lepidoptera, Choreutidae). Zoologica Scripta 41(5):536–546
Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464
Sihvonen MP et al (2011) Comprehensive molecular sampling yields a Robust Phylogeny for Geometrid Moths (Lepidoptera: Geometridae). PLoS One 6:e20356
Stamatakis A (2015) Using RAxML to Infer Phylogenies. Curr Protoc Bioinformatics 51:6.14.1–6.14.14
Strimmer K, von Haeseler A (1997) Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proc Natl Acad Sci USA 94(13):6815–6819
Tagliacollo V, Lanfear R (2018) Estimating improved partitioning schemes for ultraconserved elements (UCEs). Mol Biol Evol 35(7):1798–1811
Vinh LS, von Haeseler A (2004) IQPNNI: Moving fast through tree space and stopping in time. Mol Biol Evol 21(8):1565–1571
Wahlberg N et al (2014) Revised systematics and higher classification of pierid butterflies (Lepidoptera: Pieridae) based on molecular data. Zool Scr 43:641–650
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699
Wu S, Edwards S, Liang L (2018) Genome-scale DNA sequence data and the evolutionary history of placental mammals. Data Brief 18:1972–1975
Yang Z. (1993) Maximum-likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol Biol Evol 10(6):1396–1401
Yang Z (1996) Maximum-likelihood models for combined analyses of multiple sequence data. J Mol Evol 42(5):587–596
Zahiri Z et al (2013) Relationships among the basal lineages of Noctuidae (Lepidoptera, Noctuoidea) based on eight gene regions. Zool Scr 42:488–507
Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 102.01.2019.06.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling Editor: Arndt von Haeseler.
Rights and permissions
About this article

Cite this article
Le Kim, T., Le Sy, V. mPartition: A Model-Based Method for Partitioning Alignments. J Mol Evol 88, 641–652 (2020). https://doi.org/10.1007/s00239-020-09963-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00239-020-09963-z