
Abstract
Coronavirus disease (COVID-19) pandemic is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). COVID-19 is a global health emergency and no clinically approved vaccines or antiviral drugs available to date. Intensive research on SARS-CoV-2 is urgently warranted to understand its pathogenesis and virulence mechanisms and to discover target-based antiviral therapeutics. Among various research logics, current bioinformatics highlights novel testable hypotheses for systematic drug repositioning and designing against COVID-19. A total of 121 articles related to bioinformatics facets of this virus were collected from the PubMed Central. The content of each investigation was comprehensively reviewed, manually curated, and included herein. Interestingly, 109 COVID-19-related literature published in 2020 (January–June) were included in this review. The present article emphasizes novel resource development on its genome structure, evolution, therapeutic targets, drug designing, and drug repurposing strategies. Genome organization, the function of coding genes, origin, and evolution of SARS-CoV-2 is described in detail. Genomic insights into understanding the structure–function relationships of drug targets including spike, main protease, and RNA-dependent RNA polymerase of SARS-CoV-2 are discussed intensively. Several molecular docking and systems pharmacology approaches have been investigated some promising antiviral drugs against SARS-CoV-2 based on its genomic characteristics, pathogenesis mechanism, and host specificity. Perhaps, the present genomic insights of this virus will provide a lead to the researchers to design or repurpose of antiviral drugs soon and future directions to control the spread of COVID-19.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The coronavirus disease 19 (COVID-19) is a global epidemic with high morbidity and mortality. It is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a new strain isolated from the Huanan Seafood Market, Wuhan, China in December 2019 (Fox 2020; Khan et al. 2020b). Horseshoe bat is a primary reservoir, but the intermediate source of origin and transfer to humans is fully understood (Guo et al. 2020; Singhal 2020). Human coronaviruses (Coronaviridae family; Nidovirales order) can cause respiratory, gastrointestinal, hepatic, and central nervous system diseases. SARS-CoV-2 is the seventh coronavirus of which two α-coronaviruses (HCoV-229E and HCoV-NL63) and two β-coronaviruses (HCoV-OC43 and HCoV-HKU1) are causing only mild self-limiting upper respiratory diseases. SARS-CoV-2, SARS-CoV, and MERS-CoV can cause severe diseases (Li et al. 2020a). Human-to-human contact and travel-related cases are transmission mechanisms for the SARS-CoV-2 outbreak (Ralph et al. 2020). The fecal–oral transmission is a possible transmission mechanism of SARS-CoV-2 infection in children (Zhang et al. 2020c). SARS-CoV-2 is sensitive to heat and UV rays and can be effectively destroyed with the use of 75% ethanol, p-acetic acid, and chlorine-containing disinfectants (Zhou et al. 2020b).
On 11 March 2020, the World Health Organization (WHO) declared the outbreak a pandemic. As of 20 July 2020, 14.6 million cases have been confirmed in more than 210 countries, with 608k deaths. The United States, Spain, Italy, and Germany have registered more cases than China, where the outbreak started (https://www.worldometers.info/coronavirus/). Consequently, the WHO declared a state of global health emergency to coordinate scientific and medical efforts to rapidly develop a cure for patients (Sohrabi et al. 2020). The development of more targeted inhibitors is highly desirable, but no effective antiviral drugs or vaccines available to date (Li et al. 2020b). The source of origin, transmission, and pathogenesis mechanisms of this virus are not yet known. It hampers further investigation of safe and valuable antiviral medications. The massive research works on its genomics perspectives provide a warrant to discover antivirals against COVID-19 infectivity.
Genomic insights of SARS-CoV-2
Genome structure
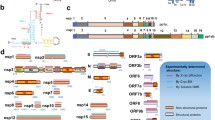
SARS-CoV-2 belongs to the Sarbecovirus subgenus (β-Coronavirus genus). It is an enveloped virus containing a single positive‐stranded RNA (Chan et al. 2020a; Lu et al. 2020; Licastro et al. 2020; Yu et al. 2020; Zhou et al. 2020a, b). The first genome sequence (accession: NC_045512.2) was completed for SARS-CoV-2 Wuhan-Hu-1 isolated from Wuhan, Hubei province, China (Wu et al. 2020). Continuous genome sequencing attempts of different laboratories have been produced 930 GenBank sequences and 288 next-generation sequences to date. The genome size of this virus is 29.9 kb with 11 open reading frames (Orfs). ORF1ab (266–21555 nts.) encodes replicase polyprotein 1ab. After cleaved by two proteases, ORF1ab (266–13483 nts.) encodes replicase polyprotein 1a with multiple functions. Gene-coding proteins are organized as 5′-leader-UTR-replicase-ORFab-S (spike)-E (envelope)-M (membrane)-N (nucleocapsid)-3′UTR-poly (A) tail-3′-UTR end (Kandeel et al. 2020; Zhou et al. 2020a) (Table 1).
The ORF1ab gene encodes a polyprotein consisting of 15 nonstructural proteins (Nsp1-16) (Fig. 1). This polyprotein is auto-proteolytically cleaved into multiple enzymes that form replicase-transcriptase machinery consisting of RNA-dependent RNA polymerase (RdRp), helicase, 3′–5′ exonuclease, endoRNAse and 2′-O-ribose methyltransferase. These enzyme components are essential to viral genome replication and nucleic acid metabolism (Gordon et al. 2020). The replicase-transcriptase system assembles at the host endoplasmic reticulum where structural proteins assembled to make essential cellular components (capsid).

Genomic organization and gene neighborhood (a) of emerging SARS-CoV-2 Wuhan-Hu-1 isolate. The function of ORF1ab gene-coding proteins is presented in (b)
A nonstructural protein 1 (Nsp1) is a host translation inhibitor that facilitates efficient viral gene expression in infected cells and evasion from the host immune response by interacting with the 40S ribosomal subunit. ORF1ab gene contains coronavirus frameshifting stimulation element stem-loop 1 and 2 in the position of 13476–13503 and 13488–13542 nucleotides, respectively. ORF10 gene harbors coronavirus 3′ UTR pseudo-knot stem-loop 1 (29609–29644 nucleotides) and 2 (29629–29657 nucleotides). The size of the stem-loop structure is ranged from 27 to 54 nucleotides. The genomic position 29740–29758 nucleotides form a noncanonical C: T base pair. The homologous positions form a highly conserved C: G base pair (Ashour et al. 2020; Kandeel et al. 2020; Li et al. 2020d).
Genome evolution
The genome of SARS-CoV-2 is about 82%, 96%, and 86.9% identical to the SARS-CoV, bat-CoV-RaTG13, and bat-SL-CoVZC45, respectively (Zhou et al. 2020b). However, the SARS-CoV-2 genome is genetically distinct from SARS-CoV and has a relatively long branch length to the bat-CoV-RaTG13 and bat-SL-CoVZXC21. SARS-CoV-2 genome is 99.98% identical across the SARS-CoV-2 genomes obtained from different patients (Lu et al. 2020). The genome-coding potential is closely related to those of the bat, civet, and HCoVs (Chan et al. 2020a). Genetic recombination events are complex more likely occurring in bat-CoV-RaTG13 and bat-SL-CoVZXC21than in SARS-CoV-2 (Lu et al. 2020; Paraskevis et al. 2020). A phylogenetic network analysis of 160 complete SARS-CoV-2 genomes found three central mutational variants distinguished by amino acid changes. Accordingly, it has classified into A, B, and C ancestral types. The synonymous mutations T29095C and T8782C are identified in type A and type B, respectively. The non-synonymous mutations C28144T (Leu to Ser) and G26144T (Gly to Val) are detected in type B and type C, respectively (Forster et al. 2020). Therefore, the genetic diversity of SARS-CoV-2 has been expanded in human hosts due to the establishment of hyper-variable genomic hotspot its population (Wen et al. 2020).
Hyper-variable genomic hotspots
Some mutational hotspots identified from structural and nonstructural genes had deleterious effects on the functional evolution of coding proteins, host specificity, and infectivity of SARS-CoV-2 (Li et al. 2020c; Phan 2020). SARS-CoV-2 mutational hotspots are located at positions 1397, 2891, 14408, 17746, 17857, 18060, 23403, and 28881, where a mutation at position 14408 is adjacent to the drugs targeting RdRp hydrophobic clef (Pachetti et al. 2020). The spike glycoprotein of this virus contains a receptor-binding domain (RBD), which shares only a 40% amino acid identity with other SARS-related coronaviruses (Chan et al. 2020a). It has the non-synonymous mutation sites that slow down the development of therapeutics apart from biologics and macrocyclic peptides (Morse et al. 2020). The RBD has three possible mutation hotspots include D614G, G476S, and V483A of which V483A repeated more frequently followed by G476S. A variant D614G could bring a potentially crucial change in a protein sequence due to aspartic acid is a negatively charged and acidic amino acid compared to glycine (Banerjee et al. 2020).
The envelope and nucleocapsid proteins contain two evolutionarily conserved regions with a sequence identity of 89–96% to the SARS-CoV. ORF3b and ORF8 proteins are structurally different from those of SARS-CoV (Zhou et al. 2020b). The endosome-associated-protein-like domain in sp2 protein had a stabilizing mutation. The Nsp3 protein is imposed by a destabilizing mutation. It suggests a possible mechanism of differentiating SARS-CoV-2 from SARS-related coronaviruses (Angeletti et al. 2020). The hyper-variable genomic hotspot (Ser/Lue) is found in ORF8 protein (Ceraolo and Giorgi 2020). The mutations identified in Nsp1, Nsp3, and Nsp15 might facilitate human adaptation and infection (Wen et al. 2020). The intrinsic disorder predisposition in the nonstructural proteins during evolution may also provide important information to explore its infectivity (Fahmi et al. 2020; Goh et al. 2020). Since, finding mutational variants in viral target proteins are important to assess possible drug-resistance phenotypes, leading to modulate the clinical presentation of the COVID-19 worldwide.
The high-frequency SNP mutation sites and hyper-variable hotspots are important for designing a COVID-19 vaccine and rapid detection of different genotypes of SARS-CoV-2. Most recently, a genotypic method has been established for monitoring and tracing SARS-CoV-2 mutations (Yin 2020). The 2019 Novel Coronavirus Resource (2019nCoVR, https://bigd.big.ac.cn/ncov) was built for studies on viral taxonomy, genome evolution, molecular diagnosis, and drug development (Zhao et al. 2020). The Genome Detective Coronavirus Typing Tool has been developed for accurate tracking of new viral mutations in SARS-CoV-2 genome sequences (Cleemput et al. 2020). These resources would allow us to agitate the drug discovery process for the COVID-19 infectivity.
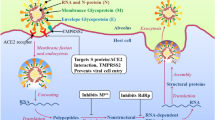
Pathogenomics
The functions of structural and nonstructural proteins are associating with pathophysiology and virulence of SARS-CoV-2. The envelope protein has a critical role in viral assembly and releases exerting viral pathogenicity and other nonstructural proteins have not yet been described (Wang et al. 2020b). The surface glycoprotein contains the S2 subunit comprising a fusion peptide, a transmembrane domain, and a cytoplasmic domain (Rabaan et al. 2020). These domains are guiding the link to host receptors. An in silico mutational study suggests a higher affinity of the SARS-Cov-2 spike protein favorable to the human ACE2 receptor, compared to the Bat-CoV spike protein (Hussain et al. 2020; Ortega et al. 2020). The N82 in ACE2 is closer contact with spike protein than M82 in human ACE2 (Luan et al. 2020). Structural genomics analysis on spike protein provides important insights to design an optimized ACE2 for SARS-CoV-2 infection. The structural variations in spike protein (Ser19Pro; Glu329Gly) and the transmembrane helical segments of nsp2 and nsp3 proteins (Ser723Gly; Pro1010Iso) may determine the host specificity of SARS-CoV-2. Such mutation hotspots in spike protein and nonstructural proteins of SARS-CoV-2 might affect molecular recognition and specificity of antivirals or vaccigenic candidates. These mutations can also influence the binding efficiency of PCR primers or probes during the molecular diagnosis of clinical samples. Consequently, evidence of such mutation hotspots could explain how SARS-CoV-2 has a mechanism for disease relapses in a host (Angeletti et al. 2020; Korber et al. 2020).
SARS-CoV-2 infection is capable of producing an excessive immune reaction in the host. The IL-6 is produced during infection by activated leukocytes and acts on a large number of cells and also stimulates the production of acute-phase proteins important to the thermoregulation. Subsequently, B lymphocyte differentiation has been elevated for inhibition of further virus replication in a host (Cascella et al. 2020; Prompetchara et al. 2020; Rabaan et al. 2020). SARS-CoV accessory protein ORF6 antagonizes the antiviral activity of the STAT1 transcription factor by sequestering IMPα/β1 on the rough ER/Golgi membrane (Frieman et al. 2007). Ivermectin is the US Food and Drug Administration (FDA)-approved broad-spectrum antiparasitic agent and HIV-1 integrase, which reported as an effective nuclear transport inhibitor against SARS-CoV-2 in vitro (Caly et al. 2020). The density of the expression levels of ACE2 in neurological tissue also suggests the possible contribution of neurological tissue damage to the morbidity and mortality caused by COIVD-19 (Baig et al. 2020).
SARS-CoV-2 requires host cellular factors for successful replication during infection. The mechanisms of its infection can be elucidated with a systematic analysis of virus-host protein–protein interactions (PPIs) (Bösl et al. 2019; Rothan and Byrareddy 2020). It is hypothesized that a host protein that functionally associates with this virus is localized in the corresponding sub-network within the comprehensive human interactome network. The host dependency factors mediating virus infection and effective molecular targets should be identified for developing broad-spectrum antiviral drugs for COVID-19. The previous virus-host proteome interaction studies identified the key host-specific proteins and interactions pathways including DNA replication, epigenetic and gene expression regulators, vesicle trafficking, lipid modification, ubiquitin ligases, and nuclear transport machinery (Table 2). Orf10 of SARS-CoV-2 has interacted with the human ubiquitin system with multiple members of the Culin-2 E3 Ligase complex (Gordon et al. 2020).
Structural genomics on drug targets
Structural genomics analysis reveals the SARS-CoV-2 genome consisting of four structural proteins (S–E–M–N) and two nonstructural proteins (main protease and RdRp) (Kandeel et al. 2020). Spike, main protease (3CLpro), papain-like protease (PLpro), and RdRp are promising therapeutic targets for SARS-CoV infections (Li and De Clercq 2020; Yoshimoto 2020). The structural and functional characteristics of these proteins are summarized here and represented in Table 3. Viral exoribonuclease greatly affects the cell’s transcriptome by RNA processing and degradation.
Spike glycoprotein
SARS-CoV-2 infects ciliated bronchial epithelial cells and type-II pneumocytes where an envelope-anchored spike protein binds to a host receptor angiotensin-converting enzyme 2 (ACE2) (Lan et al. 2020; Li et al. 2020e; Zhou et al. 2020a). The spike protein is cleaved via acid-dependent proteolysis by furin protease into an N-terminal S1 subunit (residue 14–685) and a C-terminal S2 region (residue 686–1273), followed by fusion of the viral envelop to the cellular membranes (Coutard et al. 2020). It has a functional polybasic (furin) cleavage site at the S1–S2 boundary through the insertion of 12 nucleotides. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2. Insertion of a furin cleavage site at the S1–S2 junction enhances cell–cell fusion without affecting viral entry (Hasan et al. 2020; Rabaan et al. 2020; Walls et al. 2020).The function of the predicted O-linked glycans could create a ‘mucin-like domain’ that helps SARS-CoV-2 to utilize mucin-like domains as glycan shields involved immunoevasion (Bagdonaite and Wandall 2018).
Crystallographic studies reveal the structure of the six-helical bundle core of the HR1 and HR2 domains in the S2 subunit (Fig. 2). It has a defined RBD in an N-terminal S1 subunit that specifically recognizes its receptor in humans (Lan et al. 2020; Lu et al. 2020; Xia et al. 2020). The RBD contains a core structure and a receptor-binding motif (RBM) that binds to the ACE2 receptor-binding region, integrin (Cagliani et al. 2020; Sigrist et al. 2020). The involvement of glucose-regulated protein-78 and RBD are important in recognition of the host ACE2 receptor. The binding is more favorable between regions III (C391–C525) and IV (C480–C488) of the spike protein and glucose-regulated protein-78 (Ibrahim et al. 2020).

3D structural view for the representation of SARS-CoV-2 spike glycoprotein (a) in complex with ACE2 (b) and sodium-dependent neutral amino acid transporter (c)
The binding efficiency of RBD on ACE2 can be enhanced by the presence of specific amino acids at the 442, 472, 479, 480, and 487 positions. Gln493 and Asn501 residues in the RBM provide possible interactions with ACE2. SARS-CoV-2 has acquired some capacity for human cell infection and human-to-human transmission by super binding affinity of Gln493 and Asn501 residues in RBM with ACE2 (Wan et al. 2020). It is most likely the result of natural selection on a human (Andersen et al. 2020; Zhang et al. 2020a).
The RBM recognizes directly the integrins of the ACE2 receptor-binding region and RBD has a critical impact on the cross-reactivity of neutralizing antibodies. It suggests a promising therapeutic target. EK1C4 is the most potent pan-coronavirus fusion inhibitor targeting its spike protein (Xia et al. 2020). ACE2 blocker is repurposed to control COVID-19 from gaining entry into the host cell (Phadke and Saunik 2020). Therefore, it is still necessary to develop novel monoclonal antibodies targeting RBD and fusion inhibitors targeting the S2 subunit, which will be effective therapeutics against COVID-19 (Sigrist et al. 2020; Shanmugaraj et al. 2020; Tian et al. 2020).
RNA‐dependent RNA polymerase
The RdRp is a crucial viral enzyme in the life cycle of RNA viruses that catalyzes the replication of RNA from the RNA template. ORF1ab gene-encoding polyprotein can be cut by the main protease of the virus to form RdRp and helicase (Yang and Wang 2020). The structural genomics studies found the active site of this enzyme highly conserved with two successive and surface-accessible aspartates in a beta-turn structure (Elfiky 2020a, 2020b). It has conserved polymerase motifs A–G in the palm domain having an active site chamber. It has a unique N-terminal β-hairpin at its N-terminal (Gao et al. 2020b). Crystallographic structure (PDB ID: 6M71) and homology model of this enzyme offer a way to find efficient RdRp inhibitors to treat COVID-19 (Elfiky 2020a) (Fig. 3). Molecular docking studies found theaflavin as a potential RdRp inhibitor that binds to the catalytic pocket of RdRp (−9.11 kcal/mol) (Lung et al. 2020). Hydrophobic and π-cation interactions are contributed significantly to binding between theaflavin and Arg553 side chain of RdRp (Lung et al. 2020). Several rapid drug repurposing efforts have been made for the identification of promising antiviral drugs (Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir) against COVID-19 (Elfiky 2020b; Gordon et al. 2020; Reina 2020; Shah et al. 2020; Wang et al. 2020a). Hence, drug repurposing strategy is an effective one to discover the FDA-approved drugs targeting RdRp.

3D structural view for the representation of SARS-CoV-2 proteins targeting for drug designing process
Papain-like protease
This enzyme helps to assemble a virally induced cytoplasmic double-membrane vesicle along with Nsp4 and then cleaves at the N-terminus of the replicase polyprotein for viral replication. It also removes ubiquitin and IFN-stimulated gene 15 from cellular proteins through the processing of Lys-48- and Lys-63-linked polyubiquitin chains (Clasman et al. 2020). It can be antagonized IFN-I induced innate immunity and also prevented host NF-kappa-B signaling by blocking subsequent nuclear translocation of host IFN regulatory factor-3. The structural information of this enzyme provides new insights as a valuable target for drug repositioning to treat SARS-CoV-2 infections (Wu et al. 2020). Some potential inhibitors targeting papain-like protease are identified by using a virtual screening approach using the CDOCKER program (Ma et al. 2020).
Main protease
The main protease (3CL-PRO) is a cysteine protease responsible for the processing of ORF1ab polyprotein at 11 sites. It recognizes a motif sequence [ILMVF]-Q-[SGACN] in the ORF1ab polyprotein and also binds an ADP-ribose-1-phosphate moiety. As shown in Fig. 3, a crystal structure of this enzyme contains six-stranded antiparallel β-barrels harboring a substrate-binding site located between chymotrypsin-3C protease-like domains I and picornavirus 3C protease-like domains II (Zhang et al. 2020b). Like SARS-CoV, a globular cluster of five helices is located in domain III (residues 198–303) regulating the dimerization process through a salt-bridge interaction between Glu290 of one protomer and Arg4 of the other (Shi and Song 2006; Chen et al. 2020; Chatterjee et al. 2020). Covalent main protease inhibitors are targeting the catalytic dyad (His41 and Cys145) of this enzyme (Paasche et al. 2014; Li et al. 2020c). The α-ketoamide 13b, the main protease inhibitor binds to the shallow substrate-binding site. A thiohemiketal is formed in a reversible reaction through the nucleophilic attack of the catalytic Cys145 onto the α-keto group of α-ketoamide 13b (Zhang et al. 2020b).
Several inhibitors include paritaprevir, raltegravir, velpatasvir, ledipasvir, vinyl sulfone, lopinavir, and ritonavir are identified as repurposable drugs for inhibiting 3C-like protease of SARS-CoV-2 (Chen et al. 2020; Cao et al. 2020; Morse et al. 2020; Nutho et al. 2020; Khan et al. 2020a). The FDA has already approved these antivirals for the systematic treatment of chronic hepatitis C virus and human immunodeficiency virus infections (De Clercq and Li 2016). Jeon et al. (2020) have screened and identified two FDA-approved drugs such as niclosamide and ciclesonide against SARS-CoV-2. However, no structural based inhibitors against SARS-CoV-2 infection are not yet completed a clinical trial to date (Li et al. 2020c; Liu et al. 2020a; Xu et al. 2020b).
Helicase
SARS-CoV-2 helicase is an Mg2+-dependent enzyme with a zinc-binding domain in N-terminus displaying RNA and DNA duplex-unwinding activities with 5′–3′ polarity. The putative recombination patterns identified in helicase of SARS-CoV-2 are important for evolutionary survival allowing for genotype adjustment and adaptations in rapidly changing environments (Rehman et al. 2020). Helicase is considered a better choice for the development of molecular diagnostic kits than antivirals (Chan et al. 2020b; Prajapat et al. 2020).
Exoribonuclease
The nonstructural protein Nsp15 encodes a nidoviral uridylate-specific endoribonuclease (NendoU). Like other coronaviruses, it plays an important role in viral infection and pathogenesis by interacting with the Nsp7/Nsp8 complex (Zhang et al. 2018). The high-resolution crystal structure of endoribonuclease Nsp15/NendoU from SARS-CoV-2 was solved and described its catalytic domain and binding sites, which provide structural and functional evidence for developing antiviral drugs (Kim et al. 2020).
2′-O-Methyltransferase
The nonstructural protein Nsp16 encodes cap-specific mRNA 2′-O-methyltransferase responsible for the RNA cap formation and methylation process of SARS-CoV-2. It binds to the N7-methyl guanosine cap and methylates the ribose 2′-O position of the first and second nucleotide of viral mRNA, which is essential to evade the immune system. Dolutegravir and bictegravir are important inhibitors of 2′-O-ribose methyltransferase identified by using molecular docking simulations (Khan et al. 2020a).
Envelope protein
Envelope protein from SARS-CoV-2 acts as a viroporin and self-assembles in host membranes forming ion channels. Envelope protein plays a central role in virus morphogenesis and assembly. It also induces apoptosis and IL-1β overproduction in the host cells, leading to pathogenesis. The envelope protein of SARS-CoV-2 is evolutionarily conserved with higher gene expression efficiency in the hosts (Kandeel et al. 2020; Zhou et al. 2020b). Generally, viral envelope proteins act as therapeutic targets for vaccine designing and engineering.
Nucleocapsid protein
The helical nucleocapsid interacts with spike, envelope, and membrane proteins to form the assembled virion (Zumla et al. 2016). The N-terminal RNA-binding domain of nucleocapsid protein (PDB ID: 6M3M) was purified and the crystallographic structure solved for further target-based drug discovery (Fig. 3). The structure, as well as the function of this protein, was studied and then tested its interaction with various phytochemicals for identification suitable drug candidates (Gupta et al. 2020). Still, more study on this target is needed for further drug discovery to treat COVID-19.
Development of anti-SARS-CoV-2 drugs
Among therapeutic strategies, systemic corticosteroids and unselective or inappropriate administration of antibiotics for the treatment of COVID-19 are not recommended. There is no promising antiviral treatment is currently available. The WHO has approved several medications for the treatment of COVID-19 patients (Wang et al. 2020a) (Table 4). They are: lopinavir/ritonavir, chloroquine, hydroxychloroquine, and α-interferon. The development of antivirals that specific to the SARS-CoV-2 is more effective for the current treatment of the COVID-19 pandemic. Molecular modeling studies have been predicted the protein’s structures from SARS-CoV-2 and also identified numerous antivirals (Cortegiani et al. 2020; Nabirotchkin et al. 2020; Ghosh et al. 2020). The constructed structural genomics and interactomics roadmaps could describe the molecular mechanisms behind the viral infection to design a vaccine or antivirals against COVID-19 (Srinivasan et al. 2020). Current knowledge of the SARS-CoV-2 genomic information will thus advance the further development of anti-SARS-CoV-2 drugs.
Structure-based drug designing
Conventional docking is a too computationally expensive and slow process for structure-based drug designing (Chellapandi and Prisilla 2018; Prathiviraj et al. 2019; Bharathi and Chellapandi 2019; Prathiviraj and Chellapandi 2020b). To address this challenge, a deep docking platform has been developed for the accelerated screening of large chemical libraries, consisting of billions of entities (Liu et al. 2020c; Ton et al. 2020). This platform utilizes quantitative structure–activity relationship models to screen out the top 1000 compounds targeting the main protease of which ZINC000541677852 predicted as a potent SARS-CoV-2 protease inhibitor (docking score −11.32 kcal/mol). Hydroxychloroquine is reported as a safe and effective drug against SARS-CoV-2 infection in vitro. Molecular docking combined with the physiologically based pharmacokinetic models has been used to identify its anti-SARS-CoV-2 activity (Yao et al. 2020). Hydroxychloroquine can also significantly inhibit the main protease and also decrease the production of cytokines during SARS-CoV-2 infection (Kandeel and Al-Nazawi 2020; Liu et al. 2020b). The crystal structures of SARS-CoV-2 main protease in complex with a SARS-CoV-inhibitor N3 and a potent broad-spectrum non-covalent inhibitor X77 were solved and deposited in the protein databank (PDB ID: 6LU7; 6W63). A docking view of the interaction between inhibitors (N3 and X77) and SARS-Cov-2 main protease is depicted in Fig. 4a.

3D structural view for the representation of SARS-CoV-2 target proteins in complex with main protease inhibitors (a) and human antibody CR3022 (b). The target protein is represented as a cartoon model (green) and inhibitors are represented as a ball and stick model
Structure-based vaccine designing
The structural and functional characterization of spike glycoprotein, polyprotein, envelope protein, and nucleocapsid protein is important for vaccine development. A crystal structure of the SARS-CoV-2 RBD in complex with human antibody CR3022 (PDB ID: 6W41) was solved recently, which guides vaccine designing or epitope engineering for COVID-19 (Fig. 4b). The immunoinformatics approach was used to identify significant cytotoxic T lymphocyte and B cell epitopes in the SARS-CoV-2 surface glycoprotein, which facilitates vaccine design of high priority (Baruah and Bose 2020; Grifoni et al. 2020). A similar approach has been applied to propose a specific synthetic vaccine epitope and peptidomimetic agent against SARS-CoV-2 based on the sequence motif KRSFIEDLLFNKV and surrounding variations (Robson 2020).
Systems pharmacology for drug repurposing
Systems biology offers considerable promise in uncovering novel pathways by which microbial pathogens interact with host PPI networks to mediate disease severity (Chellapandi et al. 2019; Murugan et al. 2019; Prathiviraj and Chellapandi 2020a). Systems pharmacology attempts to model the effects of drug action by simultaneously modulating multiple proteins in a network. This approach can identify cellular antiviral targets from virus-host PPIs for the development of effective treatments for viral infections. The drug-gene-metabolism-disease network models are used to identify effective repurposable drugs and drug combinations for viral infections by quantification of the interplay between the virus-host interactome and drug targets in the human PPIs network. The potential drug combination is rationally predicted from the topological relationship between two drug-target modules that reflects biological and pharmacological relationships. The suitable drug targets and repurposed drugs are evaluated with molecular docking and validated with drug-induced gene signatures and virus-induced transcriptomics in human cell lines. Thus, drug repurposing comes into sight as a novel drug discovery process to yield efficient therapies against COVID-2019 rapidly due to time consuming, safety profiles, clinical trials, and FDA approvals of newly discovery drugs (Ahn et al. 2020; Ke et al. 2020; Tu et al. 2020).
Several repurposed drugs have already been tested against COVID-2019 (Harrison 2020). Systems pharmacology-based network medicine platform has been developed for the identification of disease-related genes, whether using genomic data and expression data or data directly collected from the scientific literature (Fu et al. 2020; Zhou et al. 2020a). It is also used for the rapid identification of candidate repurposable drugs and potential drug combinations targeting SARS-CoV-2 using disease-related molecular networks. A systematic drug repurposing approach has been developed to identify promising inhibitors (Raltegravir, Paritaprevir, Bictegravir, and Dolutegravir) against 3C-like protease and 2′-O-ribose methyltransferase from SARS-CoV-2 (Khan et al. 2020a). The SCAR protocol has been developed to repurpose 6 covalent drugs targeting the main protease (3CLpro) of SARS-CoV-2 (Liu et al. 2020c). The proteomic-chemoinformatic approach has been developed for the detection of 66 druggable human proteins or host factors targeted by 69 existing FDA-approved drugs or compounds being investigated (Gordon et al. 2020).
Conclusions
The outbreak of COVID-19 poses a serious threat to global public health and local economies. The future evolution, adaptation, and spread of this virus warrant urgent investigation. It is imperative to discover affordable drugs to control and diminish the pandemic. The present review will provide new insights for those drugs currently ongoing clinical studies and also possible new strategies for drug designing or repositioning to treat SARS-CoV-2 infections. Compared to de novo drug discovery and randomized clinical trials, drug repurposing strategy could significantly shorten the time and reduce the cost of antiviral drugs. Nevertheless, experimental approaches for drug repurposing are costly and time consuming. It can be resolved by systems pharmacology approaches because targeting single virus proteins often has a high risk of drug resistance by the rapid evolution of virus genomes. Perhaps, the repositioning of launched or even failed drugs to viral diseases will provide unique translational opportunities. Genome-scale systems biological perspective of SARS-CoV-2-host interactions can guide the further in vitro and in vivo clinical studies on validation of newly identified drugs for COVID-19 infectivity. Genomic insights into developing and deploying drugs of antiviral drugs for COVID-19 will offer a substantially higher probability of success to the market and a significantly reduced cost and timeline to clinical availability. The currently established animal models are not very promising for the studies of pathogenesis and treatment of COVID-19 and therefore, testing the drugs for COVID-19 requires suitable animal models before their use in humans.
References
Ahn DG, Shin HJ, Kim MH, Lee S, Kim HS, myoung J, Kim BT, Kim SJ (2020) Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease 2019 (COVID-19). J Microbiol Biotechnol 30:313–324
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med. https://doi.org/10.1038/s41591-020-0820-9
Angeletti S, Benvenuto D, Bianchi M, Giovanetti M, Pascarella S, Ciccozzi M (2020) COVID-2019: the role of the nsp2 and nsp3 in its pathogenesis. J Med Virol. https://doi.org/10.1002/jmv.25719
Arabi YM, Deeb A, Al-Hameed, Mandourah Y, Almekhlafi GA, Sindi AA, Al-Omari A, Shalhoub S, Mady A, Alraddadi B, Almotairi A, Al Khatib K, Abdulmomen A, Qushmaq I, Solaiman O, Al-Aithan AM, Al-Raddadi R, Ragab A, Al Harthy A, Kharaba A, Jose J, Dabbagh T, Fowler RA, Balkhy HH, Merson L, Hayden FG; Saudi Critical Care Trials group (2019) Macrolides in critically ill patients with Middle East Respiratory Syndrome. Int J Infect Dis 81:184–190.
Ashour HM, Elkhatib WF, Rahman MM, Elshabrawy HA (2020) Insights into the recent 2019 novel coronavirus (SARS-CoV-2) in light of past human coronavirus outbreaks. Pathogens 9:186
Bagdonaite I, Wandall HH (2018) Global aspects of viral glycosylation. Glycobiology 28:443–467
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: TIssue Distribution, Host-virus Interaction, and Proposed Neurotropic Mechanisms. ACS Chem Neurosci 11:995–998
Banerjee AK, Begum F, Ray U (2020) Mutation hot spots in Spike protein of COVID-19. Preprints 2020:2020040281. https://doi.org/10.20944/preprints202004.0281.v1
Baruah V, Bose S (2020) Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J Med Virol 92:495–500
Bharathi M, Chellapandi P (2019) Comparative analysis of differential proteome-wide protein-protein interaction network of Methanobrevibacter ruminantium M1. Biochem Biophysics Rep 20:100698
Bösl K, Ianevski A, Than TT, Andersen PI, Kuivanen S, Teppor M, Zusinaite E, Dumpis u, Vitkauskiene A, Cox RJ, Kallio-Kokko H, Bergqvist A, Tenson T, Merits A, Oksenych V, Bjoras M, Anthonsen MW, Shum D, Kaarbo M, Vapalahti O, Windisch MP, Superti-Furga G, Snijder B, Kainov D, Kandasamy RK (2019) Common nodes of virus-host interaction revealed through an integrated network analysis. Front Immunol 10:2186
Cagliani R, Forni D, Clerici M, Sironi M (2020) Computational inference of selection underlying the evolution of the novel coronavirus, SARS-CoV-2. J Virol. https://doi.org/10.1128/JVI.00411-20
Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM (2020) The FDA-approved drug Ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res 104787
Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, Ruan L, Song B, Cai Y, Wei M, Li X, Xia J, Chen N, Xiang J, Yu T, Bai T, Xie X, Zhang L, Li C, Yuan Y, Chen H, Li H, Huang H, Tu S, Gong F, Liu Y, Wei Y, Dong C, Zhou F, Gu X, Xu J, Liu Z, Zhang Y, Li H, Shang L, Wang K, Li K, Zhou X, Dong X, Qu Z, Lu S, Hu X, Ruan S, Luo S, Wu J, Peng L, Cheng F, Pan L, Zou J, Jia C, Wang J, Liu X, Wang S, Wu X, Ge Q, He J, Zhan H, Qiu F, Guo L, Huang C, Jaki T, Hayden FG, Horby PW, Zhang D, Wang C (2020) A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2001282
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R (2020). Features, evaluation and treatment coronavirus (COVID-19). In: StatPearls. StatPearls Publishing, Treasure Island, FL
Ceraolo C, Giorgi FM (2020) Genomic variance of the 2019-nCoV coronavirus. J Med Virol 92:522–528
Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, Yuen KY (2020a) Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9:221–236
Chan JF, Yip CC, To KK, Tang TH, Wong SC, Leung k, Fung AY, Ng AC, Zou Z, Tsoi h, Choi GK, Tam AR, Cheng VC, Chan KH, Tsang OT, Yuen K (2020b) Improved molecular diagnosis of COVID-19 by the novel, highly sensitive and specific COVID-19-RdRp/Hel real-time reverse transcription-polymerase chain reaction assay validated in vitro and with clinical specimens. J Clin Microbiol. https://doi.org/10.1128/JCM.00310-20
Chatterjee S, Maity A, Chowdhury S, Islam MA, Muttinini RK, Sen D (2020) In silico analysis and identification of promising hits against 2019 novel coronavirus 3C-like main protease enzyme. J Biomol Struct Dyn. 1-14
Chellapandi P, Prisilla A (2018) Clostridium botulinum type A-virulome-gut interactions: a systems biology insight. Human Microbiome. Hum Microb J 7:15–22
Chellapandi P, Prathiviraj R, Prisilla A (2019) Deciphering structure, function and mechanism of Plasmodium IspD homologs from their evolutionary imprints. J Comput Aided Mol Des 33:419–436
Chen YW, Yiu CB, Wong KY (2020) Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL pro) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Res 9:129
Clasman JR, Everett RK, Srinivasan K, Mesecar AD (2020) Decoupling deISGylating and deubiquitinating activities of the MERS virus papain-like protease. Antivir Res 174:104661
Cleemput S, Dumon W, Fonseca V, Karim WA, Giovanetti M, Alcantara LC, Deforche K, de Oliveira T (2020) Genome detective coronavirus typing tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa145
Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S (2020) A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J Crit Care 57:279–283
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E (2020) The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res 176:104742
De Clercq E, Li G (2016) Approved antiviral drugs over the past 50 years. Clin Microbiol Rev 29:695–747
Elfiky AA (2020a) Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci 248:117477
Elfiky AA (2020b) Ribavirin, remdesivir, sofosbuvir, galidesivir, and tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci 253:117592
Fahmi M, Kubota Y, Ito M (2020) Nonstructural proteins NS7b and NS8 are likely to be phylogenetically associated with evolution of 2019-nCoV. Infect Genet Evol 81:104272
Forster P, Forster L, Renfrew C, Forster M (2020) Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci USA 117:9241–9243
Fox D (2020) What you need to know about the Wuhan coronavirus. Nature. https://doi.org/10.1038/d41586-020-00209-y
Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS (2007) Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol 81:9812–9824
Fu Y, Cheng Y, Wu Y (2020) Understanding SARS-CoV-2-mediated inflammatory responses: from mechanisms to potential therapeutic tools. Virol Sin. https://doi.org/10.1007/s12250-020-00207-4
Gao J, Zhenxue T, Yang X (2020a) Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci Trends 14:72–73
Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, Wang T, Sun Q, Ming Z, Zhang L, Ge J, Zheng L, Zhang Y, Wang H, Zhu Y, Zhu C, Hu T, Hua T, Zhang B, Yang X, Li J, Yang H, Liu Z, Xu W, Guddat LW, Wang Q, Lou Z, Rao Z (2020b) Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 368:779–782
Ghosh AK, Brindisi M, Shahabi D, Chapman ME, Mesecar AD (2020) Drug development and medicinal chemistry efforts toward SARS-Coronavirus and Covid-19 therapeutics. ChemMedChem 15:907–932
Goh GK, Dunker AK, Foster JA, Uversky VN (2020) Rigidity of the outer shell predicted by a protein intrinsic disorder model sheds light on the COVID-19 (Wuhan-2019-nCoV) infectivity. Biomolecules 10:331
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, O’Meara MJ, Guo JZ, Swaney DL, Tummino TA, Hüttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Naing ZZC, Zhou Y, Peng S, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Shen W, Shi Y, Zhang Z, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Ramachandran R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Lin Y, Wankowicz SA, Bohn M, Trenker R, Young JM, Cavero D, Hiatt J, Roth T, Rathore U, Subramanian A, Noack J, Hubert M, Roesch F, Vallet T, Meyer B, White KM, Miorin L, Agard D, Emerman M, Ruggero D, García-Sastre A, Jura N, Zastrow MV, Taunton J, Schwartz O, Vignuzzi M, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor S, Fraser JS, Gross J, Sali A, Kortemme T, Beltrao P, Shokat K, Shoichet BK, Krogan NJA (2020) SARS-CoV-2-Human protein-protein interaction map reveals drug targets and potential drug repurposing. bioRxiv preprint. https://doi.org/10.1101/2020.03.22.002386
Grifoni A, Sidney J, Zhang Y, Scheuermann RH, Peters B, Sette A (2020) A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 27:671–680
Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, Tan KS, Wang DY, Yan Y (2020) The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil Med Res 7:11
Gupta MK, Vemula S, Donde R, Gouda G, Behera L, Vadde R (2020) In-silico approaches to detect inhibitors of the human severe acute respiratory syndrome coronavirus envelope protein ion channel. J Biomol Struct Dyn 1–17. https://doi.org/10.1080/07391102.2020.1751300
Harrison C (2020) Coronavirus puts drug repurposing on the fast track. Nat Biotechnol. https://doi.org/10.1038/d41587-020-00003-1.
Hasan A, Paray BA, Hussain A, Qadir FA, Attar F, Aziz FM, Sharifi M, Derakhshankhah H, Rasti B, Mehrabi M, Shahpasand K, Saboury AA, Falahati M (2020) A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J Biomol Struct Dyn 1–9. https://doi.org/10.1080/07391102.2020.1754293.
Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, Aziz B (2020) Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. https://doi.org/10.1002/jmv.25832
Ibrahim IM, Abdelmalek DH, Elshahat ME, Elfiky AA (2020) COVID-19 spike-host cell receptor GRP78 binding site prediction. J Infect 80:554–562
Jeon S, Ko M, Lee J, Choi I, Byun SY, Park S, Shum D, Kim S (2020) Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob Agents Chemother 64:e00819–20.
Kandeel M, Al-Nazawi M (2020) Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci 117627. https://doi.org/10.1016/j.lfs.2020.117627
Kandeel M, Ibrahim A, Fayez M, Al-Nazawi M (2020) From SARS and MERS CoVs to SARS-CoV-2: moving toward more biased codon usage in viral structural and nonstructural genes. J Med Virol 92:660–666
Ke YY, Peng TT, Yeh TK, Huang WZ, Chang SE, Wu SH, Hung HC, Hsu TA, Lee SJ, Song JS, Lin WH, Chiang TJ, Lin JH, Sytwu HK, Chen CT (2020) Artificial intelligence approach fighting COVID-19 with repurposing drugs. Biomed J. https://doi.org/10.1016/j.bj.2020.05.001
Khan RJ, Jha RK, Amera GM, Jain M, Singh E, Pathak A, Singh RP, Muthukumaran J, Singh AK (2020a) Targeting SARS-CoV-2: a systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2’-O-ribose methyltransferase. J Biomol Struct Dyn 1–14. https://doi.org/10.1080/07391102.2020.1753577
Khan S, Siddique R, Shereen MA, Ali A, Liu J, Bai Q, Bashir N, Xue M (2020b) The emergence of a novel coronavirus (SARS-CoV-2), their biology and therapeutic options. J Clin Microbiol 58. https://doi.org/10.1128/JCM00187-20
Kim Y, Jedrzejczak R, Maltseva NI, Wilamowski M, Endres M, Godzik A, Michalska K, Joachimiak A (2020) Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Sci 29:1596–1605. https://doi.org/10.1002/pro.3873
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, de Silva TI, McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, Montefiori DC; Sheffield COVID-19 Genomics Group (2020) Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. https://doi.org/10.1016/j.cell.2020.06.043
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X (2020) Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–220
Li G, De Clercq (2020) Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov 19:149–150
Li H, Zhou Y, Zhang M, Wang H, Zhao Q, Liu J (2020b) Updated Approaches against SARS-CoV-2. Antimicrob Agents Chemother 64:e00483–20
Li JY, You Z, Wang Q, Zhou ZJ, Qiu Y, Luo R, Ge XY (2020a) The epidemic of 2019-novel-coronavirus (2019-nCoV) pneumonia and insights for emerging infectious diseases in the future. Microbes Infect 22:80–85
Li X, Wang W, Zhao X, Zai J, Zhao Q, Li Y, Chaillon A (2020c) Transmission dynamics and evolutionary history of 2019-nCoV. J Med Virol 92:501–511
Li Y, Zhang J, Wang N, Li H, Shi Y, Guo G, Liu K, Zeng H, Zou Q (2020d) Therapeutic drugs targeting 2019-nCoV main protease by high-throughput screening. bioRxiv Preprint. https://doi.org/10.1101/2020.01.28.922922
Li Y, Zhou W, Yang L, You R (2020e) Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharm Res 157:104833
Licastro D, Rajasekharan S, Dal Monego S, Segat L, D’Agaro P, Marcello A (2020) Isolation and full-length genome characterization of SARS-CoV-2 from COVID-19 cases in Northern Italy. J Virol 94:e00543–20. https://doi.org/10.1128/JVI.00543-20
Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, Li Y, Hu Z, Zhong W, Wang M (2020b) Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov 6:16
Liu S, Zheng Q, Wang Z (2020c) Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics. https://doi.org/10.1093/bioinformatics/btaa224
Liu X, Zhang B, Jin Z, Yang H, Rao Z (2020a) The crystal structure of COVID-19 main protease in complex with an inhibitor N3. PDB https://doi.org/10.2210/pdb6lu7/pdb
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574
Luan J, Lu Y, Jin X, Zhang L (2020) Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem Biophys Res Commun 526:165–169
Lung J, Lin YS, Yang YH, Chou YL, Shu LH, Cheng YC, Liu HT, Wu CY (2020) The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J Med Virol 92:693–697
Ma J, Huo XQ, Chen X, Zhu WX, Yao MC, Qiao YJ, Zhang YL (2020) Study on screening potential traditional Chinese medicines against 2019-nCoV based on Mpro and PLP. Zhongguo Zhong Yao Za Zhi 45:1219–1224
Morse JS, Lalonde T, Xu S, Liu WR (2020) Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem 21:730–738
Murugan R, Prathiviraj R, Mothay Dipti, Chellapandi P (2019) Substrate-imprinted docking of Agrobacterium tumefaciens uronate dehydrogenase for increased substrate selectivity. Int J Biol Macromol 140:1214–1225
Nabirotchkin S, Peluffo AE, Rinaudo P, Yu J, Hajj R, Cohen D (2020) Next-generation drug repurposing using human genetics and network biology. Curr Opin Pharmacol 11:1–15.
Nutho B, Mahalapbutr P, Hengphasatporn K, Pattaranggoon NC, Simanon N, Shigeta Y, Hannongbua S, Rungrotmongkol T (2020) Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry 59:1769–1779
Ortega JT, Serrano ML, Pujol FH, Rangel HR (2020) Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. EXCLI J 19:410–417
Paasche A, Zipper A, Schäfer S, Ziebuhr J, Schirmeister T, Engels B (2014) Evidence for substrate binding-induced zwitter ion formation in the catalytic Cys-His dyad of the SARS-CoV main protease. Biochemistry 53:5930–5946
Pachetti M, Marini B, Benedetti F, Giudici F, Mauro E, Storici P, Masciovecchio C, Angeletti S, Ciccozzi M, Gallo RC, Zella D, Ippodrino R (2020) Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med 18:179
Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, Tsiodras S (2020) Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol 79:104212
Phadke M, Saunik S (2020) COVID-19 treatment by repurposing drugs until the vaccine is in sight. Drug Dev Res. https://doi.org/10.1002/ddr.21666
Phan T (2020) Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol 81:104260
Prajapat M, Sarma P, Shekhar N, Avti P, Sinha S, Kaur H, Kumar S, Bhattacharyya A, Kumar H, Bansal S, Medhi B (2020) Drug targets for corona virus: A systematic review. Indian J Pharmacol 52:56–65
Prathiviraj R, Chellapandi P (2020a) Deciphering molecular virulence mechanism of Mycobacterium tuberculosis Dop isopeptidase based on its sequence-structure-function link. Protein J 39:33–45
Prathiviraj R, Chellapandi P (2020b) Modelling a global regulatory network of Methanothermobacter thermautotrophicus strain ΔH. Netw Model Anal Health Inf Bioinform 9:17
Prathiviraj R, Berchmans Sheela, Chellapandi P (2019) Analysis of modularity in proteome-wide protein interaction networks of Methanothermobacter thermautotrophicus strain ΔH across metal-loving bacteria. J Proteins Proteom 10:179–190
Prompetchara E, Ketloy C, Palaga T (2020) Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac J Allergy Immunol 38:1–9
Rabaan AA, Al-Ahmed SH, Haque S, Sah R, Tiwari R, Malik YS, Dhama K, Yatoo MI, Bonilla-Aldana DK, Rodriguez-Morales AJ (2020) SARS-CoV-2, SARS-CoV, and MERS-COV: a comparative overview. Infez Med 28:174–184
Ralph R, Lew J, Zeng T, Francis M, Xue B, Roux M, Toloue Ostadgavahi A, Rubino S, Dawe NJ, Al-Ahdal MN, Kelvin DJ, Richardson CD, Kindrachuk J, Falzarano D, Kelvin AA (2020) 2019-nCoV (Wuhan virus), a novel coronavirus: human-to-human transmission, travel-related cases, and vaccine readiness. J Infect Dev Ctries 14:3–17
Rehman SU, Shafique L, Ihsan A, Liu Q (2020) Evolutionary trajectory for the emergence of novel coronavirus SARS-CoV-2. Pathogens 9:240
Reina J (2020) Remdesivir, the antiviral hope against SARS-CoV-2. Rev Esp Quimioter 33:176–179
Robson B (2020) Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput Biol Med 119:103670
Rothan HA, Byrareddy SN (2020) The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimmun 109:102433
Shah B, Modi P, Sagar SR (2020) In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci 252:117652
Shanmugaraj B, Siriwattananon K, Wangkanont K, Phoolcharoen W (2020) Perspectives on monoclonal antibody therapy as potential therapeutic intervention for Coronavirus disease-19 (COVID-19). Asian Pac J Allergy Immunol 38:10–18
Shi J, Song J (2006) The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J 273:1035–1045
Sigrist CJ, Bridge A, Le Mercier P (2020) A potential role for integrins in host cell entry by SARS-CoV-2. Antivir Res 177:104759
Singhal T (2020) A review of coronavirus disease-2019 (COVID-19). Indian J Pediatr 87:281–286
Sohrabi C, Alsafi Z, O’Neill N, Khan M, Kerwan A, Al-Jabir A, Iosifidis C, Agha R (2020) World Health Organization declares global emergency: a review of the 2019 novel coronavirus (COVID-19). Int J Surg 76:71–76
Srinivasan S, Cui H, Gao Z, Liu M, Lu S, Mkandawire W, Narykov O, Sun M, Korkin D (2020) Structural genomics of SARS-CoV-2 indicates evolutionary conserved functional regions of viral proteins. Viruses 12:360
Tian X, Li C, Huang A, Xia S, Lu S, Shi Z, Lu L, Jiang S, Yang Z, Wu Y, Ying T (2020) Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect 9:382–385
Ton AT, Gentile F, Hsing M, Ban F, Cherkasov A (2020) Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 Billion compounds. Mol Inform. https://doi.org/10.1002/minf.202000028
Tu YF, Chien CS, Yarmishyn AA, Lin YY, Luo YH, Lin YT, Lai WY, Yang DM, Chou SJ, Yang YP, Wang ML, Chiou SH (2020) A review of SARS-CoV-2 and the ongoing clinical trials. Int J Mol Sci 21:2657
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–292
Wan Y, Shang J, Graham R, Baric RS, Li F (2020) Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 94:e00127–20. https://doi.org/10.1128/JVI.00127-20
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G (2020a) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen KY, Wang Q, Zhou H, Yan J, Qi J (2020b) Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 181:894–904.e9. https://doi.org/10.1016/j.cell.2020.03.045
Wen F, Yu H, Guo J, Li Y, Luo K, Huang S (2020) Identification of the hyper-variable genomic hotspot for the novel coronavirus SARS-CoV-2. J Infect 80:671–693
Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ (2020) A new coronavirus associated with human respiratory disease in China. Nature 579:265–269
Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, Qi F, Bao L, Du L, Liu S, Qin C, Sun F, Shi Z, Zhu Y, Jiang S, Lu L (2020) Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 30:343–355
Xu X, Han M, Li T, Sun W, Wang D, Fu B, Zhou Y, Zheng X, Yang Y, Li X, Zhang X, Pan A, Wei H (2020a) Effective treatment of severe COVID-19 patients with tocilizumab. Proc Natl Acad Sci USA 117:10970–10975
Xu Z, Peng C, Shi Y, Zhu Z, Mu K, Wang X, Zhu W (2020b) Nelfinavir Was Predicted to Be a Potential Inhibitor of 2019-NCov main protease by an integrative approach combining homology modelling, molecular docking and binding free energy calculation. bioRxiv. https://doi.org/10.1101/2020.01.27.921627
Yang P, Wang X (2020) COVID-19: a new challenge for human beings. Cell Mol Immunol 17:555–557
Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, Liu X, Zhao L, Dong E, Song C, Zhan S, Lu R, Li H, Tan W, Liu D (2020) In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin Infect Dis ciaa237. https://doi.org/10.1093/cid/ciaa237
Yin C (2020) Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics 112:3588–3596
Yoshimoto FK (2020) The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J 39:198–216
Yu H, Wang XC, Li J, Qian X, Yu XF, Sun Z, Chen JF, Kao QJ, Wang HQ, Pan JC. (2020) Genomic analysis of a 2019-novel coronavirus (2019-nCoV) strain in the first COVID-19 patient found in Hangzhou. Zhonghua Yu Fang Yi Xue Za Zhi 54:486-490. Chinese. https://doi.org/10.3760/cma.j.cn112150-20200217-00128
Zhang L, Li L, Yan L, Ming Z, Jia Z, Lou Z, Rao Z (2018) Structural and biochemical characterization of endoribonuclease Nsp15 encoded by middle east respiratory syndrome coronavirus. J Virol 92:e00893–18. https://doi.org/10.1128/JVI.00893-18
Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R (2020b) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368:409–412
Zhang T, Cui X, Zhao X, Wang J, Zheng J, Zheng G, Guo W, Cai C, He S, Xu Y (2020c) Detectable SARS-CoV-2 Viral RNA in feces of three children during recovery period of COVID-19 pneumonia. J Med Virol 92:909–914
Zhang T, Wu Q, Zhang Z (2020a) Probable pangolin origin of SARS-CoV-2 Associated with the COVID-19 outbreak. Curr Biol 30:1346–1351.e2
Zhao WM, Song SH, Chen ML, Zou D, Ma LN, Ma YK, Li RJ, Hao LL, Li CP, Tian DM, Tang BX, Wang YQ, Zhu JW, Chen HX, Zhang Z, Xue YB, Bao YM (2020) The 2019 novel coronavirus resource. Yi Chuan 42:212–221
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL (2020b) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F (2020a) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 6:14
Zumla A, Chan JFW, Azhar EI, Hui DSC, Yuen KY (2016) Coronaviruses-drug discovery and therapeutic options. Nat Rev Drug Discov 15:327–347
Acknowledgements
The authors would like to thank the Science and Engineering Research Board, Department of Science and Technology (MSC/2020/000438), New Delhi, India, for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chellapandi, P., Saranya, S. Genomics insights of SARS-CoV-2 (COVID-19) into target-based drug discovery. Med Chem Res 29, 1777–1791 (2020). https://doi.org/10.1007/s00044-020-02610-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-020-02610-8