
Abstract
Forest management may have significant effects on forest connectivity and natural population sizes. Harvesting old-growth single trees may also change natural patterns of genetic variation and spatial genetic structure. This study evaluated the impacts of forest management using a silvicultural system of seed trees on the genetic diversity and spatial genetic structure of Eremanthus erythropappus (DC.) MacLeish. A complete survey of 275 trees on four plots was undertaken out to compare the genetic variation of a managed stand with an unmanaged stand. We genotyped all adult and juvenile individuals 60 months after the management and compared the genetic diversity and the spatial genetic structure parameters. Genetic diversity was considered high because of an efficient gene flow between stands. There were no genetic differences between stands and no evidence of inbreeding. Genetic clustering identified a single population (K = 1), indicating no genetic differentiation between managed and unmanaged stands. Adult and juvenile individuals of the unmanaged stand were more geographically structured than individuals from the managed one. There was a tendency of coancestry among juveniles at the first class of distance of the managed stand, suggesting a drift of genetic structure possibly caused by management. Understanding early responses to management on genetic diversity and stand structure is a first step to ensuring the effectiveness of conservation practices of tree species. The sustainability of forest management of E. erythropappus on genetic diversity, and more accurately, on spatial genetic structure needs evaluation over time to promote effective conservation of the population size and genetic variability.
Similar content being viewed by others


Introduction
Sustainable forest management uses appropriate silvicultural techniques to produce different goods and services and simultaneously promotes biodiversity conservation (Petrokofsky et al. 2015). The reduction in tree density inherent to forest management practices may result in increased levels of inbreeding and erosion of evolutionary potential (Crnokrak and Roff 1999; Frankham et al. 2002; Quéméré et al. 2010). Among the main impacts, silvicultural management may affect species selection and spatial genetic structure (Hawley et al. 2005; Lacerda et al. 2008). However, although natural regeneration allows for the conservation of genetic information to the next generation, it does not avoid adaptive and non-adaptive changes in genetic structure (Rajora and Pluhar 2003).
Tropical forest regeneration is a natural process that is important for the conservation of ecosystems (Hardwick and Elliot 2015). The maintenance of the regeneration potential is essential to ensure the sustainability of silvicultural management, promote forest succession dynamics, and consequently preserve the natural patterns of resilience in disturbed environments (Ackzell 1993; Folke 2006). Due to the fragility of tropical biomes, it is fundamental to analyze their genetic diversity to define strategies of management and conservation, especially for those species whose distribution determined by high altitudes, normally presenting a high risk of extinction (Avise and Hamrick 1996; Brown and Kappelle 2001).
Forest management practices can modify stand density and age class structure at different developmental stages (Bergeron et al. 2017). Changes in the frequency and distribution of tree species may alter the genetic structure of forest populations (Sjölund and Jump 2015; González-Díaz et al. 2017). Several patterns of genetic response to forest management have been found. The main impacts of thinning on natural populations are related to frequency changes and loss of alleles or alternate gene forms (Schaberg et al. 2008; Ortega et al. 2010). Forest management processes may also commonly cause a loss of alleles between old-growth individuals and juveniles (Soliani et al. 2016), but can also be responsible for enhancing genetic diversity parameters by increasing gene flow (Cloutier et al. 2007; Wickneswari 2011). However, numerous studies have not detected any significant influence on genetic diversity as a result of forest management (Robledo-Arnuncio et al. 2004; Martins et al. 2016; Borrell et al. 2018). Therefore, it is important to detect early genetic responses in different categories of succession to evaluate management sustainability (García-Gil et al. 2015).
Genetic diversity provides the elementary condition for the evolution of tree species (Cavers and Cottrell 2014). The maintenance of natural genetic variation is essential to the continuity and evolution of populations through selective forces (Schaberg et al. 2008). Nevertheless, anthropogenic impacts can disrupt the frequency and distribution of forest genetic resources and may generate severe impacts for species genetic conservation (Young et al. 1996; Dixo et al. 2009). The most common consequences of habitat change induced by human activities are related to variations in pollen and seed dispersal which may influence genetic diversity and spatial genetic structure (SGS) (Ghanzoul 2005). Additionally, temporal environmental variation may decrease reproductive fitness, disease resistance, population adaptation and protect the forest against threats (Telford et al. 2014).
SGS is influenced by gene flow, genetic drift, and selection (Loiselle et al. 1995). Some studies have already found significant effects of silvicultural management on SGS within populations, even when overall levels of variation are maintained, allowing the exploration of the genetic legacy of forest management (Sjölund and Jump 2015; González-Díaz et al. 2017). According to Smouse and Peakall (1999), changes in genetic structure may alter local breeding and the evolution of species. In addition, clonal propagation can also have an intense effect on SGS by improving the density of regeneration and, consequently raising the incidence of clonal individuals (Sjölund and Jump 2013). Pollen and seed dispersal are the main factors responsible for driving gene flow among populations and for driving SGS (Vekemans and Hardy 2004; Cavers et al. 2005). Therefore, sustainable forest management should proactively evaluate the impact of harvesting large trees on tree genetic diversity and the SGS of regenerating individuals (Aravanopoulos 2018).
Eremanthus erythropappus is a species found in Minas Gerais State, Brazil (Scolforo et al. 2012), and is used for fence production because of its high durability and for the extraction of α-bisabolol, an important essential oil highly prized by the pharmaceutical and cosmetic industries (Oliveira et al. 2009). There have been several studies on its structure (Scolforo et al. 2012), growth dynamics (Silva et al. 2008; Scolforo et al. 2015), economic aspects (Cruz 2006; Andrade 2009; Santos et al. 2017), silviculture (Braga 2006; Borrell et al. 2018) and genetic analysis (Pádua et al. 2016; Rocha et al. 2020a).
Further analyses are necessary to evaluate the genetic sustainability as a result of forest management of exploited populations. In this paper, the effect of a silvicultural system of seed trees on the genetic diversity and spatial genetic structure of a managed stand of E. erythropappus in southern Minas Gerais State, Brazil is analyzed. We aim to obtain information that can be used to maintain the genetic variability of the species to evaluate the following hypotheses: (1) that managed stands present similar levels of genetic diversity as unmanaged stands; and (2) that the spatial genetic structure of the regeneration may be affected by thinning. Correlation between the frequency of alleles and the management strategy will be determined. Additionally, the effects of changes on genetic structure as well as the importance of gene flow for maintaining the genetic variability in the managed stand will be analyzed.
Materials and methods
Study site
The study site is located in Itamonte, southern Minas Gerais State. Two stands 300 m apart were designated as managed (22°16′45″S; 22°16′44.6″S) and unmanaged (44°46′24″W; 44°46′25.6″W) (Fig. 1). Before harvesting, the managed stand was composed only of E. erythropappus. The sites are within the Serra da Mantiqueira Environmental Protected Area (APA Serra da Mantiqueira), a mountain range that reaches 2798 m (9180.79 ft). The control or unmanaged stand was selected considering its proximity, environmental relatedness and structural similarity to the managed stand, and had no tree removals.

Location of the managed stand in Itamonte, Minas Gerais state, Brazil
Edaphoclimatic classification of the sampling sites revealed a predominance of humic cambisol soils showing extensive soil compaction with low amounts of organic matter but little vulnerability to environmental contamination (Curi et al. 2008). The climate is temperate humid (Cwa) with dry winters and hot summers (Köppen and Geiger 1928). Average annual precipitation and temperature are approximately 1700 mm and 16 °C, respectively. Stand thinning occurred in 2009 with the removal of 1918 mature trees, leaving the number remaining at 3197 trees ha−1. Standing timber volume before thinning was 165.1 m3 ha−1, with 99.1 m3 ha−1 remaining. The managed stand was scarified immediately after thinning (circles with 60-cm radius distributed throughout the area). The residual stand was thinned 40 months after initial thinning to reduce competition between trees.
Data collection
The sampling intensity was 1.5% of the total area in each stand. Ribeiro (2009), studying the natural regeneration of a managed population of E. erythropappus, also employed a silvicultural system of seed trees, and found 5364 seedlings per hectare 48 months after thinning. Four 100-m2 plots, each 10 m × 10 m, were established 50-m apart inside the managed and unmanaged stands (Fig. 1). Each plot was centered on a seed tree to analyze the occurrence of clonal individuals. However, a maximum distance of 5-m from the original point was identified to maintain the original position between plots. Each plot was subdivided into four 5 m × 5 m subplots to facilitate sampling (Fig. 2).

Sampling at Itamonte, southern Minas Gerais state;dark squares with dots represent plot distribution (1, 2, 3 and 4), and light shaded areas the managed stand
Sampling was carried out January 2014, 60 months post-thinning. Leaf tissues were collected from juvenile cohorts and old-growth mature trees (seed trees) in both managed and unmanaged stands. The sampled trees were classified as adults and juveniles to analyze the genetic diversity parameters. Trees > 1.5 m height and > 5 cm diameter at breast height (DBH) were classified as adults. Trees < 1.5 m in height and < 5 cm in DBH were juveniles.
Based on DBH and height, the two life stages (adults and juveniles) were divided into four ontogenetic stages to analyze SGS: juveniles (seedlings), large juveniles (nonreproductive trees), adults (reproductive adults), and large (reproductive) adults.
Microsatellite analysis
DNA extraction was carried out using the CTAB protocol (Doyle and Doyle 1987), quantified using a NanoVue Plus™ spectrophotometer, and its purity checked using the A260/A280 ratio. Noncorrelated microsatellite primers were used for the PCR process (Ere02, Ere03, Ere04, Ere07, Ere08 and Ere14) developed for E. erythropappus (Rocha et al. 2020b) according to specific conditions (see supplementary Table S1). The total volume of each sample was 15.0 μL: approximately 30 ng of DNA was added to 12.0 μL of reaction mixture (3.33 mM IB Phoneutria buffer consisting of 100 mM Tris–HCl pH 8.4; 500 mM KCl, 1% Triton X-100, 15 mM MgCl2), 1.5 mM MgCl2, 0.28 mM of each dNTP, 1 U Taq polymerase, 0.22 mM each primer (Forward and Reverse) and the final volume was achieved with ultrapure water.
The PCR conditions consisted of 3 min of initiation at 94 °C, followed by 30 cycles of denaturation at 94 °C for 30 s, annealing temperature for 30 s, extension at 72 °C for 1 min and a final extension at 72 °C for 7 min. PCR products were subjected to capillary electrophoresis in an ABI 3730 Automatic Sequencer (Life Technologies, Carlsbad, CA, United States of America) with the GS500 LIZ size standard reference.
Data analysis
Genetic diversity
Fragment analysis was scored using GeneMarker 1.9 V (AppliedBiosystems). The genetic diversity parameters in the managed and unmanaged stands were analyzed to obtain the average number of observed alleles (AO), the average of exclusive alleles (A), the allelic richness (AR), effective (AE) and private (AP) alleles, and the expected (HE) and observed (HO) heterozygosities using GenAlEx 6.501 (Peakall and Smouse 2006). The frequency of null alleles and genotyping errors were analyzed using Micro-Checker v. 2.2.3. (Van Oosterhout et al. 2004).
where FIS was obtained using FSTAT 2.9.3.2 (Goudet 2002) with Bonferroni correction for multiple comparisons.
The differentiation index (FST) between stands and life stages was estimated using ARLEQUIN v 3.5 (Excoffier and Lischer 2010) to analyze the differences in allele frequencies among stands. We assumed that adult and regenerating individuals had the same pattern of mating (Nei 1978) using a distance method and an analysis locus-by-locus with pairwise differences for the polymorphic loci with 1000 permutations was performed. The gene flow among populations was obtained according to Crow and Aoki (1984):
where α = [n/(n – 1)2] and “n” is the number of populations.
Spatial genetic structure
The model-based clustering algorithm was first implemented (Pritchard et al. 2000; Falush et al. 2003) and the empirical statistic K determined the number of subpopulations in each stand for both juveniles and adults using the software STRUCTURE v.2.3.3. Default model parameters were used with K varying from 1 to 5. Each run consisted of 20 replicates of 250,000 burn-in repetitions and 750,000 data collection iterations. The number of representative populations was identified according to the ΔK method (Evanno et al. 2005), considering the log probability among successive K values (ΔK) and the mean of the log-likelihoods of 20 runs of each K. We used the STRUCTURE HARVESTER (Earl 2012) to visualize likelihood parameters across the K values and visualize the number of genetic groups that best represented the data.
Old-growth adults and juvenile cohorts in both stands were georeferenced to evaluate SGS between life stages. Coordinates were correlated with genetic data to create a spatial autocorrelation analysis. The presence of inbreeding was analyzed using the kinship coefficient (Ritland 1996). In addition, the SGSMAX was calculated (Jump et al. 2012), which is defined as the greatest distance at which the mean kinship coefficient (Fd) is significant at p < 0.05. We assumed a minimum proportion of 50% for each individual present at least once in each distance class, and the coefficient of variation for the number of times that each individual was expressed in each class was defined as ≤ 1 (Hardy and Vekemans 2002). SGS was carried out using 10,000 permutations for each life stage in both stands for the full distance range. We plotted the average pairwise estimates of the genetic relatedness as a function of distance to generate spatial autocorrelograms.
The kinship statistic, also known as coancestry, analyzes the probability of identifying two sampled alleles from homologous genes. Particularly, it is the probability of identifying a descending allele by gene comparison. The kinship coefficient is defined as
where Qij is the probability that the random samples of individuals i and j are identical by ancestry and Qm is the probability that random samples taken in the population are identical by descent. The kinship coefficient was estimated using SPAGeDi, version 1.1 (Hardy and Vekemans 2002). The mean standard error was obtained by a resampling jackknife test. The confidence intervals were analyzed using 95% probability for each class of distance.
The Sp statistic was calculated (Vekemans and Hardy 2004) to provide a comparison of the SGS between stands. The following formula was applied:
where bF is the regression slope curve of the kinship coefficient (Fij) on distance classes and F1 is the mean kinship coefficient of the first class of distance (0–10 m). Finally, the analysis of molecular variance (AMOVA) among and within stands were also calculated using GenAlEx 6.501 (Peakall and Smouse 2006).
Results
Genetic diversity
Genetic diversity parameters were considered high but not statistically different between the managed and unmanaged stands (Table S2; Table S3). Overall, the total number of alleles was 18, considering all six microsatellite loci. The number of alleles per locus (AO) ranged from 1 to 5, with an average of alleles (A) equal to 2.667 and 2.334 for the managed and unmanaged stands, respectively (Table S4). Allele richness (AR) ranged from 1.000 to 4.733 and 1.000 to 4.239 for the managed and unmanaged stands, respectively. The number of effective alleles (AE) was 1.972 and 1.879 for the managed and unmanaged stands, respectively. We found a tendency for private alleles in the managed stands (AP = 0.500) and unmanaged stands (AP = 0.167). Expected heterozygosity (HE) did not significantly differ between life stages and had a value of 0.437 for the managed stand for both life stages (Table S5). For the unmanaged stand, HE was 0.402 and 0.418 for the juveniles and adults, respectively. The observed heterozygosity (HO) values were 0.675 and 0.700 in the managed stand and 0.622 and 0.631 in the unmanaged stand for the juveniles and adults, respectively (Table 1). FIS did not statistically differ (p < 0.05) for managed and unmanaged stands. There were no null alleles, linkage disequilibrium, stuttering, or large allele dropout. The FIS values ranged from − 0.588 to − 0.540 in the managed stand, and from − 0.506 to − 0.503 in the unmanaged one both for juvenile and adults, respectively. This pattern indicates an excess of heterozygotes within both stands. Additionally, FST values revealed low differentiation between the two stands (FST = 0.01051; p value ± 0.0001) and among different life stages for the managed (FST = 0.00797; p-value = 0.04985 ± 0.00682) and the unmanaged (FST = 0.00307; p-value = 0.04985 ± 0.00682) stands. The mean number of migrants per generation (NM) was 5.88 in both stands.
Spatial genetic structure (SGS)
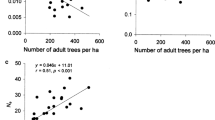
Bayesian clustering suggested an optimum genetic cluster based on delta K (ΔK) = 1 (see supplementary Fig. S1). The inferred cluster did not show a contrasting pattern between managed and unmanaged stands, indicating evident genetic similarity and sufficient gene flow among stands. We found a significant fine-scale SGS in the managed (Fig. 3) and unmanaged (Fig. 4) stands between life stages. SGSMAX revealed a significant structuring of adult individuals in the unmanaged stand, showing a more intense genetic similarity than the expected range from 16 to 25 m but also less similarity for larger distances ranging from 76 to 83 m (Fig. 3a).

Spatial genetic structure autocorrelograms for Eremanthus erythropappus managed stand: Adult (a) and Juvenile (b) based on the kinship coefficient Fij, estimated from six microsatellite loci. Shaded areas represent 95% confident intervals obtained from 10,000 permutations of genotypes among locations. Black bars around mean kinship (Fij) values represent standard errors derived through jackknifing over loci

Spatial genetic structure autocorrelograms for Eremanthus erythropappus unmanaged stand: Adult (a) and Juvenile (b) based on the kinship coefficient Fij, estimated from six microsatellite loci. Shaded areas represent 95% confident intervals obtained from 10,000 permutations of genotypes between locations. Black bars around mean kinship (Fij) values represent standard errors derived through jackknifing over loci
The kinship coefficient for the first class of distance (F1) was statistically significant for this life stage. There was a positive SGS for juvenile individuals ranging from 19 to 27 m and less genetic similarity with an SGS from 115 to 137 m in the managed stand (Fig. 3b). Additionally, the kinship coefficient F(1) and the Sp statistic also indicated a positive association between SGS and the management of juveniles in the managed stand (Table 1).
Comparing SGS between life stages, the Sp value was higher for adults in both stands. In addition, the F(1) value was higher for juveniles in the managed stand. On the other hand, juvenile individuals from the unmanaged stand had a negative F(1) value, indicating the absence of endogamy for this life stage (Table 1). The hierarchical AMOVA also supported the hypothesis of a single genetic cluster (94%, p < 0.001), since the component of variance between the two population groups was statistically equal to zero (see supplementary Table S6).
Discussion
Microsatellite markers were used to analyze the effects of forest management on the genetic diversity and SGS of E. erythropappus, an overexploited species of the Brazilian Savanna and Atlantic Forest region. This approach may be useful for discovering the possible impacts of altering forest structure on genetic diversity parameters. Although the mean levels of genetic diversity were high and not significantly different, there was a positive relationship between SGS and forest management for juvenile cohorts, indicating an effect of management on spatial genetic structure. Moreover, there was a tendency for increasing allelic richness in both life stages in the managed stand. The higher patterns of genetic diversity may be an effect of increasing pollen/seed flow among different populations. Further analysis should be performed in different fields to better understand the complexity of genetic interactions after management.
Impact of forest management on genetic diversity parameters
These parameters were relatively high, suggesting that the population remnants are enough to inhibit the effects of gene drift resulting from forest management. As expected, higher values of expected (HE = 0.437) and observed (HO = 0700 and 0.675) heterozygosity were found in the managed stand for juveniles and adults, respectively. According to Scolforo et al. (2012), E. erythropappus produces abundant small seeds that can travel long distances. Therefore, effective seed distribution may intensify gene flow and prevent the effects of genetic drift. In addition, these results may be a response of an intensive incidence of sunlight. The canopy opening generally intensifies the growth of regenerating individuals of E. erythropappus. In addition, the presence of canopy gaps may also allow the introduction of seeds from different populations and accelerate the colonization process. The effective gene flow in the managed stand was confirmed by the high number of migrants (NM = 5.88). According to Slatkin and Barton (1989), NM values higher than 1.0 indicate gene flow sufficient for preventing genetic drift.
In general, early successional species exhibit higher genetic divergence among their populations than late-successional species (Nybom and Bartish 2000; Nybom 2004). In outcrossing species that predominantly belong to advanced successional stages, a higher genetic divergence is expected within rather than among populations due to intense gene flow. On the other hand, species that self-fertilize, belonging to initial successional stages or are annual species, are expected to exhibit low diversity within populations and high divergence among populations. The longevity of individuals, mating systems, pollination strategies, seed dispersal, and clonal reproduction are the main factors involved in the maintenance and distribution of genetic variation levels within and between populations of plant species (Loveless and Hamrick 1984).
Seed dispersal by E. eryhtropappus is anemochory or wind dispersed (Scolforo et al. 2012). According to Vieira et al. 2012, this species is mainly pollinated by the western honeybee, Apis mellifera L., and Trigona spp. (Hymenoptera, Apidae) and have hermaphroditic flowers with capitula at the end of the branches, a special characteristic of the Asteraceae family. Therefore, the high patterns of genetic diversity patterns may be due to the high production of seeds by this species that maintain genetic cohesion among populations.
Although forest fragmentation can modify natural patterns of genetic diversity, gene flow through pollen or seed dispersion may reduce the effects of biodiversity loss and prevent gene drift (Couvet 2002). Barreira et al. (2006) used isoenzyme markers to analyze the effects of forest management in E. erythropappus populations. The mating system was predominantly allogamous, with high rates of multi-locus outcrossing (tm = 0.963). Similarly, forest management also caused a positive but not statistically significant increase in genetic diversity by comparison between unmanaged (HO = 0.357; HE = 0.403) and managed (HO = 0.423; HE = 0.425) stands as a result of the increase of outcrossing fertilization among genetically different individuals. According to Hamrick and Godt (1990), tree species with a long-life cycle, efficient pollen and seed flow, a mixed reproduction system and a wide geographic distribution should have high levels of genetic diversity. These high levels are due to a combination of many factors, such as widespread pollination by animals, high out-crossing rates and complex breeding systems, allowing their persistence even if their populations undergo an abrupt decline (Hamrick et al. 1992; Cascante et al. 2002).
Genetic diversity parameters within tropical tree species may be influenced by characteristics such as size, longevity, fecundity, breeding systems and geographic distribution (Hamrick and Godt 1990). There were a relatively low number of alleles (N = 18) when compared with different microsatellite analyses for tropical tree species, indicating a low polymorphism rate and high genetic similarity among the six microsatellite loci. In fact, both stands analyzed in this study are located at high elevations and therefore, the species could be influenced by topographic isolation as a result of restricted geographic distribution. Furthermore, Castilla et al. (2015) found a positive influence due to elevation in Miconia affinis DC., a pioneer tropical tree species from the mountains of South America, where geographic distance significantly increased genetic differentiation between populations.
Considering that the six microsatellite loci were distributed throughout the genome of E. erythropappus, genetic interactions revealed by our analysis within the investigated population are representative and very important for the genetic conservation of E. erythropappus. Similarly, Rocha et al. (2020a) used cpDNA to analyze the genetic diversity of two Eremanthus species (E. erythropappus and Eremanthus incanus Less.). These researchers found three haplotypes shared by both species with relatively low intensity and total genetic diversity, indicating that these species may be very susceptible to habitat fragmentation and future climate change.
The relatively low allelic richness may be an intrinsic characteristic of the analyzed species. Additionally, the isolation by distance may also cause the occurrence of a low number of alleles in segregated populations (Wright 1931, 1946). In addition, considering that E. erythropappus is an endemic species that occurs in isolated mountains, even with high seed and pollen dispersal, the species may not be able to overcome the gaps, effectively isolating the populations. Furthermore, even when there is little exchange of migrants or alleles, gene flow is able to contribute to the cohesion among populations (Wright 1931; Morjan and Rieseberg 2004). Nevertheless, further analyses should integrate landscape genetics to analyze possible patterns of isolation among nearby populations, considering that the effects of management practices in isolated populations may induce evolutionary pressure.
The impacts of forest management on tree genetic diversity may differ, even when adopting the same type or intensity of harvesting. Therefore, similar silvicultural harvesting has different genetic effects among and within populations. The main factors influencing different responses in forest management are biological and ecological attributes intrinsic to each species and population (Ratnam et al. 2014). In general, we found very similar genetic diversity parameters between stands. Nevertheless, the averages of genetic diversity parameters were considerably higher than those in other studies for E. erythropappus populations (Estopa et al. 2006; Freitas et al. 2008; Pádua et al. 2016). This may be related to the higher levels of polymorphism normally found in microsatellite markers (Varshney et al. 2005). The high genetic diversity may be inherent to intrinsic ecological characteristics such as natural occurrence in environments with unfavorable conditions, which normally hinder the development of interspecific competition. According to Kisdi and Stefan (1999), populations adapted to extreme conditions may have a higher probability of incorporating new alleles by mutation than small populations from homogeneous environments.
Furthermore, the overall number of alleles per locus (A) and rarefied allelic richness (AR) were not significantly different in the managed stand. However, adult individuals from the managed stand showed a tendency to higher allelic richness (AR = 0.500) than juvenile individuals (AR = 0.167). Although forest management may lead to a range of losses in the total number of alleles in tropical species (Silva et al. 2008; Vinson 2009; Carneiro et al. 2011), surrounding populations may mitigate the effects of harvesting by the reintroduction of lost alleles from regenerating individuals. Barreira et al. (2006) found a number of effective alleles per locus (A) ranging from 1.67 to 1.93. Pádua et al. (2016) used ISSR markers in natural populations of E. erythropappus in Minas Gerais state and found the number of alleles per locus ranged from 1.46 to 1.68.
Finally, the observed heterozygosity showed higher levels compared to expected, suggesting an excess of heterozygotes according to the Hardy–Weinberg equilibrium. The maintenance and even a slight increase in natural levels of genetic diversity in the managed stand of E. erythropappus indicates that this species has efficient systems of gene flow and seed dispersion to maintain the levels of natural genetic diversity when subjected to thinning. Furthermore, endemic and rare species may have developed strategies that allow them to survive and promote their conservation even under self-fertilization, low genetic diversity and high inbreeding rates (Lande and Schemske 1985; Oostermeijer et al. 1994).
Long-lived and outcrossing species show most of the genetic variability within populations (Nybom 2004). In fact, several studies have found a tendency for high genetic variation within populations of tropical outbreeding tree species (Duarte et al. 2015; Brito et al. 2016; Dias et al. 2017). AMOVA results showed that most of the genetic variance is retained within populations (96%). Accordingly, the low differentiation indexes (FST) corroborate the hypothesis of high gene flow between the stands. These results confirm the theory of effective gene flow among populations because genetic divergence is considerably low (6%) among stands. Moura (2005), studying the genetic diversity of E. erythropappus populations using isoenzyme and RAPD markers, found a similar pattern of variation (96.5%) within populations.
Impacts of forest management on spatial genetic structure
Tropical forests commonly present contrasting patterns of regeneration that differentiate based on the sources available at the site, such as the seed bank, stem and root sprouts and remnant vegetation, and on the colonization process from seeds outside the site (Guariguata and Ostertag 2001). Our results from Bayesian clustering confirm that the managed and unmanaged stands are composed of a single genetic population, presenting a single genetic cluster structure (K = 1). These results confirm that the managed and unmanaged stands are controlled by intense pollen and/or seed flow between stands and life stages. Consequently, forest thinning did not differentiate the stands as two genetic populations due to the relatively small distance between them.
On the other hand, we detected an influence on the SGS of the managed stand, indicating the presence of coancestry in the first class of distance for juvenile individuals (SGSMAX = 25 m; 100 m) from the managed stand and for adult individuals (SGSMAX = 21 m) from the unmanaged stand. These results indicate the presence of more genetically similar individuals in the first class of distance. We did not find a pattern of SGS (except for some scatter) for the other classes, suggesting that individuals may have a random genetic distribution. Likewise, Sp values were similar to those of previous studies for outcrossing, wind-dispersed tropical tree species (Vekemans and Hardy 2004).
The positive SGS for juvenile individuals as an impact of forest management is confirmed by the lowest value of Sp (0.0070) and by a relatively high and statistically significant value of F(1)(0.00690; p < 0.05), indicating that the juveniles are less structured than adults. Additionally, the SGS for adults from the managed stand was also confirmed by F(1)(0.01467), which was the highest and was statistically significant. In addition, the Sp value for adult individuals in the managed stand was the highest (0.0156), indicating a more structured SGS. Generally, Sp values were slightly higher in the unmanaged stand. There was an increase in Sp values from juveniles to adults within both stands, meaning that distant pairs of juvenile individuals from both stands are more genetically similar than they used to be by chance. The absence of any effect on SGS for adult trees in the unmanaged stand may be related to the old growth of the seed trees sampled, which might not be genetically related.
Barreira et al. (2006) also found an effect of forest management on the SGS of E. erythropappus within 180 m of distance. Positive results for coancestry may be a result of vegetative propagation but also a direct effect of inefficient seed dispersion and the regeneration of brothers and half-brothers near the mother trees, creating a nonrandom distribution of alleles and genotypes among populations (Loveless and Hamrick 1984). The intensity of gene flow and seed dispersal are the main factors responsible for determining the extent of gene dispersion in tropical rain forests (Dick et al. 2008). According to Melo et al. (2012), E. erythropappus tends to sprout from roots and stems after the harvest of adult trees, which may induce the growth of clonal individuals. Because the stands analyzed here appeared to have efficient patterns of gene flow, confirmed by the similar levels of genetic diversity between populations, vegetative propagation may be a trend influencing the SGS of E. erythropappus. Pádua et al. (2016) used inter simple sequence repeat markers (ISSR) in nine populations of E. erythropappus throughout Minas Gerais. In addition, they also identified an SGS for reproductive trees. Moreover, five populations showed a random spatial distribution of the genotypes, and another four had significant genetic structuring (p > 0.05), indicating a positive and significant coancestry.
Genetic structuring may be an induced response to changes in seed dispersion methods and the pioneering behavior of regenerating individuals. According to Aldrich and Hamrick (1998), the intrinsic conditions of a population can promote changes in the type and abundance of seed dispersers that can affect dispersal distances and the probability of the establishment of maternal families and their spatial distribution within a population. Although there was no evident effect on genetic diversity, we emphasize that the managed stand was relatively recently thinned, and there was not enough time to perform a complete analysis. Therefore, the effects of forest management on the genetic structure may be more evident in subsequent generations.
Conclusions
Comparison between a managed stand using a silvicultural system of seed trees and an unmanaged stand, 300-m distance from each other, was carried out at the Serra da Mantiqueira Environmental Protected Area (APA Serra da Mantiqueira), Itamonte, Minas Gerais. Although old-growth adults and juvenile cohorts from both stands showed low allelic richness, overall high levels of genetic diversity were found. Both life stages from the managed stand did not show a statistically significant increase in genetic variability. In addition, outcrossing tree species normally show a high genetic diversity that can be maintained by long-distance pollen flow in fragmented landscapes. Previous studies of E. erythropappus populations found high patterns of genetic diversity (Moura 2005; Estopa et al. 2006; Mori et al. 2009; Carvalho et al. 2012), and genetic variability may not be influenced by forest management (Barreira et al. 2006). Nevertheless, our results indicate that forest management using a silvicultural system of seed trees may have affected the fine-scale spatial genetic structure in juveniles from the managed stand and may increase the genetic relatedness of individuals in the long-term.
References
Ackzell L (1993) A comparison of planting, sowing and natural regeneration for Pinus sylvestris L. in boreal Sweden. For Ecol Manag 61:229–245. https://doi.org/10.1016/0378-1127(93)90204-Z
Aldrich PR, Hamrick JL (1998) Reproductive dominance of pasture trees in a fragmented tropical forest mosaic. Science 281:103–105. https://doi.org/10.1126/science.281.5373.103
Andrade IS (2009) Avaliação técnica e econômica de sistemas de manejo de candeais nativos. D. Phil. thesis, Universidade Federal de Lavras
Aravanopoulos F (2018) Do silviculture and forest management affect the genetic diversity and structure of long-impacted forest tree populations? Forests 9(6):355. https://doi.org/10.3390/f9060355
Avise JC, Hamrick JL (1996) Conservation genetics, case histories from nature. Chapman & Hall, 512 pp
Barreira S, Sebbenn AM, Scolforo JRS, Kageyama PY (2006) Diversidade genética e sistema de reprodução em população nativa de Eremanthus erythropappus (DC.) MacLeish sob exploração. Sci For 71:119–130
Bergeron Y, Vijayakumar DBIP, Ouzennou H, Raulier F, Leduc A, Gauthier S (2017) Projections of future forest age class structure under the influence of fire and harvesting: implications for forest management in the boreal forest of eastern Canada. Int J For Res 90(4):485–495. https://doi.org/10.1093/forestry/cpx022
Borrell JS, Wang N, Nichols RA, Buggs RJ (2018) Genetic diversity maintained among fragmented populations of a tree undergoing range contraction. Heredity 121(4):304. https://doi.org/10.1038/s41437-018-0132-8
Braga EA (2006) Substratos e fertilização na produção de mudas de candeia Eremanthus erythropappus (DC.) McLeisch, em tubetes. Dissertation, Universidade Federal de Lavras
Brito VL, Mori GM, Vigna BB, Azevedo-Silva M, Souza AP, Sazima M (2016) Genetic structure and diversity of populations of polyploid Tibouchina pulchra Cogn. (Melastomataceae) under different environmental conditions in extremes of an elevational gradient. Tree Genet Genomes 12(6):101. https://doi.org/10.1007/s11295-016-1059-y
Brown AD, Kappelle M (2001) Introdducción a los bosques nublados del neotrópico: una síntesis regional. In: Kappell M, Brown AD (eds) Bosques nublados del neotrópico. Instituto Nacional de Biodiversidad, Santo Domingo de Heredia, pp 25–40
Carneiro FS, Lacerda AEB, Leme MR, Gribel R, Kanashiro M, Wadt LHO, Sebbenn AM (2011) Effects of selective logging on the mating system and pollen dispersal of Hymenaea courbaril L. (leguminosae) in the Eastern Amazon as revealed by microsatellite analysis. For Ecol Manag 262:1758–1765. https://doi.org/10.1016/j.foreco.2011.07.023
Carvalho D, Barreira S, Moura MC (2012) Genética, manejo e conservação. In: Scolforo JRS, Oliveira AD, Davide AC (eds) O manejo sustentável da candeia: o caminhar de uma nova experiência florestal em Minas Gerais. Editora UFLA, Lavras, pp 237–246
Cascante A, Quesada M, Lobo JJ, Fuchs EA (2002) Effects of dry tropical forest fragmentation on the reproductive success and genetic structure of the tree Samanea saman. Conserv Biol 16(1):137–147. https://doi.org/10.1046/j.1523-1739.2002.00317.x
Castilla AR, Pope N, Jha S (2015) Positive density-dependent reproduction regulated by local kinship and size in an understory tropical tree. Ann Bot 117(2):319–329. https://doi.org/10.1093/aob/mcv170
Cavers S, Cottrell JE (2014) The basis of resilience in forest tree species and its use in adaptive forest management in Britain. Int J For Res 88(1):13–26. https://doi.org/10.1093/forestry/cpu027
Cavers S, Degen B, Caron H, Lemes MR, Margis R, Salgueiro F, Lowe AJ (2005) Optimal sampling strategy for estimation of spatial genetic structure in tree populations. Heredity 95(4):281–289. https://doi.org/10.1038/sj.hdy.6800709
Cloutier D, Kanashiro M, Ciampi AY, Schoen DJ (2007) Impacts of selective logging on inbreeding and gene dispersal in an Amazonian tree population of Carapa guianensis Aubl. Mol Ecol 16:797–809. https://doi.org/10.1111/j.1365-294x.2006.03193.x
Couvet D (2002) Deleterious effects of restricted gene flow in fragmented populations. Conserv Biol 16(2):369–376. https://doi.org/10.1046/j.1523-1739.2002.99518.x
Crnokrak P, Roff DA (1999) Inbreeding depression in the wild. Heredity 83:260–270. https://doi.org/10.1038/sj.hdy.6885530
Crow JF, Aoki K (1984) Group selection for a polygenic behavioral trait: estimating the degree of population subdivision. Proc Natl Acad Sci 81(19):6073–6077. https://doi.org/10.1073/pnas.81.19.6073
Cruz ES (2006) Dinâmica, modelagem do crescimento e cadeia de comercialização da candeia Eremanthus incanus (Less.) Less. D. Phil. thesis, Universidade Federal de Lavras
Curi N, Marques JJG, Marques AFSM, Fernandes EI (2008) Solos, relevo, geologia e mineração. In: Scolforo JR, Carvalho LMT, Oliveira AD (eds) Zoneamento Ecológico Econômico de Minas Gerais: componentes geofísico e biótico. Editora UFLA, Lavras, pp 73–88
Dias ACC, Serra AC, Sampaio DS, Borba EL, Bonetti AM, Oliveira PE (2017) Unexpectedly high genetic diversity and divergence among populations of the apomictic Neotropical tree Miconia albicans. Plant Biol 20(2):244–251. https://doi.org/10.1111/plb.12654
Dick CW, Hardy OJ, Jones FA, Petit RJ (2008) Spatial scales of pollen and seed-mediated gene flow in tropical rain forest trees. Trop Plant Biol 1(1):20–33. https://doi.org/10.1007/s12042-007-9006-6
Dixo M, Metzger JP, Morgante JS, Zamudio KR (2009) Habitat fragmentation reduces genetic diversity and connectivity among toad populations in the Brazilian Atlantic Coastal. Biol Conserv 142(8):1560–1569. https://doi.org/10.1016/j.biocon.2008.11.016
Doyle JJ, Doyle JL (1987) Isolation of plant DNA from fresh tissue. Focus 12(1):13–15
Duarte JF, Carvalho D, Vieira FA (2015) Genetic conservation of Ficus bonijesulapensis in a dry forest on limestone outcrops. Biochem Syst Ecol 59:54–62. https://doi.org/10.1016/j.bse.2015.01.008
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Estopa RA, Souza AD, Moura MCO, Botrel MCG, Mendonça EG, Carvalho D (2006) Diversidade genética em populações naturais de candeia (Eremanthus erythropappus (DC.) MacLeish). Sci Forestalis 70:97–106
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294x.2005.02553.x
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Folke C (2006) Resilience: the emergence of a perspective for social-ecological systems analyses. Glob Environ Change 16(3):253–267. https://doi.org/10.1016/j.gloenvcha.2006.02.003
Frankham R, Ballou J, Briscoe D (2002) An introduction to conservation genetics. Cambridge University Press, p 642
Freitas VLO, Lemos-Filho JP, Lovato MB (2008) Contrasting genetic diversity and differentiation of populations of two successional stages in a neotropical pioneer tree (Eremanthus erythropappus, Asteraceae). Genet Mol Res 7(2):388–398. https://doi.org/10.4238/vol7-2gmr429
García-Gil MR, Floran V, Östlund L, Gull BA (2015) Genetic diversity and inbreeding in natural and managed populations of Scots pine. Tree Genet Genomes 11(2):1–12. https://doi.org/10.1007/s11295-015-0850-5
Ghanzoul J (2005) Pollen and seed dispersal among dispersed plants. Biol Rev 80:413–443. https://doi.org/10.1017/S1464793105006731
González-Díaz P, Jump AS, Perry A, Wachowiak W, Lapshina E, Cavers S (2017) Ecology and management history drive spatial genetic structure in Scots pine. For Ecol Manag 400:68–76. https://doi.org/10.1016/j.foreco.2017.05.035
Goudet J (2002) Fstat Vision (1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Guariguata MR, Ostertag R (2001) Neotropical secondary forest succession: changes in structural and functional characteristics. For Ecol Manag 148:185–206. https://doi.org/10.1016/S0378-1127(00)00535-1
Hamrick JL, Godt MJW (1990) Allozyme diversity in plant species. In: Brown AHD, Clegg MT, Kahler AL, Weir BS (eds) Plant population genetics, breeding and genetic resources. Sinauer, Sunderland, pp 44–64
Hamrick JL, Godt JWM, Sherman-Broyles SL (1992) Factors influencing levels of genetic diversity in woody plant species. New For 6:95–124. https://doi.org/10.1007/BF00120641
Hardwick KA, Elliot S (2015) Second growth: the promise of tropical forest regeneration in an age of deforestation. Restor Ecol 24(1):137. https://doi.org/10.1111/rec.12320
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyze spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hawley GJ, Schaberg PG, DeHayes DH, Brissette JC (2005) Silviculture alters the genetic structure of an eastern hemlock forest in Maine. USA. Can J For Res 35:143–150. https://doi.org/10.1139/x04-148
Jump AS, Rico L, Coll M, Penuelas J (2012) Wide variation in spatial genetic structure between natural populations of the European beech (Fagus sylvatica) and its implications for SGS comparability. Heredity 108(6):633–639. https://doi.org/10.1038/hdy.2012.1
Kisdi E, Stefan AHG (1999) Adaptive dynamics in allele space: evolution of genetic polymorphism by small mutations in a heterogeneous environment. Evolution 53(4):993–1008. https://doi.org/10.1111/j.1558-5646.1999.tb04515.x
Köppen W, Geiger R (1928) Klimate der Erde. Gotha: Verlag Justus Perthes. Wall-map 150 cm × 200 cm
Lacerda AEB, Kanashiro M, Sebbenn AM (2008) Effects of reduced impact logging on genetic diversity and spatial genetic structure of a Hymenaea courbaril population in the Brazilian Amazon Forest. For Ecol Manag 255:1034–1043. https://doi.org/10.1016/j.foreco.2007.10.009
Lande R, Schemske DW (1985) The evolution of self-fertilization and inbreeding depression in plants. I. Genetic models. Evolution 39(1):24–40. https://doi.org/10.1111/j.1558-5646.1985.tb04077.x
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82(11):1420–1425. https://doi.org/10.2307/2445869
Loveless MD, Hamrick JL (1984) Ecological determinants of genetic-structure in plant populations. Annu Rev Ecol Syst 15:65–95. https://doi.org/10.1146/annurev.es.15.110184.000433
Martins K, Kimura RK, Francisconi AF, Gezan S, Kainer K, Christianini AV (2016) The role of very small fragments in conserving genetic diversity of a common tree in a hyper fragmented Brazilian Atlantic forest landscape. Conserv Genet 17:509–520. https://doi.org/10.1007/s10592-015-0800-7
Melo LA, Davide AC, Teixeira LAT (2012) Metodologia para resgate de matrizes e enraizamento de estacas de Eremanthus erythropappus. Cerne 18(4):631–638. https://doi.org/10.1590/S0104-77602012000400013
Mori CLSO, Brito JO, Scolforo JRS, Vidal EJ, Mendes LM (2009) Influence of altitude, age and diameter on yield and alpha-bisabolol content of candeia trees (Eremanthus erythropappus). Cerne 15(3):339–345
Morjan CL, Rieseberg LH (2004) How species evolve collectively: implications of gene flow and selection for the spread of advantageous alleles. Mol Ecol 13:1341–1356. https://doi.org/10.1111/j.1365-294X.2004.02164.x
Moura MCO (2005) Distribuição da variabilidade genética em populações naturais de Eremanthus erythropappus (DC) MacLeish por isoenzimas e RAPD. D. Phil thesis, Universidade Federal de Lavras
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89(3):583–590
Nybom H (2004) Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol Ecol 13:1143–1155. https://doi.org/10.1111/j.1365-294X.2004.02141.x
Nybom H, Bartish IV (2000) Effects of life history traits and sampling strategies on genetic diversity estimates obtained with RAPD markers in plants. Perspect Plant Ecol Syst 3(2):93–114. https://doi.org/10.1078/1433-8319-00006
Oliveira ADD, Ribeiro SAI, Scolforo JRS, Mello JMD, Acerbi Junior FW, Camolesi JF (2009) Market chain analysis of candeia timber (Eremanthus erythropappus). Cerne 15(3):257–264
Oostermeijer JGB, Van Eijck MW, Den Nijs HCM (1994) Offspring fitness in relation to population size and genetic variation in the rare perennial plant species Gentiana pneumonanthe (Gentianaceae). Oecologia 97:289–296. https://doi.org/10.1007/BF00317317
Ortega J, Bonal R, Muñoz A (2010) Genetic consequences of habitat fragmentation in long-lived tree species: the case of the mediterranean holm oak (Quercus ilex, L.). J Hered 101(6):717–726. https://doi.org/10.1093/jhered/esq081
Pádua JAR, Brandão MM, Carvalho D (2016) Spatial genetic structure in natural populations of the overexploited tree Eremanthus erythropappus (DC.) MacLeish (Asteraceae). Biochem Syst Ecol 66:307–311. https://doi.org/10.1016/j.bse.2016.04.015
Peakall ROD, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6(1):288–295
Petrokofsky G, Sist P, Blanc L, Doucet JL, Finegan B, Gourlet-Fleury S, Healey JR, Livoreil B, Nasi R, Peña-Claros M, Putz FE (2015) Comparative effectiveness of silvicultural interventions for increasing timber production and sustaining conservation values in natural tropical production forests. Environ Evid 4(8):1–7. https://doi.org/10.1186/s13750-015-0034-7
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Quéméré E, Crouau-Roy B, Rabarivola C, Louis EE, Chikhi L (2010) Landscape genetics of an endangered lemur (Propithecus tattersalli) within its entire fragmented range. Conserv Genet 19:1606–1621. https://doi.org/10.1111/j.1365-294x.2010.04581.x
Rajora OP, Pluhar SA (2003) Genetic diversity impacts of forest fires, forest harvesting, and alternative reforestation practices in black spruce (Picea mariana). Theor Appl Genet 106(7):1203–1212. https://doi.org/10.1007/s00122-002-1169-9
Ratnam W, Rajora OP, Finkeldey R, Aravanopoulos F, Bouvet JM, Vaillancourt RE, Kanashiro M, Fady B, Vinson C (2014) Genetic effects of forest management practices: global synthesis and perspectives. For Ecol Manag 333:52–65. https://doi.org/10.1016/j.foreco.2014.06.008
Ribeiro ISA (2009) Avaliação técnica e econômica de sistemas de manejo de candeais nativos. D. Phil thesis, Universidade Federal de Lavras
Ritland K (1996) Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res 67(2):175–185. https://doi.org/10.1017/S0016672300033620
Robledo-Arnuncio JJ, Alia R, Gil L (2004) Increased selfing and correlated paternity in a small population of a predominantly outcrossing conifer, Pinus sylvestris. Mol Ecol 13(9):2567–2577. https://doi.org/10.1111/j.1365-294X.2004.02251.x
Rocha LF, Carmo IEP, Póvoa JRS, Carvalho D (2020a) Effects of climate changes on distribution of Eremanthus erythropappus and E. incanus (Asteraceae) in Brazil. J For Res 31:353–364. https://doi.org/10.1007/s11676-019-00968-z
Rocha LF, Paula NR, Nazareno AG, Carvalho D (2020b) Development and characterization of nuclear microsatellite markers for Eremanthus erythropappus and their transferability across related species. Biol Res 53(30):1–5. https://doi.org/10.1186/s40659-020-00298-z
Santos KA, Frohlich PC, Hoscheid J, Tiuman TS, Gonçalves JE, Cardozo-Filho L, Silva EA (2017) Candeia (Eremanthus erythroppapus) oil extraction using supercritical CO2 with ethanol and ethyl acetate cosolvents. J Supercrit Fluids 128:323–330. https://doi.org/10.1016/j.supflu.2017.03.029
Schaberg PG, DeHayes DH, Hawley GJ, Nijensohn SE (2008) Anthropogenic alterations of genetic diversity within tree populations: implications for forest ecosystem resilience. For Ecol Manag 256(5):855–862. https://doi.org/10.1016/j.foreco.2008.06.038
Scolforo JRS, Loeuille BFP, Altoé TF (2012) O manejo sustentável da candeia: o caminhar de uma nova experiência florestal em Minas Gerais. Editora UFLA
Scolforo HF, Scolforo JRS, Mello JM, Rossoni DF, Altoé TF, Oliveira AD, Lima RR (2015) Autoregressive spatial analysis and individual tree modeling as strategies for the management of Eremanthus erythropappus. J For Res 27(3):595–603. https://doi.org/10.1007/s11676-015-0185-y
Silva MB, Kanashiro M, Ciampi AY, Thompson I, Sebbenn AM (2008) Genetic effects of selective logging and pollen gene flow in a low-density population of the dioecious tropical tree Bagassa guianensis in the Brazilian Amazon. For Ecol Manag 255:1548–1558. https://doi.org/10.1016/j.foreco.2007.11.012
Sjölund MJ, Jump AS (2013) The benefits and hazards of exploiting vegetative regeneration for forest conservation management in a warming world. Forestry 86(5):503–513. https://doi.org/10.1093/forestry/cpt030
Sjölund MJ, Jump AS (2015) Coppice management of forests impacts spatial genetic structure but not genetic diversity in European beech (Fagus sylvatica L.). For Ecol Manag 336:65–71. https://doi.org/10.1016/j.foreco.2014.10.015
Slatkin M, Barton NH (1989) A comparison of three indirect methods for estimating average levels of gene flow. Evolution 43(7):1349–1368. https://doi.org/10.1111/j.1558-5646.1989.tb02587.x
Smouse PE, Peakall R (1999) Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82(5):561–573. https://doi.org/10.1046/j.1365-2540.1999.00518.x
Soliani C, Vendramin GG, Gallo LA, Marchelli P (2016) Logging by selective extraction of best trees: does it change patterns of genetic diversity? The case of Nothofagus pumilio. For Ecol Manag 373:81–92. https://doi.org/10.1016/j.foreco.2016.04.032
Telford A, Cavers S, Ennos RA, Cottrell JE (2014) Can we protect forests by harnessing variation in resistance to pests and pathogens? Int J For Res 88(1):3–12. https://doi.org/10.1093/forestry/cpu012
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538
Varshney RK, Graner A, Sorrells ME (2005) Genic microsatellite markers in plants: features and applications. Trends Biotechnol 23(1):48–55. https://doi.org/10.1016/j.tibtech.2004.11.005
Vekemans X, Hardy OJ (2004) New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13(4):921–935. https://doi.org/10.1046/j.1365-294X.2004.02076.x
Vieira FA, Fajardo CG, Carvalho D (2012) Biologia Floral de candeia (Eremanthus erythropappus, Asteraceae). Pesqui Florest Bras 32(72):477–481. https://doi.org/10.4336/2012.pfb.32.72.477
Vinson CC (2009) The impact of selective logging on inbreeding and gene flow in two Amazonian timber species with contrasting ecological and reproductive characteristics. D. Phil. thesis, University of Oxford
Wickneswari R (2011) Threats to genetic viability of Southeast Asian forest species. In: Wickneswari R, Cannon CH (eds) Managing the future of Southeast Asia’s valuable tropical rainforests, a practitioner’s guide to forest genetics. Springer, Dordrecht Heidelberg, pp 69–82
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Wright S (1946) Isolation by distance under diverse systems of mating. Genetics 31(1)
Young A, Boyle T, Brown T (1996) The population genetic consequences of habitat fragmentation for plants. Trends Ecol Evol 11(10):413–418. https://doi.org/10.1016/0169-5347(96)10045-8
Acknowledgements
The authors acknowledge Elisângela Monteiro Coser and the FIOCRUZ (Instituto René Rachou - Belo Horizonte/MG) for DNA sequencing. We would like to thank Alison Gonçalves Nazareno and Eduardo van den Berg for review of the paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Project funding: The work was supported by the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) [CRA APQ-02641-14] and in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior−Brasil (CAPES)−Finance Code 001.
The online version is available at http://www.springerlink.com.
Corresponding editor: Tao Xu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rocha, L.F., Paula, N.R.d. & De Carvalho, D. Fine-scale analysis reveals a potential influence of forest management on the spatial genetic structure of Eremanthus erythropappus. J. For. Res. 32, 1567–1578 (2021). https://doi.org/10.1007/s11676-020-01204-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11676-020-01204-9