
Abstract
Parkinson’s disease (PD) pathogenesis may involve the epigenetic control of enhancers that modify neuronal functions. Here, we comprehensively examine DNA methylation at enhancers, genome-wide, in neurons of patients with PD and of control individuals. We find a widespread increase in cytosine modifications at enhancers in PD neurons, which is partly explained by elevated hydroxymethylation levels. In particular, patients with PD exhibit an epigenetic and transcriptional upregulation of TET2, a master-regulator of cytosine modification status. TET2 depletion in a neuronal cell model results in cytosine modification changes that are reciprocal to those observed in PD neurons. Moreover, Tet2 inactivation in mice fully prevents nigral dopaminergic neuronal loss induced by previous inflammation. Tet2 loss also attenuates transcriptional immune responses to an inflammatory trigger. Thus, widespread epigenetic dysregulation of enhancers in PD neurons may, in part, be mediated by increased TET2 expression. Decreased Tet2 activity is neuroprotective, in vivo, and may be a new therapeutic target for PD.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


Data availability
All sequencing data generated in this study are available from the NCBI Gene Expression Omnibus (GEO) database under the accession number GSE136010. DNA methylation padlock-seq and RNA-seq data used in this study are publicly available under the GEO accession number GSE135037. Hi-C data are available at GSM2322542. RNA-seq data from Parkinson’s disease models in Extended Data Fig. 7a–d are available under the GEO accession numbers GSE54795, GSE108370, GSE125239 and GSE130752. https://github.com/LeeLMarshall/Epigenomic-analysis-of-Parkinson-s-disease-neurons-identifies-Tet2-loss-as-neuroprotectiveSource data are provided with this paper.
Code availability
Code used for the analyses in this study is provided in the Supplementary Software and is available at https://github.com/LeeLMarshall/Epigenomic-analysis-of-Parkinson-s-disease-neurons-identifies-Tet2-loss-as-neuroprotective.
References
GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 17, 939–953 (2018).
Wirdefeldt, K., Gatz, M., Reynolds, C. A., Prescott, C. A. & Pedersen, N. L. Heritability of Parkinson disease in Swedish twins: a longitudinal study. Neurobiol. Aging 32, 1923.e1–1923.e8 (2011).
Gomez-Esteban, J. C. et al. Factors influencing the symmetry of Parkinson’s disease symptoms. Clin. Neurol. Neurosurg. 112, 302–305 (2010).
Baldereschi, M. et al. Parkinson’s disease and parkinsonism in a longitudinal study: two-fold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 55, 1358–1363 (2000).
Labbe, C., Lorenzo-Betancor, O. & Ross, O. A. Epigenetic regulation in Parkinson’s disease. Acta Neuropathol. 132, 515–530 (2016).
Jakubowski, J. L. & Labrie, V. Epigenetic biomarkers for Parkinson’s disease: from diagnostics to therapeutics. J. Parkinsons Dis. 7, 1–12 (2017).
Masliah, E., Dumaop, W., Galasko, D. & Desplats, P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics 8, 1030–1038 (2013).
Kaut, O., Schmitt, I. & Wullner, U. Genome-scale methylation analysis of Parkinson’s disease patients’ brains reveals DNA hypomethylation and increased mRNA expression of cytochrome P450 2E1. Neurogenetics 13, 87–91 (2012).
Young, J. I. et al. Genome-wide brain DNA methylation analysis suggests epigenetic reprogramming in Parkinson disease. Neurol. Genet. 5, e342 (2019).
Braak, H. et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003).
Guo, J. U. et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 14, 1345–1351 (2011).
Feng, J. et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430 (2010).
Li, X. et al. Ten-eleven translocation 2 interacts with forkhead box O3 and regulates adult neurogenesis. Nat. Commun. 8, 15903 (2017).
Hon, G. C. et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell 56, 286–297 (2014).
Lister, R. et al. Global epigenomic reconfiguration during mammalian brain development. Science 341, 1237905 (2013).
Price, A. J. et al. Divergent neuronal DNA methylation patterns across human cortical development reveal critical periods and a unique role of CpH methylation. Genome Biol. 20, 196 (2019).
Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Dao, L. T. M. et al. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat. Genet. 49, 1073–1081 (2017).
Dong, X. et al. Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat. Neurosci. 21, 1482–1492 (2018).
Soldner, F. et al. Parkinson-associated risk variant in distal enhancer of ɑ-synuclein modulates target gene expression. Nature 533, 95–99 (2016).
Fernandez-Santiago, R. et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol. Med. 7, 1529–1546 (2015).
Li, P. et al. Hemispheric asymmetry in the human brain and in Parkinson’s disease is linked to divergent epigenetic patterns in neurons. Genome Biol. 21, 61 (2020).
Narayanan, N. S., Rodnitzky, R. L. & Uc, E. Y. Prefrontal dopamine signaling and cognitive symptoms of Parkinson’s disease. Rev. Neurosci. 24, 267–278 (2013).
Luo, C. et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017).
Farley, J. E. et al. Transcription factor Pebbled/RREB1 regulates injury-induced axon degeneration. Proc. Natl Acad. Sci. USA 115, 1358–1363 (2018).
Schmitt, A. D. et al. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Rep. 17, 2042–2059 (2016).
Lio, C. J. & Rao, A. TET enzymes and 5hmC in adaptive and innate immune systems. Front. Immunol. 10, 210 (2019).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
Figge, D. A., Eskow Jaunarajs, K. L. & Standaert, D. G. Dynamic DNA methylation regulates levodopa-induced dyskinesia. J. Neurosci. 36, 6514–6524 (2016).
Risso, D., Ngai, J., Speed, T. P. & Dudoit, S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 32, 896–902 (2014).
Donega, V. et al. Transcriptome and proteome profiling of neural stem cells from the human subventricular zone in Parkinson’s disease. Acta Neuropathol. Commun. 7, 84 (2019).
Johnson, M. E., Stecher, B., Labrie, V., Brundin, L. & Brundin, P. Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci. 42, 4–13 (2019).
Qin, L. et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462 (2007).
Brichta, L. et al. Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat. Neurosci. 18, 1325–1333 (2015).
Sodersten, E. et al. A comprehensive map coupling histone modifications with gene regulation in adult dopaminergic and serotonergic neurons. Nat. Commun. 9, 1226 (2018).
Wu, T. T. et al. TET2-mediated Cdkn2A DNA hydroxymethylation in midbrain dopaminergic neuron injury of Parkinson’s disease. Hum. Mol. Genet. 29, 1239–1252 (2020).
Kriaucionis, S. & Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930 (2009).
Kozlenkov, A. et al. A unique role for DNA (hydroxy)methylation in epigenetic regulation of human inhibitory neurons. Sci. Adv. 4, eaau6190 (2018).
Szulwach, K. E. et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616 (2011).
Herrup, K. & Yang, Y. Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378 (2007).
Ellison, E. M., Abner, E. L. & Lovell, M. A. Multiregional analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of Alzheimer’s disease. J. Neurochem. 140, 383–394 (2017).
Stoger, R., Scaife, P. J., Shephard, F. & Chakrabarti, L. Elevated 5hmC levels characterize DNA of the cerebellum in Parkinson’s disease. NPJ Parkinsons Dis. 3, 6 (2017).
Carrillo-Jimenez, A. et al. TET2 regulates the neuroinflammatory response in microglia. Cell Rep. 29, e698 (2019).
Jain, N. et al. Global modulation in DNA epigenetics during pro-inflammatory macrophage activation. Epigenetics 14, 1183–1193 (2019).
Zhang, Q. et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393 (2015).
Pronier, E. et al. Inhibition of TET2-mediated conversion of 5-methylcytosine to 5-hydroxymethylcytosine disturbs erythroid and granulomonocytic differentiation of human hematopoietic progenitors. Blood 118, 2551–2555 (2011).
Ichiyama, K. et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 42, 613–626 (2015).
Izzo, F. et al. DNA methylation disruption reshapes the hematopoietic differentiation landscape. Nat. Genet. 52, 378–387 (2020).
Gagne, J. J. & Power, M. C. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology 74, 995–1002 (2010).
Peter, I. et al. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 75, 939–946 (2018).
Caligiore, D. et al. Parkinson’s disease as a system-level disorder. NPJ Parkinsons Dis. 2, 16025 (2016).
Weintraub, D. et al. Neurodegeneration across stages of cognitive decline in Parkinson disease. Arch. Neurol. 68, 1562–1568 (2011).
Kordower, J. H. et al. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136, 2419–2431 (2013).
Li, P. et al. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 10, 2246 (2019).
Pai, S. et al. Differential methylation of enhancer at IGF2 is associated with abnormal dopamine synthesis in major psychosis. Nat. Commun. 10, 2046 (2019).
He, Y. & Wang, T. EpiCompare: an online tool to define and explore genomic regions with tissue or cell type-specific epigenomic features. Bioinformatics 33, 3268–3275 (2017).
Ernst, J. & Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 12, 2478–2492 (2017).
Labrie, V. et al. Lactase nonpersistence is directed by DNA-variation-dependent epigenetic aging. Nat. Struct. Mol. Biol. 23, 566–573 (2016).
Diep, D. et al. Library-free methylation sequencing with bisulfite padlock probes. Nat. Methods 9, 270–272 (2012).
Krueger, F. & Andrews, S. R. Bismark: a flexible aligner and methylation caller for Bisulfite-seq applications. Bioinformatics 27, 1571–1572 (2011).
The 1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Mo, A. et al. Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015).
Chen, B., Khodadoust, M. S., Liu, C. L., Newman, A. M. & Alizadeh, A. A. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol. Biol. 1711, 243–259 (2018).
Akalin, A. et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012).
Kwon, A. T., Arenillas, D. J., Worsley Hunt, R. & Wasserman, W. W. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-seq datasets. G3 (Bethesda) 2, 987–1002 (2012).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Mishra, A. & Hawkins, R. D. Three-dimensional genome architecture and emerging technologies: looping in disease. Genome Med. 9, 87 (2017).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Res. 4, 1310 (2015).
Forcato, M. et al. Comparison of computational methods for Hi-C data analysis. Nat. Methods 14, 679–685 (2017).
Levy-Leduc, C., Delattre, M., Mary-Huard, T. & Robin, S. Two-dimensional segmentation for analyzing Hi-C data. Bioinformatics 30, i386–i392 (2014).
McLean, C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010).
Reimand, J., Kull, M., Peterson, H., Hansen, J. & Vilo, J. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35, W193–W200 (2007).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Reimand, J. et al. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 14, 482–517 (2019).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Diaz, A., Park, K., Lim, D. A. & Song, J. S. Normalization, bias correction, and peak calling for ChIP-seq. Stat. Appl. Genet. Mol. Biol. 11, 9 (2012).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Yu, Q. & He, Z. Comprehensive investigation of temporal and autism-associated cell type composition-dependent and independent gene expression changes in human brains. Sci. Rep. 7, 4121 (2017).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Konnova, E. A. & Swanberg, M. Animal models of Parkinson’s disease. in Parkinson’s Disease: Pathogenesis and Clinical Aspects (eds Stoker, T. B. & Greenland, J. C.) (Codon Publications, 2018).
Caiazzo, M. et al. Direct generation of functional dopaminergic neurons from mouse and human fibroblasts. Nature 476, 224–227 (2011).
Cheng, L. et al. Gene dysregulation is restored in the Parkinson’s disease MPTP neurotoxic mice model upon treatment of the therapeutic drug Cu(II)(atsm). Sci. Rep. 6, 22398 (2016).
Chen, X. et al. Parkinson’s disease-linked D620N VPS35 knockin mice manifest tau neuropathology and dopaminergic neurodegeneration. Proc. Natl Acad. Sci. USA 116, 5765–5774 (2019).
Maco, B. et al. Semiautomated correlative 3D electron microscopy of in vivo-imaged axons and dendrites. Nat. Protoc. 9, 1354–1366 (2014).
McQuin, C. et al. CellProfiler 3.0: next-generation image processing for biology. PLoS Biol. 16, e2005970 (2018).
Acknowledgements
We thank the Van Andel Institute Flow Cytometry, Genomics, Bioinformatics and Biostatistics, Pathology and Vivarium Cores. We also thank the CAMH Sequencing Facility. We thank the Parkinson’s UK Brain Bank, the NIH NeuroBioBank and the Michigan Brain Bank for the brain tissue provided. This work was funded by a Department of Defense grant (grant no. W81XWH1810512) and a VAI Innovation Award to V.L. V.L. is also supported by grants from the National Institute of Neurological Disorders and Stroke (grant no. 1R21NS112614-01, grant no. 1R01NS114409-01A1 and grant no. 1R01NS113894-01A1), the Farmer Family Foundation Parkinson’s Research Initiative and a Gibby & Friends vs. Parky Award. S.J. and M.W. are supported by the Richard and Helen DeVos Foundation.
Author information
Authors and Affiliations
Contributions
L.L.M. contributed to experimental design, computational analyses, immunohistochemistry experiments and cell culture study. B.A.K. performed neuronal nuclei isolations and the bisulfite padlock probe sequencing of human patient samples, and was involved in the immunohistochemistry and behavioral analyses in mice. E.E. performed the TET2 mRNA analysis in patient samples and RNA isolation of mouse samples, and contributed to the immunohistochemistry experiments. P.L. was involved in the Hi-C analysis and experimental design of computational approaches. W.C. performed the bisulfite padlock probe sequencing of the cell culture study. K.X.L. contributed to the immunohistochemistry analysis. M.W. and S.J. were involved in the flow sorting of neuronal nuclei. J.G. contributed to the hMEDIP analysis. G.A.C. contributed to experimental design. J.M. and X.W. contributed to the analysis of mouse models. V.L. was involved with study design and overseeing the experiments. The manuscript was written by V.L. and L.L.M. and commented on by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Neuroscience thanks Schahram Akbarian and Tiago Outeiro for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Analysis of sample distribution and genomic context of modified cytosine sites at enhancers.
a, Dendrogram showing unsupervised clustering of samples based on cytosine sites. DNA methylation at top 10,000 most variable cytosine sites. Clustering of corresponding technical (nuclei sorting and/or library preparation replicated for select DNA samples) and sequencing replicates (sequencing replicated across lanes/flow cells for select libraries) are shown in pink and purple, respectively. Pearson’s correlation of technical replicates is 0.972 ± 0.0012 s.e.m. and of sequencing replicates is 0.977 ± 0.0010 s.e.m., supporting a high technical reproducibility. b, The proportion of each CpH subtype for the differentially methylated cytosine sites in PD neurons (significant: orange) as compared to the total number of cytosine sites profiled (background: gray). CpH context for cytosine sites in enhancers in this study was similar to that of previous whole genome studies in neurons24. c, The proportion of differentially modified cytosine sites in PD located within an enhancer or promoter. Differentially modified cytosines in neurons of PD patients relative to controls were identified by logistic regression after adjusting for age, sex, postmortem interval, and neuronal subtypes (n = 57 PD, 48 controls; q < 0.05, multiple testing corrected). d, The proportion of 21 specific neuronal subtypes present in the prefrontal cortex of PD patients (orange) and controls (gray). There are 12 glutamatergic and 9 GABAergic neuronal types in the NeuN+ nuclei from PD and controls (n = 57 and 48 individuals, respectively). Neuronal proportions were calculated by computational deconvolution, using neuronal subtype specific markers (n = 563 cytosine sites)24. No significant differences were determined between PD and controls, as determined by a repeated-measures ANOVA and post hoc Tukey HSD test. Error bars represent s.e.m. e, Proportion of glutamatergic neurons and GABAergic neurons in PD and controls. Neuronal proportions determined by the sum of all glutamatergic or GABAergic neuronal subtypes identified by computational deconvolution in panel a. Averaged 76.95% glutamate neuronal subtype proportion ± 0.814 s.e.m. There were no significant differences in glutamatergic and GABAergic neuronal proportions between PD patients and controls, as determined by a repeated-measures ANOVA and post hoc Tukey HSD test. Boxplot center line is the mean, the bounds of the box are the lower and upper quartiles, and the whiskers extend to minimum and maximum data points within 1.5 times the interquartile range.
Extended Data Fig. 2 Genome-wide hypermethylation of differentially methylated enhancer regions in PD neurons.
Differentially methylated regions at enhancers in prefrontal cortex neurons of 57 PD patients and 48 controls were identified (methylKit, sliding window: 1000 bp length, 50 bp step increment). a, Manhattan plot showing differentially methylated regions at enhancers in PD neurons (q < 0.05, logistic regression, adjusting for age, sex, postmortem interval, and neuronal subtypes). -log10(PValue) refers to the significance of region methylation change in PD, with the sign corresponding to the direction of change (red: hypermethylated, blue: hypomethylated region in PD). Threshold for genome-wide significance is q < 0.05 (black line, multiple testing corrected). Differentially methylated region in an enhancer of TET2 in PD neurons is highlighted. b, Proportion of hypermethylated (red) and hypomethylated (blue) regions at enhancers in PD. Enrichment determined by Fisher’s exact test with OR referring to the odds ratio and P = 10-38 significance of hypermethylation enrichment. Gray is all regions in the analysis (background). c, Intersection of the genes with differentially methylated cytosines or regions at enhancers in PD neurons. P = 10−16 is the significance of overlap determined by Fisher’s exact test.
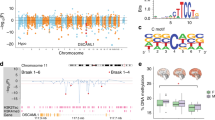
Extended Data Fig. 3 Epigenetic changes in the prefrontal cortex precede the arrival of PD pathology.
Differentially methylated cytosines in neurons of the PD prefrontal cortex before the arrival of Lewy pathology (PD Braak stage 3–4, n = 22 individuals, green), after the arrival of Lewy pathology (PD Braak stage 5–6, n = 34 individuals, yellow) and in the combined PD group (n = 57 individuals, red), in comparison to healthy controls (n = 48 individuals). a, b, Manhattan plot showing differentially methylated cytosines in PD neurons before (a) and after (b) the arrival of Lewy pathology. -log10(PValue) refers to the significance of cytosine methylation change in PD, with the sign corresponding to the direction of change (red: hypermethylated, blue: hypomethylated cytosine in PD). Threshold for genome-wide significance is q < 0.05 (black line, multiple testing corrected). c, Density plot of differentially methylated cytosines in PD prefrontal cortex neurons before and after arrival of Lewy pathology. Enrichment determined by Fisher’s exact test with OR referring to the odds ratio and p-value is significance of hypermethylation enrichment. d, Intersection of differentially methylated cytosines in PD prefrontal cortex neurons in the combined PD group and before or after arrival of Lewy pathology.
Extended Data Fig. 4 Independent validation of DNA methylation changes at enhancers, genome-wide, in PD neurons.
Differential methylation at enhancers in PD prefrontal cortex neurons identified in the discovery cohort were validated with an independent replication cohort of prefrontal cortex neurons from 27 PD patients and 31 controls. a, Manhattan plot showing differentially methylated cytosines at enhancers in PD neurons, identified by logistic regression after adjusting for age, sex, postmortem interval, and neuronal subtypes. -log10(PValue) refers to the significance of cytosine methylation change in PD, with the sign corresponding to the direction of change (red: hypermethylated, blue: hypomethylated cytosine in PD). Threshold for genome-wide significance is q < 0.05 (black line, multiple testing corrected). b, Proportion of hypermethylated (red) and hypomethylated (blue) cytosines at enhancers in PD replication cohort. Enrichment determined by Fisher’s exact test with OR referring to the odds ratio and P = 10−6 is the significance of hypermethylation enrichment. Gray is all cytosines in the analysis (background). c, Correlation between the discovery and replication cohort in the DNA methylation fold changes at enhancer cytosine sites in PD neurons. P = 10−111 is the significance of Pearson’s correlation by two-tailed t-test. d, Intersection of the genes with differentially methylated cytosines at their enhancers in the discovery and replication cohort. P = 10−79 is the significance of overlap determined by Fisher’s exact test.
Extended Data Fig. 5 Medications are not a major contributor to the PD-associated DNA methylation changes identified in this study.
a, Heatmap correlations and unsupervised clustering of patient covariates (yellow: positively correlated, blue: negatively correlated; Pearson’s correlation). b, Venn diagram showing the overlap of differentially methylated cytosines at enhancers in PD patients (n = 57 PD, 48 controls) with differentially methylated cytosines related to PD patients taking dopamine agonists, COMT inhibitors, and/or MAO-B inhibitors. Differentially methylated cytosines related to PD medications were identified by logistic regression, after adjusting for age, sex, postmortem interval, and neuronal subtypes (q < 0.05, multiple testing corrected). c, Intersection of genes with differential methylation in PD and with differential methylation in response to levodopa (L-Dopa) in the brain of a rat model of PD (dorsal striatum of 6-OHDA–treated rats receiving L-Dopa)9,29. Enrichment determined by Fisher’s exact test.
Extended Data Fig. 6 The prefrontal cortex of PD patients does not exhibit differences in the proportion of brain cell types.
Cell-type deconvolution in 24 PD and 12 control prefrontal cortex transcriptomes was performed using 834 cell-type-specific gene signatures63,81. a, Cell-type proportions in the prefrontal cortex of PD patients (orange) and controls (gray). There were no significant differences between PD patients and controls in brain cell types, as determined by a repeated-measures ANOVA and post hoc Tukey HSD test. Boxplot center line is the mean, the bounds of the box are the lower and upper quartiles, and the whiskers extend to minimum and maximum data points within 1.5 times the interquartile range. b, Correlation of neuronal proportions in transcriptomic data with the correction for sources of unknown variation. Statistical analysis of transcriptomic changes in the prefrontal cortex of PD patients was corrected for unknown sources variation, as determined by RUVseq30. P = 10−9 is the significance of Pearson’s correlation by two-tailed t-test, with 95% confidence intervals shown in gray. c, Comparison of statistical models that identifies differentially expressed genes in the PD prefrontal cortex after controlling for unknown sources of variation or neuronal proportion. Differentially expressed genes in the prefrontal cortex of PD patients were identified using a generalized linear model, adjusting for age, sex, RIN, and neuronal proportion or sources of unknown variation (q < 0.05, absolute logFC≥0.5, multiple testing corrected). The outcome of both models is comparable, showing significant overlap of differentially expressed genes in PD (P = 10−228, Fisher’s exact test). d, TET1 and TET3 transcript levels in the prefrontal cortex of PD patients relative to controls. Differentially expressed genes in the prefrontal cortex of PD patients were identified using a generalized linear model, adjusting for age, sex, RIN, and sources of unknown variation. There are no significant changes in TET1 and TET3 transcript levels between PD patients and controls (nominal P = 0.96 and 0.09, respectively). Boxplot center line is the mean, the bounds of the box are the lower and upper quartiles, and the whiskers extend to minimum and maximum data points within 1.5 times the interquartile range. a‒d, n = 12 controls, 24 PD. e, Increased TET2 transcript levels are correlated with reduced TH protein levels in the prefrontal cortex. n = 11 individuals. P = 0.03 is the significance of inverse association between TET2 mRNA and TH protein levels by linear regression. The 95% confidence intervals are shown in gray.
Extended Data Fig. 7 TET2 expression is increased in neurons of PD-relevant models and PD patient neurons.
a, TET2 transcript levels in induced dopaminergic (iDA) neurons of PD patients and controls (n = 8 control fibroblasts, 7 PD fibroblasts, 8 control iDA neurons, 9 PD iDA neurons). b, TET2 transcript levels in bulk subventricular zone (SVZ), isolated neuronal stem cells (NSCs), and isolated microglia cells from PD patients and controls (n = 3 control SVZ, 4 PD SVZ, 4 control NSCs, 5 PD NSCs, 2 control microglia, 4 PD microglia). c, d, Tet2 transcript levels in neurons of PD-relevant models. Tet2 mRNA levels in dopaminergic neurons of mice treated with (c) MPTP (n = 4 saline, 4 MPTP) or (d) 6-OHDA (n = 4 non-injected side, 4 6-OHDA). a‒d, Transcriptomic analyses of RNA-seq datasets were performed using a generalized linear model adjusting for sources of unknown variation, with contrast fit for group-wise comparisons. TET2 transcript levels in counts per million (cpm) with nominal P < 0.05 significance. Datasets: GSE54795, GSE108370, GSE125239, and GSE130752. e, TET2 mRNA depletion by siRNA in the neuronal cell model, SH-SY5Y cells. TET2 mRNA transcript levels in SH-SY5Y cells 72 h after incubation with TET2 siRNA (TET2i) or non-target control (NT). TET2 mRNA levels determined by qPCR and normalized to housekeeping genes B2M, HPRT, and RPL13 (n = 6 NT, 6 TET2i samples). ****P = 10−5, by two-tailed, unpaired equal variance t-test. a‒e, Boxplot center line is the mean, the bounds of the box are the lower and upper quartiles, and the whiskers extend to minimum and maximum data points within 1.5 times the interquartile range. f, Cytosine modification changes in response to TET2 mRNA depletion in vitro. Differentially modified cytosines induced by TET2 reduction in SH-SY5Y cells were identified using logistic regression (n = 8 NT, 8 TET2i). Enrichment in hypomethylation in response to TET2 inactivation determined by Fisher’s exact test with odds ratio (OR) and P = 10−234 for significance of enrichment. The proportion of significantly hypomethylated (blue) and hypermethylated (red) cytosines induced by TET2 inactivation is shown, with all cytosines in the analysis (background) in gray.
Extended Data Fig. 8 Immunohistochemistry analysis of microglia activation and proteinase-K resistant α-synuclein aggregates in mice with Tet2 inactivation 11 months after exposure to inflammation.
Immunohistochemistry image analysis of microglial activation in the (a) dorsal striatum and the (b) substantia nigra. Representative images of dorsal striatum and substantia nigra microglia (IBA1-positive). n = 9 wild-type + saline, 10 wild-type + LPS, 14 Tet2 knockout + saline, and 10 Tet2 knockout + LPS. Immunohistochemistry image analysis of proteinase-K resistant α-synuclein aggregates in the (c) dorsal striatum and the (d) substantia nigra. Data analyzed by two-way ANOVA followed by post hoc Tukey HSD test. n = 9 wild-type + saline, 9 wild-type + LPS, 14 Tet2 knockout + saline, and 7 Tet2 knockout + LPS. Error bars represent s.e.m. e, Transcript analysis of inflammatory marker genes in the midbrain of wild-type and Tet2 knockout mice 11 months post-LPS or saline injection. Transcript levels of IL-1β, IL-6, NFKB2, RELA and TNF-α were analyzed by qPCR and normalized to housekeeping genes HPRT and GAPDH. n = 6 wild-type + saline, 8 wild-type + LPS, 5 Tet2 knockout + saline, 10 Tet2 knockout + LPS. f, Tet2 inactivation does not induce differences in midbrain Tet1 or Tet3 transcript levels. Tet1 and Tet3 transcript levels in the midbrain of wild-type (Tet2+/+) and Tet2 knockout (Tet2-/-) mice 24 h after saline or LPS treatment (n = 4 wild-type + saline, 4 wild-type + LPS, 3 Tet2 knockout + saline, 3 Tet2 knockout + LPS). There was no significant change in Tet1 or Tet3 expression between mouse groups, as determined by a generalized linear model. Transcript levels in counts per million (cpm) from RNA-seq data. e, f, Boxplot center line is the mean, the bounds of the box are the lower and upper quartiles, and the whiskers extend to minimum and maximum data points within 1.5 times the interquartile range.
Extended Data Fig. 9 Tet2 inactivation protects against LPS induced motor learning deficits. Behavioral analysis in wild-type and Tet2 knockout mice 8 and 10 months after exposure to LPS or saline.
a, Rotarod learning assessed in wild-type and Tet2 knockout mice 8 months post-LPS or saline injection (*P = 0.04, **P < 0.01, repeated measures ANOVA). b, Rotarod analysis of wild-type and Tet2 knockout mice at 10 months post-LPS or saline injection. Open field analysis of wild-type and Tet2 knockout mice at (c) 8 months and (d) 10 months post-LPS or saline injection. Gait analysis using the CatWalk system for wild-type and Tet2 knockout mice measuring (e, f) average speed or (g, h) cadence at 8 months and 10 months post-LPS or saline injection. a‒h, All statistical analyses were performed using a two-way or repeated measures ANOVA followed by post hoc Tukey HSD test. 8 month: n = 11 wild-type + saline, 7 wild-type + LPS, 13–14 Tet2 knockout + saline, 11 Tet2 knockout + LPS; 10 month: n = 11 wild-type + saline, 7 wild-type + LPS, 11–12 Tet2 knockout + saline, 8 Tet2 knockout + LPS. Error bars representing s.e.m.
Extended Data Fig. 10 Proposed model displaying the contribution of TET2 to the development and progression of PD.
TET2 is responsible for the conversion of DNA methylation to hydroxymethylation, and TET2 inactivation results in cytosine modification changes relevant to those observed in PD neurons. In PD, there is epigenetic dysregulation of the TET2 locus and an upregulation of TET2 expression, which leads to increased hydroxymethylation. Based on our study and the existing literature13,27,43, this could have two main consequences. First, elevated TET2 levels increases susceptibility to immune activation, promoting neuroinflammation. Second, TET2 upregulation activates genes involved in neurogenesis in post-mitotic neurons. Both of these aberrant processes may lead to the neurodegeneration observed in PD patients.
Supplementary information
Supplementary Information
Supplementary Fig. 1.
Supplementary Software
Analysis code
Supplementary Table 1
Demographic and clinical information for study samples
Supplementary Table 2
Differentially methylated cytosine sites at enhancers in PD neurons
Supplementary Table 3
Differentially methylated regions at enhancers in PD neurons
Supplementary Table 4
Differentially methylated sites in PD neurons before and after the arrival of α-synuclein pathology in the prefrontal cortex
Supplementary Table 5
Differentially methylated cytosine sites at enhancers in PD neurons in replication cohort
Supplementary Table 6
Gene targets of differentially methylated enhancers in PD determined by chromatin interaction analysis in prefrontal cortex
Supplementary Table 7
Differentially methylated sites of PD medications at enhancers in PD neurons
Supplementary Table 8
Hydroxymethylation changes, genome-wide, in PD neurons determined by hMeDIP-seq
Supplementary Table 9
Hydroxymethylation changes at enhancers in PD neurons
Supplementary Table 10
Overlap of hydroxymethylation changes with differentially modified enhancers in PD
Supplementary Table 11
Differentially expressed genes in the prefrontal cortex of PD patients determined by RNA-seq
Supplementary Table 12
Pathway analysis integrating DNA methylation and transcriptomic alterations in PD brain
Supplementary Table 13
Epigenetically and transcriptionally dysregulated genes in PD
Supplementary Table 14
Differentially modified cytosines at the TET2 gene locus and surrounding genomic area
Supplementary Table 15
Differentially modified cytosines induced by TET2 knockdown in SH-SY5Y cells
Supplementary Table 16
Differentially modified cytosines induced by TET2 knockdown in vitro that overlap enhancers dysregulated in PD neurons
Supplementary Table 17
Transcriptomic alterations in wild-type and Tet2 knockout mice given saline or LPS
Supplementary Table 18
Pathways altered in wild-type and Tet2 knockout mice given saline or LPS
Supplementary Table 19
Primers
Supplementary Table 20
Antibodies
Supplementary Table 21
Computational tools used for study analyses
Source data
Source Data 1
Source data for Extended Data Fig. 6. Correlation of TET2 expression and TH protein levels.
Rights and permissions
About this article

Cite this article
Marshall, L.L., Killinger, B.A., Ensink, E. et al. Epigenomic analysis of Parkinson’s disease neurons identifies Tet2 loss as neuroprotective. Nat Neurosci 23, 1203–1214 (2020). https://doi.org/10.1038/s41593-020-0690-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41593-020-0690-y
This article is cited by
-
Inhibition of EHMT1/2 rescues synaptic damage and motor impairment in a PD mouse model
Cellular and Molecular Life Sciences (2024)
-
Epigenome-wide association study of peripheral immune cell populations in Parkinson’s disease
npj Parkinson's Disease (2023)
-
Functional genomics elucidates regulatory mechanisms of Parkinson’s disease-associated variants
BMC Medicine (2022)
-
Parkinson’s disease-associated, sex-specific changes in DNA methylation at PARK7 (DJ-1), SLC17A6 (VGLUT2), PTPRN2 (IA-2β), and NR4A2 (NURR1) in cortical neurons
npj Parkinson's Disease (2022)
-
Epigenome-wide association study of human frontal cortex identifies differential methylation in Lewy body pathology
Nature Communications (2022)
