
Abstract
Three new azaphilones, sassafrin E (1), sassafrin F (2), and sassafrinamine A (3), were isolated from the filamentous fungus Aspergillus neoglaber. The structures of the compounds were determined by nuclear magnetic resonance spectroscopy, and were found to be novel analogues of two already known compound classes; sassafrins and berkchaetoazaphilones. Sassafrin E and F were both oxygen containing, while sassafrinamine A additionally contained a nitrogen atom, originating from an aminoethanol moiety, as well as extensive conjugation resulting in an intense purple colour of the pure compound. The structure of sassafrin E was further confirmed using deuterium exchange experiments coupled with high-resolution tandem mass spectrometry.
Similar content being viewed by others


Key points
We present three new azaphilone pigments isolated from the fungus Aspergillus neoglaber. Two pigments (Sassafrin E and Sassafrin F) were yellow and the third (sassafrinamine A) purple. The structure of Sassafrin E was supported by tandem MS experiments of the deuterium labelled molecule.
Introduction
The use of natural or naturally derived pigments for the food and cosmetics industry has in recent years been of increasing interest, and filamentous fungi have long been known to produce various colourful secondary metabolites. Several of such compounds exist within the class of azaphilones, such as atrorosins, a group of red pigments that we recently characterised (Isbrandt et al. 2020). Azaphilones are common in the three large genera Aspergillus, Penicillium and Talaromyces (Osmanova et al. 2010; Samson et al. 2011; Romsdahl and Wang 2019). In Aspergillus, azaphilones have been found in the subgenera Circumdati and Nidulantes, but they are less common in subgenus Fumigati (Samson et al. 2007; Frisvad and Larsen 2016). Aspergillus fumigatus in subgenus Fumigati section Fumigati has been reported to produce epi-pinophilin B and other azaphilones (Zhang et al. 2019), and Aspergillus clavatus in subgenus Fumigati section Clavati has been reported to produce small azaphilones, such as felinone A and aspergillusone B (Wang et al. 2015).
One strategy for identifying new compounds is by looking into the secondary metabolites of underexplored species. The secondary metabolites of the filamentous fungus Aspergillus neoglaber (= Neosartorya glabra) from section Fumigati have not been well described, and only little work has been done to gain a better understanding of which compounds are produced by this species (Samson et al. 2007; Jayasuriya et al. 2009; Liu et al. 2015; Frisvad and Larsen 2016; May Zin et al. 2016). A few of the reported secondary metabolites include the bioactive glabramycins (Jayasuriya et al. 2009; Li 2015) and satoryglabrins (Liu et al. 2015), as well as various diketopiperazines and tetracyclopeptides (May Zin et al. 2016). When cultivated on solid media, we found Aspergillus neoglaber to color the surrounding media red, suggesting production of one or several red-pigmented compounds, none of which have been characterized as yet. This discovery prompted us to further investigate the nature of these predicted new metabolites, and thereby shed some light on this underexplored species.
Materials and methods
Solvents and instrumentation
All solvents were acquired from Sigma-Aldrich (St. Louis, Missouri, USA), ultra-pure water was made with a Milli-Q system (Millipore, Burlington, Massachusetts, USA).
Ultra-high Performance Liquid Chromatography-High Resolution Tandem Mass Spectrometry (UHPLC-HRMS/MS) was performed on an Agilent Infinity 1290 UHPLC system (Agilent Technologies, Santa Clara, CA, USA) equipped with a diode array detector. Separation was obtained on an Agilent Poroshell 120 phenyl-hexyl column (2.1 × 250 mm, 2.7 μm) with a linear gradient consisting of water (A) and acetonitrile (B) both buffered with 20 mM formic acid, starting at 10% B and increased to 100% in 15 min where it was held for 2 min, returned to 10% in 0.1 min and remaining for 3 min (0.35 mL/min, 60 °C). MS detection was performed in positive detection mode on an Agilent 6545 QTOF MS equipped with Agilent Dual Jet Stream electrospray ion source with a drying gas temperature of 250 °C, gas flow of 8 L/min, sheath gas temperature of 300 °C and flow of 12 L/min. Capillary voltage was set to 4000 V and nozzle voltage to 500 V. Mass spectra were recorded at 10, 20 and 40 eV as centroid data for m/z 85–1700 in MS mode and m/z 30–1700 in MS/MS mode, with an acquisition rate of 10 spectra/s. Lock mass solution in 70:30 methanol:water was infused in the second sprayer using an extra LC pump at a flow of 15 μL/min using a 1:100 splitter. The solution contained 1 μM tributylamine (Sigma-Aldrich) and 10 μM Hexakis (2,2,3,3-tetrafluoropropoxy)phosphazene (Apollo Scientific Ltd., Cheshire, UK) as lock masses. The [M + H]+ ions (m/z 186.2216 and 922.0098 respectively) of both compounds were used.
1D and 2D NMR spectra were recorded on a Bruker Avance 600 MHz or Bruker Avance 800 MHz spectrometer (Bruker, Billerica, MA, USA). NMR spectra were acquired using standard pulse sequences. The solvent used was CD3OD, and residual MeOH-d3 was used as references with signals at δH = 3.31 ppm and δC = 49.0 ppm. Data processing and analysis was done using TopSpin 3.5pl7 (Bruker). J-couplings are reported in hertz (Hz) and chemical shifts in ppm (δ).
Deuterium exchange of 1 was achieved by leaving the compound in CD3OD for 5 days at 5 °C.
Strain and purification
The strain used for this study was Aspergillus neoglaber IBT 3020, obtained from the DTU strain collection. For large scale extractions, the fungus was grown in six 2L conical flasks each with 500 mL of yeast extract sucrose (YES) medium containing only 10% of the normal amount of agar.
Biomass and media was separated by decantation, and extraction was done twice on the biomass, using ethyl acetate (EtOAc) acidified with 1% formic acid. Initial fractionation of the extract was done on an Isolera One (Biotage) flash system using a diol column eluted stepwise with dichloromethane (DCM), DCM:EtOAc (1:1), EtOAc, EtOAc:MeOH (1:1), and MeOH. Final isolation of the pure compounds was done using a semi-preparative Waters 600 Controller with a 996 photodiode array detector (Waters, Milford, MA, USA) equipped with a Luna II C18 column (250 × 10 mm, 5 μm, Phenomenex), using a H2O/acetonitrile gradient with 50 ppm TFA.
Results
Analysis by ultra-high performance liquid chromatography coupled to diode array detection and high resolution tandem mass spectrometry (UHPLC-DAD-HRMS/MS) of the ethyl acetate extract from the filamentous fungus Aspergillus neoglaber IBT 3020 = CBS 111.55, which is the ex-type culture of the species (Fig. 1), identified the major secondary metabolite as a yellow compound (1) absorbing at 345 nm with an m/z of 453.2277 Da ([M + H]+) and molecular formula C27H32O6. A second, also yellow compound (2) absorbing at 360 nm and with a mass and molecular formula corresponding to the addition of two protons to 1 was tentatively identified as a likely analogue (m/z 455.2428, [M + H]+, C27H34O6). In addition to the two yellow compounds, a third, red–purple compound (3) absorbing at 545 nm with a m/z of 494.2541, corresponding to a molecular formula of C29H36NO6, and found to be responsible for the red colour of the extract was also identified. UV–VIS spectra acquired during HPLC analysis for each of the three compounds can be found in Additional file 1: Fig. S1.

Base peak chromatogram (LC-DAD-HRMS, ESI +) of the ethyl acetate extract from A. neoglaber grown on YES medium
In order to purify the three compounds, A. neoglaber IBT 3020 was cultivated on 6 × 500 mL semi-liquid YES media. The biomass was extracted with ethyl acetate, and purification was done using normal phase flash chromatography followed by semi-preparative RP-HPLC. One- and two-dimensional NMR experiments were used in order to elucidate the structures of the compounds.
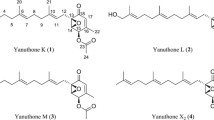
In compound 1, a total of 31 protons could be identified from the 1H-spectrum, suggesting one exchangeable proton. In combination with multiplicity edited HSQC (edHSQC), 12 CH-groups, two CH2-groups, and five CH3-groups could be identified. Eight of the CH-groups had carbon shifts matching alkenes, and one CH-group was identified to be attached to a hydroxyl group, accounting for the 32nd proton not observed in the 1H-spectrum. Both CH2-groups appeared as diastereotopic. 3J H–H couplings obtained from DQF-COSY, identified four spin systems consisting of H1 to H3, H13 to H16 and C15-CH3, H18 to H21, and H10 and H23, as well as five singlets. Correlations in the DQF-COSY, were all confirmed by H2BC. HMBC correlations linked H15, C17–CH3 and H18 to C17, and H10 and H20 to C22. Additionally, H1, H2 and H3, along with H5 was linked to C4. Ambiguity in the HMBC around C6 and C11 meant that additional, more specific experiments were needed, and 1,n- and 1,1-ADEQUATE (Reif et al. 1996) experiments were used to connect H5 and H7 to C6, H10 to C11, as well as C9-CH3 to C24 (Fig. 2). NOESY correlations around the lactone could assign relative stereochemistry to the methyl group C9-CH3 and the two protons H10 and H23. In summary, compound 1 turned out to be a novel azaphilone, with high structural similarity to groups of compounds such as sassafrins (Quang et al. 2005) and berkchaetoazaphilones (Stierle et al. 2015), and has been named sassafrin E.

Structure and key HMBC (plain arrows) and ADEQUATE (dashed arrows) correlations for sassafrin E (1)
The NMR data for compounds 2 and 3, was highly similar to that of 1, with only few variations. Compound 2 was determined to only differ from 1, by having the ketone at C22 being reduced to a hydroxyl group and has been named sassafrin F.
Compared to compound 1, compound 3 (m/z 494.2541, [M + H]+, C29H36NO6) included three additional hydrogen atoms, two more carbon atoms, as well as a nitrogen. From the edHSQC spectrum, the additional carbon atoms were determined to be two CH2-groups (C1′ and C2′). HMBC correlations from H12 to C1′ determined the two-carbon moiety to be an N-linked aminoethanol, generating an isoquinoline ring as shown in Fig. 3. Furthermore, no protons could be linked to carbons C10 and C23, and it is therefore assumed that these are connected via a double bond. Similarly, no correlations to C6 were observed. The UV–VIS spectrum for 3 was quite unique, with slight absorption all the way from 270 nm to 580 nm, with maximum at 545 nm, and the extensive conjugation is in agreement with the violet/purple colour of the pure compound. Compound 3 has been named sassarinamine A,Footnote 1 based on the incorporation of nitrogen, a key feature of azaphilones. Structures of compounds 1, 2, and 3 are shown in Fig. 3 and chemical shifts are listed in Table 1. Recorded NMR spectra for each compounds can be found in Additional file 1.

Numbered structures of sassafrin E (1), sassafrin F (2), and sassafrinamine A (3). Relative configuration is shown for C9, C10 and C23
In addition to the NMR experiments, we were able to further confirm the structure of sassafrin E by exchanging H23 with deuterium, and using the isotope labelled fragments in tandem MS experiments to generate the MS/MS spectra found in Additional file 1: Fig. S2 and S3. In this way, three destinct fragmentation pathways for the molecule could be suggested as depicted in Additional file 1: Fig. S4.
Discussion
During the initial stage of this study, 1 was suspected to be the acetylcholineesterase inhibitor arisugacin C (Otoguro et al. 1997, 2000) or an analogue hereof. However, dereplication by comparison of retention time, absorption spectra, and fragmentation pattern (Additional file 1: Fig. S5) with an extract from Penicillium echinulatum, a known producer of arisugacin C, quickly clarified that the compound in A. neoglaber was not an arisugacin, but rather belonging to an entirely different class of compounds.
The isolated compounds were found to be members of the compound class azaphilones, a diverse group of compounds, such as the ones obtained from various Monascus species, i.e. the so-called Monascus pigments (Gao et al. 2013), including the newly described subclass atrorosins of which we recently reported nineteen new analogues (Isbrandt et al. 2020). The exact biosynthetic pathway of these compounds have not been fully elucidated, although some work has been done in order to propose a possible route (Hajjaj et al. 1999; Somoza et al. 2012; Liu et al. 2014; Woo et al. 2015; Chen et al. 2017; Tolborg et al. 2017). We therefore expect that the compounds described in this study are biosynthesised in a similar fashion, as outlined in Fig. 4. However, in contrast to many azaphilone pigments, e.g. the Monascus pigments which are made from a hexaketide moiety and a 3-oxo-fatty acid, we propose sassafrin E and F, and sassafrinamine A to be constructed from two polyketides, due to the low level of reduction in the side chain (C13 to C24). For Sassafrin F, we further expect the reduction of the ketone at C22 to happen after construction of the compound backbone, as fungal PKSs are well known to be stringent with regard to the reduction pattern of their products, why it is unlikely that two different PKs would originate from the same biosynthetic PKS. This hypothesis is further strengthened when also considering the structures of the previously described sassafrins (Quang et al. 2005), all containing different levels of reduction in the side chain, as well as the berkchaetoazaphilones (Stierle et al. 2015) containing fully reduced side chains. The fusion of the lactone ring also differs from the linear fashion found in Monascus pigments and is more similar to compounds such as chaetoviridins (Winter et al. 2012).

Proposed biosynthetic pathway, for compounds 1, 2 and 3
As we have recently discovered for the compound class atrorosins (Isbrandt et al. 2020), the incorporation of nitrogen into the isochromene system can be done using various primary amine containing compounds (Tolborg et al. 2020). We speculate that the nitrogen, and additional carbons and oxygen in 3 originate from a decarboxylated serine moiety, i.e. ethanolamine which is abundant in cells as constituents in phospholipids in cell membranes (Wellner et al. 2013). Additionally, further investigation of the crude extract tentatively revealed an aminoethanol derivative of 2, as well as the non-nitrogen containing precursor of 3 to be present in lower amounts. A common feature for both the compounds characterised in this study and the previously described atrorosins, is the observed CO loss during tandem MS experiments (see Additional file 1: Fig. S4).
Availability of data and materials
Not applicable.
Notes
Low amounts of compound meant that signals for C6, C10 and C23 were not observed for 3, and the tricyclic part of the structure was determined based on similarities with the remaining signals, compared to compound 1 and 2.
References
Chen W, Chen R, Liu Q, He Y, He K, Ding X, Kang L, Guo X, Xie N, Zhou Y, Lu Y, Cox RJ, Molnár I, Li M, Shao Y, Chen F (2017) Orange, red, yellow: biosynthesis of azaphilone pigments in Monascus fungi. Chem Sci 8:4917–4925. https://doi.org/10.1039/C7SC00475C
Frisvad JC, Larsen TO (2016) Extrolites of Aspergillus fumigatus and other pathogenic species in aspergillus section fumigati. Front Microbiol 6:1–14. https://doi.org/10.3389/fmicb.2015.01485
Gao J, Yang S, Qin J (2013) Azaphilones: chemistry and biology. Chem Rev 113:4755–4811. https://doi.org/10.1021/cr300402y
Hajjaj H, Klaébé A, Loret MO, Goma G, Blanc PJ, François J (1999) Biosynthetic pathway of citrinin in the filamentous fungus Monascus ruber as revealed by 13C nuclear magnetic resonance. Appl Environ Microbiol 65:311–314
Isbrandt T, Tolborg G, Ødum A, Workman M, Larsen TO (2020) Atrorosins: a new subgroup of Monascus pigments from Talaromyces atroroseus. Appl Microbiol Biotechnol 104:615–622. https://doi.org/10.1007/s00253-019-10216-3
Jayasuriya H, Zink D, Basilio A, Vicente F, Collado J, Bills G, Goldman ML, Motyl M, Huber J, Dezeny G, Byrne K, Singh SB (2009) Discovery and antibacterial activity of glabramycin A-C from Neosartorya glabra by an antisense strategy. J Antibiot (Tokyo) 62:265–269. https://doi.org/10.1038/ja.2009.26
Li Y (2015) Structural revision of glabramycins B and C, antibiotics from the fungus Neosartorya glabra by DFT calculations of NMR chemical shifts and coupling constants. RSC Adv 5:36858–36864. https://doi.org/10.1039/C5RA01753J
Liu Q, Xie N, He Y, Wang L, Shao Y, Zhao H, Chen F (2014) MpigE, a gene involved in pigment biosynthesis in Monascus ruber M7. Appl Microbiol Biotechnol 98:285–296. https://doi.org/10.1007/s00253-013-5289-8
Liu W, Zhao H, Li R, Zheng H, Yu Q (2015) Polyketides and Meroterpenoids from Neosartorya glabra. Helv Chim Acta 98:515–519
May Zin W, Buttachon S, Dethoup T, Fernandes C, Cravo S, Pinto M, Gales L, Pereira J, Silva A, Sekeroglu N, Kijjoa A (2016) New cyclotetrapeptides and a new diketopiperzine derivative from the marine sponge-associated fungus Neosartorya glabra KUFA 0702. Mar Drugs 14:136. https://doi.org/10.3390/md14070136
Osmanova N, Schultze W, Ayoub N (2010) Azaphilones: a class of fungal metabolites with diverse biological activities. Phytochem Rev. https://doi.org/10.1007/s11101-010-9171-3
Otoguro K, Kuno F, Omura S (1997) Arisugacins, selective acetylcholinesterase inhibitors of microbial origin. Pharmacol Ther 76:45–54. https://doi.org/10.1016/S0163-7258(97)00093-4
Otoguro K, Shiomi K, Yamaguchi Y, Arai N, Sunazuka T, Masuma R, Iwai Y, Omura S (2000) Arisugacins C and D, novel acetylcholinesterase inhibitors and their related novel metabolites produced by Penicillium sp. FO-4259-11. J Antibiot (Tokyo) 53:50–57. https://doi.org/10.7164/antibiotics.53.50
Quang DN, Hashimoto T, Fournier J, Stadler M, Radulović N, Asakawa Y (2005) Sassafrins A-D, new antimicrobial azaphilones from the fungus Creosphaeria sassafras. Tetrahedron 61:1743–1748. https://doi.org/10.1016/j.tet.2004.12.031
Reif B, Köck M, Kerssebaum R, Kang H, Fenical W, Griesinger C (1996) ADEQUATE, a new set of experiments to determine the constitution of small molecules at natural abundance. J Magn Reson, Ser A 118:282–285. https://doi.org/10.1006/jmra.1996.0038
Romsdahl J, Wang CCC (2019) Recent advances in the genome mining of Aspergillus secondary metabolites (covering 2012–2018). Medchemcomm 10:840–866. https://doi.org/10.1039/C9MD00054B
Samson RA, Hong S, Peterson SW, Frisvad JC, Varga J (2007) Polyphasic taxonomy of Aspergillus section Fumigati and its teleomorph Neosartorya. Stud Mycol 59:147–203. https://doi.org/10.3114/sim.2007.59.14
Samson RA, Yilmaz N, Houbraken J, Spierenburg H, Seifert KA, Peterson SW, Varga J, Frisvad JC (2011) Phylogeny and nomenclature of the genus Talaromyces and taxa accommodated in Penicillium subgenus Biverticillium. Stud Mycol 70:159–183. https://doi.org/10.3114/sim.2011.70.04
Somoza AD, Lee K-H, Chiang Y-M, Oakley BR, Wang CCC (2012) Reengineering an azaphilone biosynthesis pathway in Aspergillus nidulans to create lipoxygenase inhibitors. Org Lett 14:972–975. https://doi.org/10.1021/ol203094k
Stierle AA, Stierle DB, Girtsman T, Mou TC, Antczak C, Djaballah H (2015) Azaphilones from an acid mine extremophile strain of a Pleurostomophora sp. J Nat Prod 78:2917–2923. https://doi.org/10.1021/acs.jnatprod.5b00519
Tolborg G, Isbrandt T, Larsen TO, Workman M (2017) Establishing novel cell factories producing natural pigments in Europe. In: Singh OV (ed) Bio-pigmentation and biotechnological implementations, 1st edn. Wiley, Hoboken, pp 23–60
Tolborg G, Ødum ASR, Isbrandt T, Larsen TO, Workman M (2020) Unique processes yielding pure azaphilones in Talaromyces atroroseus. Appl Microbiol Biotechnol 104:603–613. https://doi.org/10.1007/s00253-019-10112-w
Wang J, Bai G, Liu Y, Wang H, Li Y, Yin W, Wang Y, Lu F (2015) Cytotoxic metabolites produced by the endophytic fungus Aspergillus clavatus. Chem Lett 44:1148–1149. https://doi.org/10.1246/cl.150417
Wellner N, Diep TA, Janfelt C, Hansen HS (2013) N-acylation of phosphatidylethanolamine and its biological functions in mammals. Biochim Biophys Acta - Mol Cell Biol Lipids 1831:652–662. https://doi.org/10.1016/j.bbalip.2012.08.019
Winter JM, Sato M, Sugimoto S, Chiou G, Garg NK, Tang Y, Watanabe K (2012) Identification and Characterization of the Chaetoviridin and Chaetomugilin Gene Cluster in Chaetomium globosum reveal dual functions of an iterative highly-reducing polyketide synthase. J Am Chem Soc 134:17900–17903. https://doi.org/10.1021/ja3090498
Woo PCY, Lam C-W, Tam EWT, Lee K-C, Yung KKY, Leung CKF, Sze K-H, Lau SKP, Yuen K-Y (2015) The biosynthetic pathway for a thousand-year-old natural food colorant and citrinin in Penicillium marneffei. Sci Rep 4:6728. https://doi.org/10.1038/srep06728
Zhang Y, Peng X, Feng L, Zhu H, Cao F, Wang C-Y (2019) A new epimer of azaphilone derivative pinophilin B from the gorgonian-derived fungus Aspergillus fumigatus 14–27. Nat Prod Res. https://doi.org/10.1080/14786419.2019.1669028
Acknowledgements
We thank Kasper Enemark-Rasmussen at the Department of Chemistry, Technical University of Denmark, for running NMR experiments.
Funding
We thank the Novo Nordisk Foundation for their Grant support (NNF16O23644 and NNF18OC33388).
Author information
Authors and Affiliations
Contributions
TI and TOL conceived and designed research. TI and AM conducted experiments. TI analyzed data. TI, TOL and JCF wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Compliance with ethical standards
This article does not contain any studies with human participants or animals performed by any of the authors.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declares that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
UV-, MS/MS and NMR spectra of sassafrin E, F and sassafrinamine A, as well as dereplication and proposed MS/MS fragmentation pathway for sassafrin E.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Isbrandt, T., Frisvad, J.C., Madsen, A. et al. New azaphilones from Aspergillus neoglaber. AMB Expr 10, 145 (2020). https://doi.org/10.1186/s13568-020-01078-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-020-01078-4