Mirtazapine Reduces Adipocyte Hypertrophy and Increases Glucose Transporter Expression in Obese Mice

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Dietary Induction of Obesity, and Mirtazapine Treatment
2.2. Insulin Level, Food Intake, and Body Weight Measurement
2.3. Serum Triglyceride and Aspartate Aminotransferase Level Measurement
2.4. Intraperitoneal Glucose Tolerance Test
2.5. IR and Insulin Sensitivity Index
2.6. Morphometric and Histological Analyses of Tissues
2.7. Western Blotting
2.8. Statistical Analysis
3. Results
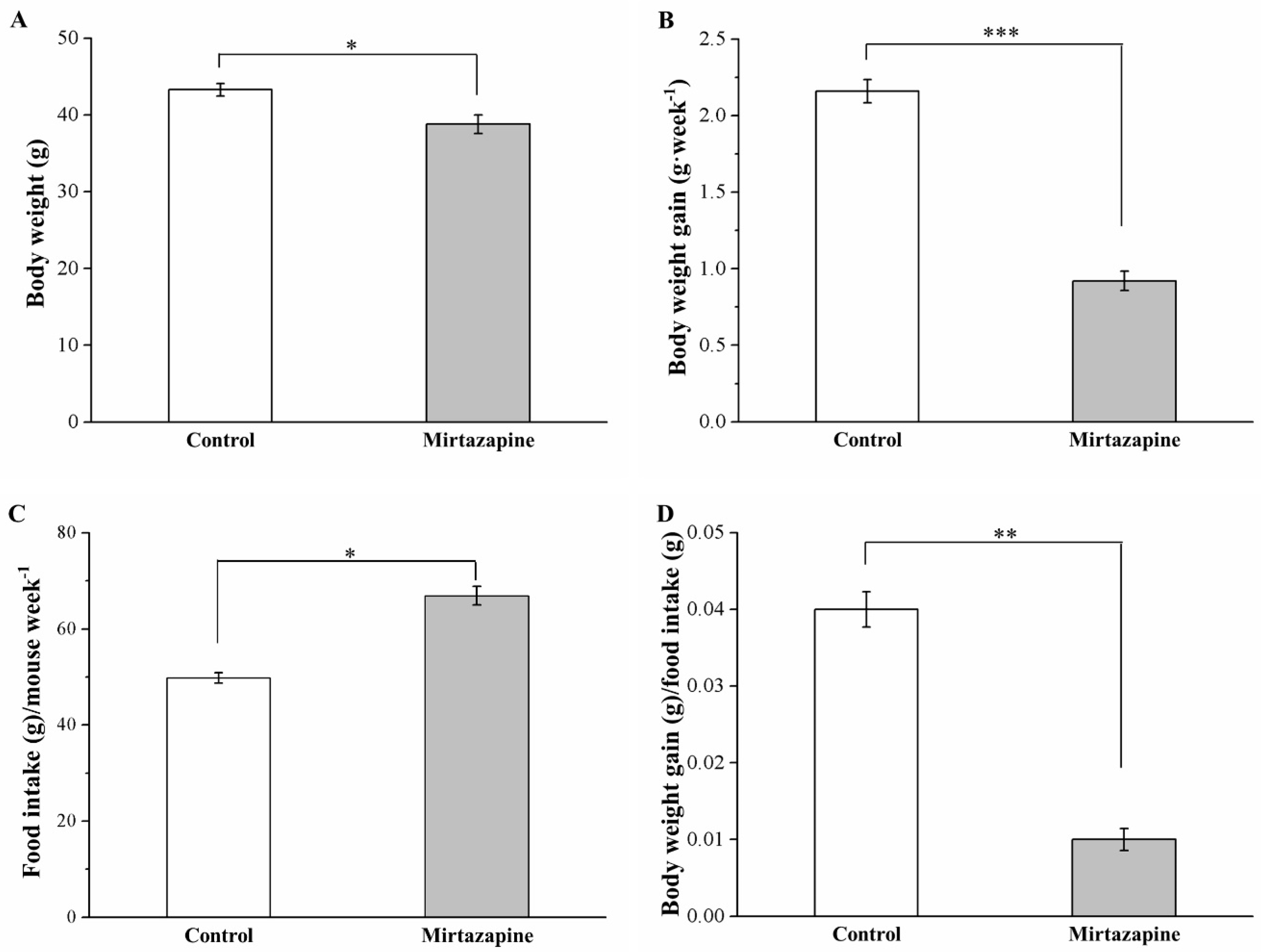
3.1. Mirtazapine Affects Morphometric Parameters and Food Intake and Efficiency
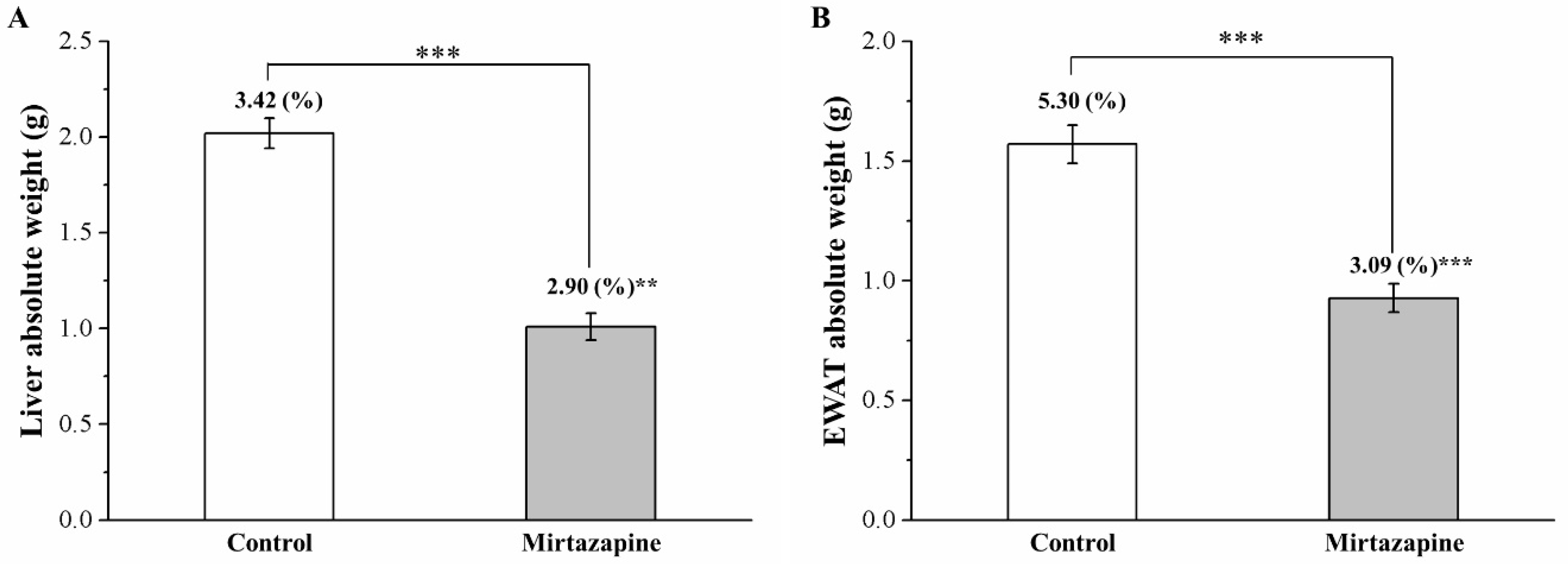
3.2. Mirtazapine Reduces Liver and Fat Pad Weights
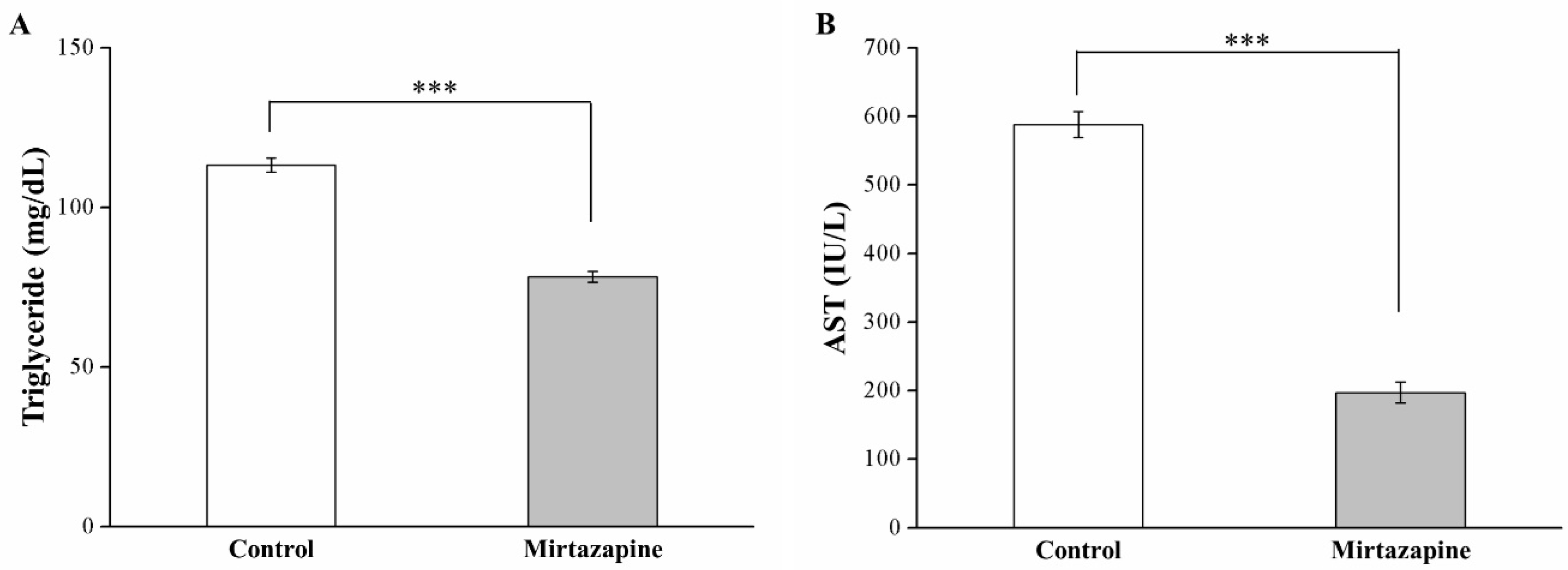
3.3. Mirtazapine Reduces Serum Triglyceride and AST Levels
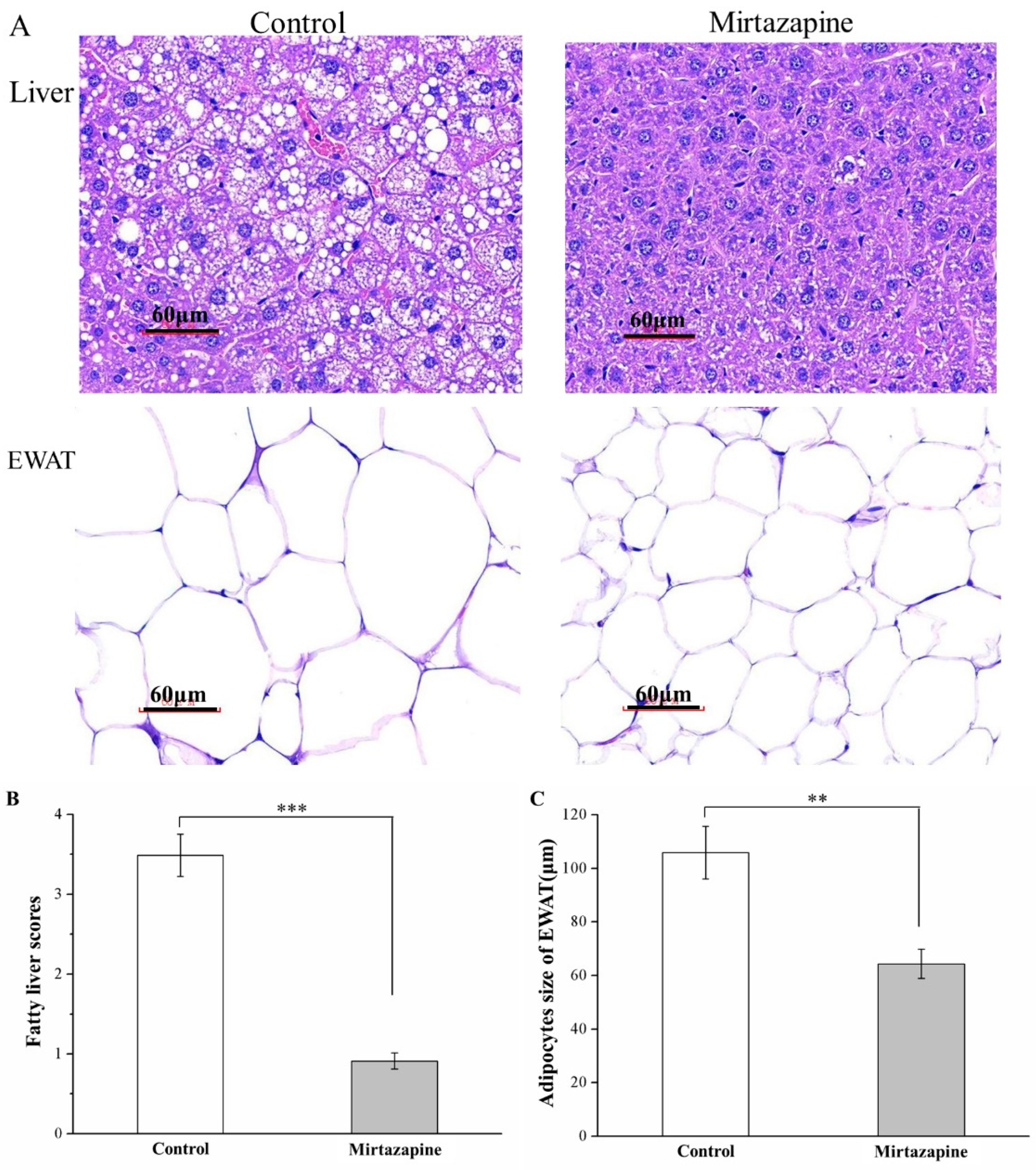
3.4. Mirtazapine Reduces Liver Fat Accumulation and Adipocyte Size
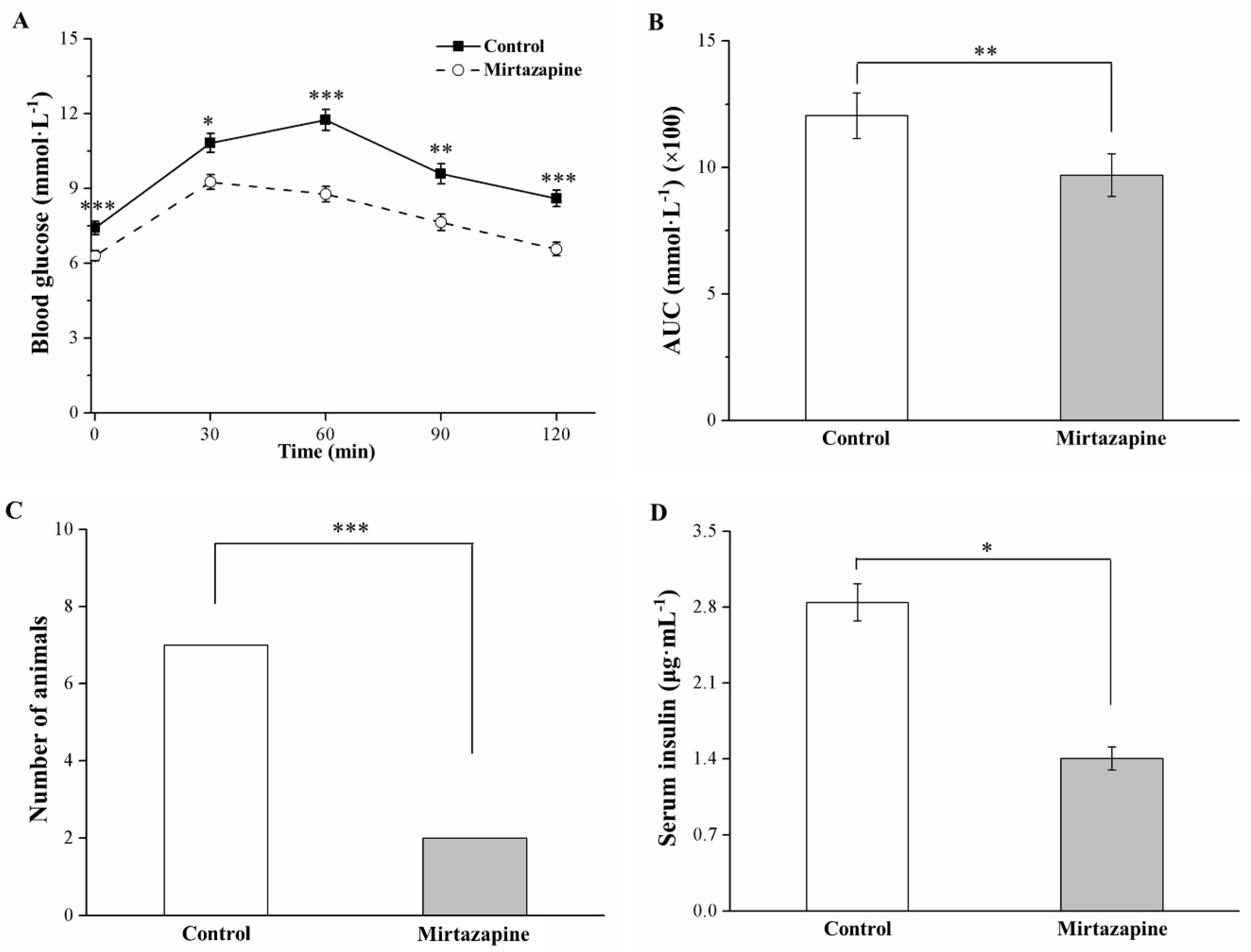
3.5. Mirtazapine Alleviates Glucose Intolerance and Reduces Insulin Level
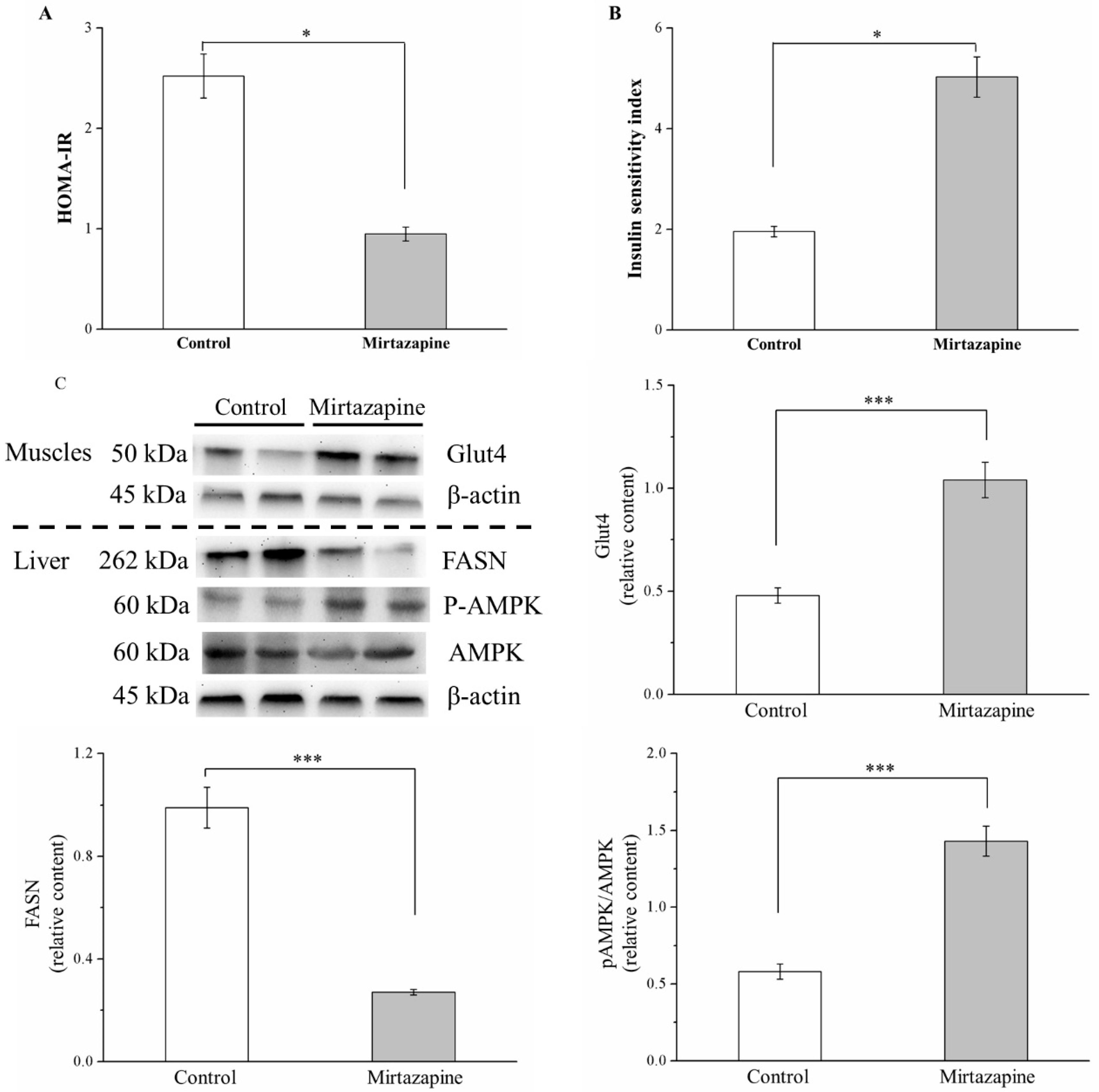
3.6. Mirtazapine Enhances IS by Altering GLUT4 Expression, Reducing FASN Expression, and Lipogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davis, R.; Wilde, M. Mirtazapine: A review of its pharmacology and therapeutic potential in the management of major depression. CNS Drugs 1996, 5, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Croom, K.F.; Perry, C.M.; Plosker, G.L. Mirtazapine: A review of its use in major depression and other psychiatric disorders. CNS Drugs 2009, 23, 427–452. [Google Scholar] [CrossRef] [PubMed]
- Blier, P. Crosstalk between the norepinephrine and serotonin systems and its role in the antidepressant response. J. Psychiatry Neurosci. 2001, 26, S3–S10. [Google Scholar] [PubMed]
- Carpenter, L.L.; Leon, Z.; Yasmin, S.; Price, L.H. Clinical Experience with Mirtazapine in the Treatment of Panic Disorder. Ann. Clin. Psychiatry 1999, 11, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Quimby, J.M.; Lunn, K. Mirtazapine as an appetite stimulant and anti-emetic in cats with chronic kidney disease: A masked placebo-controlled crossover clinical trial. Vet. J. 2013, 197, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Bays, H.E. Current and Investigational Antiobesity Agents and Obesity Therapeutic Treatment Targets. Obes. Res. 2004, 12, 1197–1211. [Google Scholar] [CrossRef]
- Grabarczyk, T.R. Observational comparative effectiveness of pharmaceutical treatments for obesity within the veterans health administration. Pharmacotherapy 2018, 38, 19–28. [Google Scholar] [CrossRef]
- Chang, G.-R.; Chiu, Y.-S.; Wu, Y.-Y.; Chen, W.-Y.; Liao, J.-W.; Chao, T.-H.; Mao, F.C. Rapamycin Protects Against High Fat Diet–Induced Obesity in C57BL/6J Mice. J. Pharmacol. Sci. 2009, 109, 496–503. [Google Scholar] [CrossRef]
- Chang, G.-R.; Chen, W.-K.; Hou, P.-H.; Mao, F.C. Isoproterenol exacerbates hyperglycemia and modulates chromium distribution in mice fed with a high fat diet. J. Trace Elem. Med. Biol. 2017, 44, 315–332. [Google Scholar] [CrossRef]
- Hou, P.-H.; Chang, G.-R.; Chen, C.-P.; Lin, Y.-L.; Chao, I.-S.; Shen, T.-T.; Mao, F.C. Long-term administration of olanzapine induces adiposity and increases hepatic fatty acid desaturation protein in female C57BL/6J mice. Iran. J Basic Med Sci 2018, 21, 495–501. [Google Scholar] [PubMed]
- Heiser, P.; Singh, S.; Krieg, J.; Vedder, H. Effects of different antipsychotics and the antidepressant mirtazapine on glucose transporter mRNA levels in human blood cells. J. Psychiatr. Res. 2006, 40, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Correll, C.U.; Lencz, T.; Malhotra, A.K. Antipsychotic drugs and obesity. Trends Mol. Med. 2011, 17, 97–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, D.S.; Pinkofsky, H.B.; Liu, Y.; Bradley, R.J. Antipsychotic drugs affect glucose uptake and the expression of glucose transporters in PC12 cells. Prog. Neuro Psychopharmacol. Biol. Psychiatry 1999, 23, 69–80. [Google Scholar] [CrossRef]
- Wojtaszewski, J.F.; Higaki, Y.; Hirshman, M.F.; Michael, M.D.; Dufresne, S.D.; Kahn, C.R.; Goodyear, L.J. Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J. Clin. Investig. 1999, 104, 1257–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dazzi, L.; Ladu, S.; Spiga, F.; Vacca, G.; Rivano, A.; Pira, L.; Biggio, G. Chronic treatment with imipramine or mirtazapine antagonizes stress- and FG7142-induced increase in cortical norepinephrine output in freely moving rats. Synapse 2002, 43, 70–77. [Google Scholar] [CrossRef]
- Rogóz, Z.; Skuza, G.; Legutko, B. Repeated treatment with mirtazepine induces brain-derived neurotrophic factor gene expression in rats. J. Physiol. Pharmacol. 2005, 56, 661–671. [Google Scholar]
- Fang, C.-K.; Chen, H.-W.; Chiang, I.-T.; Chen, C.-C.; Liao, J.-F.; Su, T.-P.; Tung, C.-Y.; Uchitomi, Y.; Hwang, J.-J. Mirtazapine Inhibits Tumor Growth via Immune Response and Serotonergic System. PLoS ONE 2012, 7, e3888. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, S.; Zhao, Z.; Zhao, P.; Chen, L.; Mei, Z. Antidiabetic activities of entagenic acid in type 2 diabetic db/db mice and L6 myotubes via AMPK/GLUT4 pathway. J. Ethnopharmacol. 2018, 211, 366–374. [Google Scholar] [CrossRef]
- Chang, G.-R.; Chiu, Y.-S.; Wu, Y.-Y.; Lin, Y.-C.; Hou, P.-H.; Mao, F.C. Rapamycin impairs HPD-induced beneficial effects on glucose homeostasis. Br. J. Pharmacol. 2015, 172, 3793–3804. [Google Scholar] [CrossRef] [Green Version]
- Chang, G.-R.; Hou, P.-H.; Chen, W.-K.; Lin, C.-T.; Tsai, H.-P.; Mao, F.C. Exercise Affects Blood Glucose Levels and Tissue Chromium Distribution in High-Fat Diet-Fed C57BL6 Mice. Molecules 2020, 25, 1658. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Huan, Y.; Huang, H.; Song, G.-M.; Sun, S.-J.; Shen, Z.-F. Atorvastatin improves insulin sensitivity in mice with obesity induced by monosodium glutamate. Acta Pharmacol. Sin. 2009, 31, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, G.-R.; Wu, Y.-Y.; Chiu, Y.-S.; Chen, W.-Y.; Liao, J.-W.; Hsu, H.-M.; Chao, T.-H.; Hung, S.-W.; Mao, F.C. Long-term Administration of Rapamycin Reduces Adiposity, but Impairs Glucose Tolerance in High-Fat Diet-fed KK/HlJ Mice. Basic Clin. Pharmacol. Toxicol. 2009, 105, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, D.; Carlsson, M.; Almgren, P.; Isomaa, B.; Taskinen, M.-R.; Tuomi, T.; Groop, L.C. Insulin secretion and insulin sensitivity in relation to glucose tolerance: Lessons from the Botnia Study. Diabetes 2000, 49, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Tanbouly, D.M.; Wadie, W.; Sayed, R.H. Modulation of TGF-β/Smad and ERK signaling pathways mediates the anti-fibrotic effect of mirtazapine in mice. Toxicol. Appl. Pharmacol. 2017, 329, 224–230. [Google Scholar] [CrossRef]
- Abosi, O.; Lopes, S.; Schmitz, S.; Fiedorowicz, J.G. Cardiometabolic effects of psychotropic medications. Horm. Mol. Biol. Clin. Investig. 2018, 36, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Raeder, M.B.; Fernø, J.; Vik-Mo, A.O.; Steen, N.E. SREBP Activation by Antipsychotic- and Antidepressant-Drugs in Cultured Human Liver Cells: Relevance for Metabolic Side-Effects? Mol. Cell. Biochem. 2006, 289, 167–173. [Google Scholar] [CrossRef]
- Ye, L.; Cao, Z.; Lai, X.; Shi, Y.; Zhou, N. Niacin Ameliorates hepatic steatosis by inhibiting de novo lipogenesis via a GPR109A-mediated PKC-ERK1/2-AMPK signaling pathway in C57BL/6 mice fed a high-fat diet. J. Nutr. 2020, 150, 672–684. [Google Scholar] [CrossRef]
- Hao, J.; Liu, S.; Zhao, S.; Liu, Q.; Lv, X.; Chen, H.; Niu, Y.; Duan, H. PI3K/Akt pathway mediates high glucose-induced lipogenesis and extracellular matrix accumulation in HKC cells through regulation of SREBP-1 and TGF-β1. Histochem. Cell Biol. 2011, 135, 173–181. [Google Scholar] [CrossRef]
- Li, J.; Gong, L.; Liu, S.; Zhang, Y.; Zhang, C.; Tian, M.; Lu, H.; Bu, P.; Yang, J.; Ouyang, C.; et al. Adipose HuR protects against diet-induced obesity and insulin resistance. Nat. Commun. 2019, 10, 2375. [Google Scholar] [CrossRef] [Green Version]
- Jocken, J.W.E.; Langin, D.; Smit, E.; Saris, W.H.M.; Valle, C.; Hul, G.B.; Holm, C.; Arner, P.; Blaak, E.E. Adipose Triglyceride Lipase and Hormone-Sensitive Lipase Protein Expression Is Decreased in the Obese Insulin-Resistant State. J. Clin. Endocrinol. Metab. 2007, 92, 2292–2299. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase To Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wei, W.; Li, N.-J.; Yuan, W.; Ding, Y.; Yang, W.-D.; Liu, J.-S.; Balamurugan, S.; Li, H.-Y. Heterogeneous expression of human PNPLA3 triggers algal lipid accumulation and lipid droplet enlargement. Algal Res. 2018, 31, 276–281. [Google Scholar] [CrossRef]
- Clarke, S.D. Regulation of fatty acid synthase gene expression: An approach for reducing fat accumulation. J. Anim. Sci. 1993, 71, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Kraus, T.; Haack, M.; Schuld, A.; Hinze-Selch, D.; Koethe, D.; Pollmächer, T. Body Weight, the Tumor Necrosis Factor System, and Leptin Production during Treatment with Mirtazapine or Venlafaxine. Pharmacopsychiatry 2002, 35, 220–225. [Google Scholar] [CrossRef]
- Schilling, C.; Gilles, M.; Blum, W.F.; Daseking, E.; Colla, M.; Weber-Hamann, B.; Lederbogen, F.; Krumm, B.; Heuser, I.; Wudy, S.A.; et al. Leptin Plasma Concentrations Increase During Antidepressant Treatment With Amitriptyline and Mirtazapine, But Not Paroxetine and Venlafaxine. J. Clin. Psychopharmacol. 2013, 33, 99–103. [Google Scholar] [CrossRef]
- Chau, M.D.L.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK–SIRT1–PGC-1α pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Wu, Z.; Yin, X.; Liu, Y.; Yan, X.; Lin, S.; Xiao, J.; Wang, X.; Feng, W.; Li, X.-K. Serum Levels of FGF-21 Are Increased in Coronary Heart Disease Patients and Are Independently Associated with Adverse Lipid Profile. PLoS ONE 2010, 5, e15534. [Google Scholar] [CrossRef]
- Sitsen, J.M.A.; Zivkov, M. Mirtazapine. CNS Drugs 1995, 4, 39–48. [Google Scholar] [CrossRef]
- Holm, K.J.; Markham, A. Mirtazapine. Drugs 1999, 57, 607–631. [Google Scholar] [CrossRef]
- Jiang, S.-M.; Wu, J.-H.; Jia, L. Intervention of Mirtazapine on gemcitabine-induced mild cachexia in nude mice with pancreatic carcinoma xenografts. World J. Gastroenterol. 2012, 18, 2867–2871. [Google Scholar] [CrossRef]
- Coccurello, R.; Moles, A. Potential mechanisms of atypical antipsychotic-induced metabolic derangement: Clues for understanding obesity and novel drug design. Pharmacol. Ther. 2010, 127, 210–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.L.; Spinowitz, N.; Karwa, M. Hypertriglyceridemia, acute pancreatitis, and diabetic ketoacidosis possibly associated with mirtazapine therapy: A case report. Pharmacotherapy 2003, 23, 940–944. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, L.M.; Ford, A.L.; Esposito, S.M.; Ekstrom, R.D.; Golden, R.N. The effects of mirtazapine on plasma lipid profiles in healthy subjects. J. Clin. Psychiatry. 2003, 64, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Urstad, A.P.L.; Semenkovich, C.F. Fatty acid synthase and liver triglyceride metabolism: Housekeeper or messenger? Biochim. Biophys. Acta 2012, 1821, 747–753. [Google Scholar] [CrossRef] [Green Version]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–883. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Salerno, A.; Ramírez, C.M.; Guo, L.; Allen, R.M.; Yin, X.; Langley, S.R.; Esau, C.; Wanschel, A.; Fisher, E.A.; et al. Long-term therapeutic silencing of miR-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol. Med. 2014, 6, 1133–1141. [Google Scholar] [CrossRef]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.-J.; et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Mancini, S.J.; White, A.D.; Bijland, S.; Rutherford, C.; Graham, D.; Richter, E.A.; Viollet, B.; Touyz, R.M.; Palmer, T.M.; Salt, I.P. Activation of AMP-activated protein kinase rapidly suppresses multiple pro-inflammatory pathways in adipocytes including IL-1 receptor-associated kinase-4 phosphorylation. Mol. Cell. Endocrinol. 2017, 440, 44–56. [Google Scholar] [CrossRef]
- Hajduch, E.; Rencurel, F.; Balendran, A.; Batty, I.H.; Downes, C.P.; Hundal, H.S. Serotonin (5-Hydroxytryptamine), a Novel Regulator of Glucose Transport in Rat Skeletal Muscle. J. Biol. Chem. 1999, 274, 13563–13568. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.; Jirák, R.; Jachymova, M.; Vecka, M.; Tvrzicka, E.; Žák, A. Leptin, adiponectin, leptin to adiponectin ratio and insulin resistance in depressive women. Neuro Endocrinol. Lett. 2009, 30, 387–395. [Google Scholar] [PubMed]
- Wade, J.M.; Juneja, P.; Mackay, A.W.; Graham, J.L.; Havel, P.J.; Tecott, L.H.; Goulding, E.H. Synergistic impairment of glucose homeostasis in ob/ob mice lacking functional serotonin 2C receptors. Endocrinology 2008, 149, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Misra, A. Hepatic Steatosis, Insulin Resistance, and Adipose Tissue Disorders. J. Clin. Endocrinol. Metab. 2002, 87, 3019–3022. [Google Scholar] [CrossRef] [PubMed]
- Edgerton, D.S.; Johnson, K.M.S.; Cherrington, A.D. Current strategies for the inhibition of hepatic glucose production in type 2 diabetes. Front. Biosci. 2009, 14, 1169–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezende, L.F.; Camargo, R.L.; Branco, R.C.S.; Cappelli, A.P.G.; Boschero, A.C.; Carneiro, E.M. Reduced insulin clearance and lower insulin-degrading enzyme expression in the liver might contribute to the thrifty phenotype of protein-restricted mice. Br. J. Nutr. 2014, 112, 900–907. [Google Scholar] [CrossRef] [Green Version]
- Berglund, E.D.; Li, C.Y.; Bina, H.A.; Lynes, S.E.; Michael, M.D.; Shanafelt, A.B.; Kharitonenkov, A.; Wasserman, D.H. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology 2009, 150, 4084–4093. [Google Scholar] [CrossRef] [Green Version]
- Bektur, N.E.; Sahin, E.; Baycu, C. Mirtazapine may show anti-hyperglycemic effect by decreasing GLUT2 through leptin and galanin expressions in the liver of type 1 diabetic rats. Iran. J. Basic Med. Sci 2019, 22, 676–682. [Google Scholar]
- Can, Ö.D.; Üçel, U.İ.; Özkay, Ü.D.; Dikmen, M. Effect of mianserin on streptozotocin-induced hyperglycemia and metabolic alterations in rats. Cukurova Med. J. 2017, 42, 103–119. [Google Scholar] [CrossRef] [Green Version]
- Al-Majed, A.; Bakheit, A.H.; Alharbi, R.M.; Abdel Aziz, H.A. Mirtazapine. Profiles Drug Subst. Excip. Relat. Methodol. 2018, 43, 209–254. [Google Scholar]
- Hennings, J.M.; Schaaf, L.; Fulda, S. Glucose metabolism and antidepressant medication. Curr. Pharm. Des. 2012, 18, 5900–5919. [Google Scholar] [CrossRef]
- Hong, Y.J.; Park, J.D.; Kim, J.G. Hyperglycemia associated with mirtazapine: A case report. J. Korean Neuropsychiatry Assoc. 2001, 40, 151–156. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Control | Mirtazapine |
|---|---|---|
| 0–40 μm (%) | 3.92 ± 1.06 | 13.69 ± 1.09 ** |
| 40–80 μm (%) | 10.17 ± 1.57 | 68.61 ± 0.65 *** |
| 80–120 μm (%) | 71.93 ± 1.75 | 17.69 ± 1.27 *** |
| >120 μm (%) | 13.97 ± 1.72 | 0 ± 0 ** |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-F.; Hou, P.-H.; Mao, F.C.; Su, Y.-C.; Wu, C.-Y.; Yang, W.-C.; Lin, C.-S.; Tsai, H.-P.; Liao, H.-J.; Chang, G.-R. Mirtazapine Reduces Adipocyte Hypertrophy and Increases Glucose Transporter Expression in Obese Mice. Animals 2020, 10, 1423. https://doi.org/10.3390/ani10081423
Wu C-F, Hou P-H, Mao FC, Su Y-C, Wu C-Y, Yang W-C, Lin C-S, Tsai H-P, Liao H-J, Chang G-R. Mirtazapine Reduces Adipocyte Hypertrophy and Increases Glucose Transporter Expression in Obese Mice. Animals. 2020; 10(8):1423. https://doi.org/10.3390/ani10081423
Chicago/Turabian StyleWu, Ching-Feng, Po-Hsun Hou, Frank Chiahung Mao, Yao-Chi Su, Ching-Yang Wu, Wei-Cheng Yang, Chen-Si Lin, Hsiao-Pei Tsai, Huei-Jyuan Liao, and Geng-Ruei Chang. 2020. "Mirtazapine Reduces Adipocyte Hypertrophy and Increases Glucose Transporter Expression in Obese Mice" Animals 10, no. 8: 1423. https://doi.org/10.3390/ani10081423