
Abstract
The complement system plays a major role in human immunity, but its abnormal activation can have severe pathological impacts. By mimicking a natural mechanism of complement regulation, the small peptide compstatin has proven to be a very promising complement inhibitor. Over the years, several compstatin analogs have been created, with improved inhibitory potency. A recent analog is being developed as a candidate drug against several pathological conditions, including COVID-19. However, the reasons behind its higher potency and increased binding affinity to complement proteins are not fully clear. This computational study highlights the mechanistic properties of several compstatin analogs, thus complementing previous experimental studies. We perform molecular dynamics simulations involving six analogs alone in solution and two complexes with compstatin bound to complement component 3. These simulations reveal that all the analogs we consider, except the original compstatin, naturally adopt a pre-bound conformation in solution. Interestingly, this set of analogs adopting a pre-bound conformation includes analogs that were not known to benefit from this behavior. We also show that the most recent compstatin analog (among those we consider) forms a stronger hydrogen bond network with its complement receptor than an earlier analog.
Similar content being viewed by others


Introduction
The complement system acts as the first line of defense in the immune system, as part of both innate and adaptive immunity [1]. It plays a major role in homeostasis by clearing foreign pathogens and compromised host cells [2]. The complement system is activated through several initiation pathways that produce strong opsonins, pro-inflammatory anaphylatoxins, and membrane attack complexes [3]. Complement activity is normally tightly regulated by various plasma and membrane proteins, such as complement receptor 1 and factor H [1]. Erroneous complement activation leads to tissue damage, causing or aggravating numerous pathological conditions, such as heart attacks, strokes, burn injuries, Alzheimer’s disease, adult respiratory distress syndrome, and various autoimmune diseases [2, 4]. To prevent or remedy these undesirable effects, several complement inhibitors have been developed, targeting different steps of complement activation [3, 5, 6].
Thanks to its excellent efficacy, high specificity, low molecular weight, and ability to inhibit complement regardless of the initiation pathway, compstatin is considered a very promising complement inhibitor [3]. It has been evaluated for its potential to modulate the complement system in degenerative diseases. For example, it has been the subject of in vitro studies, non-human primate (NHP) studies, and clinical trials for age-related macular degeneration (AMD), but unfortunately without much success [7,8,9,10]. Compstatin has prevented lung fibrosis and organ damage in NHP models of E. coli sepsis [11, 12]. It has also proven effective against paroxysmal nocturnal hemoglobinuria (PNH) and C3 glomerulopathy (C3G) in in vitro models [13, 14]. In NHP models, it has reduced inflammation and bone loss during treatment of periodontitis, both in a ligature-induced disease setting and in a natural setting [15, 16]. Other NHP studies have shown that a single dose of compstatin prior to a hemodialysis session could alleviate hemodialysis-induced inflammation throughout the treatment [17]. Therefore, it could also prove useful against conditions related to innate immunity activation triggered by biomaterials during medical procedures [18]. Recently, compstatin has been successfully used to treat a patient suffering from acute respiratory distress syndrome (ARDS) triggered by COVID-19 [19]. As a result, it is now being evaluated in a phase II clinical study [20].
Various compstatin analogs were developed over the years, with increased complement inhibition at every step [9, 21,22,23,24,25,26]. The mechanisms underlying the improved potency of each analog involve various factors that have been partially uncovered in several experimental studies (see Background section). In addition, structural models of some of these analogs, either free in solution or bound to a complement protein, have been deposited in the Protein Data Bank (PDB) [27]. However, it is not yet fully clear why recent compstatin analogs are more potent than their predecessors (see Background section).
In this paper, we perform a computational analysis of several compstatin analogs, using molecular dynamics (MD). We run MD simulations of six analogs free in solution and two complexes with compstatin bound to the core portion of C3-like complement proteins (see Background section). Our results are consistent with the view that one of the factors increasing binding affinity is the tendency of recent compstatin analogs to adopt a pre-bound conformation in solution [25]. However, we demonstrate that this tendency appeared in the lineage of compstatin analogs earlier than previously thought. In addition, we show that the latest compstatin analog (among the six we study) forms a stronger hydrogen bond network with complement proteins than an earlier analog. In this regard, our computational study complements previous experimental studies on explaining differences in behavior between compstatin analogs. This is important because compstatin is currently being developed as a drug candidate against various pathological conditions [2, 3].
Background
Complement system
The main ways to activate the complement system are the classical, lectin, and alternative pathways [1]. These recognition and activation pathways all converge at an amplification step involving the complement component 3 (C3) [2]. C3 is a large protein that belongs to the α2-macroglobulin family of host-defense molecules [1]. It is composed of 1641 amino acid residues, for a total molecular mass of 187 kDa. Upon activation by foreign pathogens or altered host cells via any of the pathways, enzymatic C3 convertase complexes are formed. Then, they transform the abundant plasma protein C3, through proteolytic cleavage, into its reactive fragments C3a (9 kDa) and C3b (177 kDa) [28]. The opsonin C3b then attaches covalently to activating surfaces, where it can form additional C3 convertases, thereby amplifying the complement response and inducing downstream events such as pro-inflammatory signaling and phagocytosis [3].
The structure of native C3 was solved by X-ray crystallography at 3.3 Å resolution [28]. It reveals a β-chain (residues 1–645) and an α-chain (residues 650–1641) of 75 and 110 kDa, respectively, arranged in 13 domains. Most of the β-chain, i.e., residues 1–534, form five so-called macroglobulin (MG) domains, denoted by MG1 to MG5. The sixth domain of the β-chain (residues 578–645) is called the linker (LNK) and runs in between domains MG1, MG4, and MG5, through the ring formed by domains MG1–MG5. The α-chain also forms six domains by itself, but one of the additional MG domains, denoted by MG6, is formed by parts of both the α-chain (residues 746–806) and the β-chain (residues 535–577). The core of C3, which is formed by domains MG1 to MG6 and LNK, is referred to as the β-ring and is known to be structurally stable [28]. That is why we will use it in our MD simulations (see Methods section).
Complement inhibitor compstatin
Compstatin is a small, cyclic, non-immunogenic peptide inhibiting the activation and amplification of the complement system [4]. It does so by binding to C3, sterically hindering the binding between substrate C3 and convertase complexes, and blocking C3’s proteolytic activation [2]. Note that this happens without major alteration to the conformation of C3 [29]. In addition, compstatin binds to several proteins derived from C3, such as C3b and C3c [4]. Interestingly, it was shown that its binding is improved by a naturally occurring point mutation of C3-like proteins observed in patients with a complement dysfunction [30].
Compstatin had been initially identified as a 13-residue disulfide-bridged peptide with a molecular mass of 1551 Da [4]. Its original amino acid sequence was Ile1-[Cys2-Val3-Val4-Gln5-Asp6-Trp7-Gly8-His9-His10-Arg11-Cys12]-Thr13-NH2, where the brackets represent the disulfide bridge between Cys2 and Cys12. Its structure in solution was then determined using 2D nuclear magnetic resonance (NMR) techniques and deposited in the PDB under code 1A1P [31]. This structure was described as a closed-loop coil with a flexible type I β-turn comprising residues Gln5–Gly8. In what follows, we refer to this original compstatin as 1A1P.
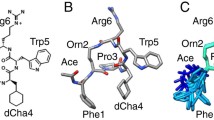
The acetylation of the amino-terminus was followed by a threefold increase in activity for the analog Ac-compstatin [21]. Then, the substitution of Val by the aromatic residue Trp at position 4 and of His by the helix promoter Ala at position 9 produced an analog with a 45-fold higher inhibitory activity [21]. We refer to this analog as 4W9A because of the replacements Val4Trp and His9Ala (i.e., V4W and H9A). Its full sequence is Ac-Ile1-[Cys2-Val3-Trp4-Gln5-Asp6-Trp7-Gly8-Ala9-His10-Arg11-Cys12]-Thr13-NH2. MD simulations of 4W9A in solution showed that it had a reduced tendency to feature the Gln5–Gly8 β-turn characterizing 1A1P [32]. It was also hypothesized that its increased flexibility could explain its higher affinity. The crystal structure of 4W9A in complex with C3c (available under PDB code 2QKI) later showed that the compstatin-binding site is formed by domains MG4 and MG5, at the bottom end of the β-ring, far from other binding sites of C3 [29]. Comparing this bound conformation with the one in solution (i.e., 1A1P) reveals that compstatin undergoes a large conformational change upon binding [29]. The β-turn involves residues Gly8–Arg11 in bound compstatin, as opposed to residues Gln5–Gly8 in free compstatin. Overall, free compstatin seems to adopt an elongated and open υ-shaped conformation, while bound compstatin seems to adopt a twisted and more closed α-shaped conformation (see Fig. 1). After superposition, the root mean square deviation (RMSD) between these two conformations is 3.7 Å, considering only Cα atoms [29].

Backbone conformations of compstatin analogs 1A1P, 4W9A, and Cp10. The conformation of 1A1P (free in solution) is elongated and υ-shaped. The conformation of 4W9A (bound to C3c, under PDB code 2QKI) is more compact and α-shaped. The conformation of Cp10 (free in solution, extracted from an NMR ensemble) is more compact than that of 1A1P. The disulfide bridge creating the cycle is represented with thin lines in all analogs
Several tryptophan analogs were then evaluated at position 4. Replacing Trp4 by a 1-methyl-tryptophan, i.e., Trp(1Me) or Trp(Me) for short, produced a compstatin analog with a 264-fold increase in inhibitory activity over the original compstatin [22]. Its full sequence is Ac-Ile1-[Cys2-Val3-Trp(Me)4-Gln5-Asp6-Trp7-Gly8-Ala9-His10-Arg11-Cys12]-Thr13-NH2. In what follows, we refer to this analog as 4MeW, although it has also been designated as POT-4 (by Potentia Pharmaceuticals) and AL-78898A (by Alcon) in past clinical trials for exudative AMD [25]. Its increase in potency and binding affinity has been attributed to more favorable hydrophobic interactions mediated by the methylated tryptophan [22].
A follow-up substitution study yielded new analogs with improved efficacy and affinity. Replacing Gly by Sar (i.e., Gly with an N-methylation, or Gly(NMe) for short) at position 8 and of Thr by Ile at position 13 produced the analog known as Cp10, with increased kinetic association rate and binding entropy [23]. It is described as Ac-Ile1-[Cys2-Val3-Trp(Me)4-Gln5-Asp6-Trp7-Sar8-Ala9-His10-Arg11-Cys12]-Ile13-NH2. It was shown that the N-methylation of Gly8 improved hydrophobic interactions and stabilized the bound-like β-turn structure [23]. This suggested that the improved binding affinity of Cp10 might be due to a higher similarity between its free and bound conformations, leading to faster ligand recognition and complex formation. This was later supported by the solution structure of Cp10 elucidated using NMR (which has not been deposited in the PDB) [25]. It is more compact than the solution structure of the original compstatin because of the presence of two β-turns involving residues Cys2–Gln5 and Trp(Me)4–Trp7 (see Fig. 1).
The N-methylation of Ile (denoted by Ile(Me) for short) at position 13 yielded the analog known as Cp20, with an improved dissociation rate [23]. Its sequence is Ac-Ile1-[Cys2-Val3-Trp(Me)4-Gln5-Asp6-Trp7-Sar8-Ala9-His10-Arg11-Cys12]-Ile(Me)13-NH2. Cp20 shows a 1000-fold increase in inhibitory potency and binding affinity for C3 compared to the original compstatin [23]. However, the high flexibility of residue 13 makes it difficult to elucidate the mechanisms underlying this improved activity and affinity.
The effects of replacing the N-terminal acetyl moiety (called position 0 for consistency of residue numbering with other analogs) by amino acids, including non-proteinogenic ones, were subsequently investigated [25]. The analog involving the D-isoform of tyrosine (DTyr) showed the highest inhibitory potency and the slowest dissociation rate. Its full amino acid residue sequence is DTyr0-Ile1-[Cys2-Val3-Trp(Me)4-Gln5-Asp6-Trp7-Sar8-Ala9-His10-Arg11-Cys12]-Ile(Me)13-NH2. This analog also featured an improved solubility and more favorable pharmacokinetic properties. It was the first analog with sub-nanomolar binding affinity (KD = 0.5 nM, which is 5600-fold stronger than the original compstatin) and is known as Cp40. It is currently developed as a candidate drug by Amyndas (under the designation AMY-101) and has received orphan drug status from the European Medicines Agency and the US Food and Drug Administration for both PNH and C3G [33, 34]. This analog has recently been used to successfully cure a patient suffering from ARDS resulting from COVID-19 pneumonia [19]. It is currently the subject of a phase II clinical study [20].
Methods
Structural models of compstatin analogs
We ran MD simulations of the six compstatin analogs (free in solution) presented in the Background section, whose amino acid sequences are recapitulated in Table 1. This required obtaining or creating all-atom structural models of these analogs. For the original compstatin analog, we used the first of the 21 NMR models of free compstatin reported under PDB code 1A1P. For 4W9A, we extracted chain G from the crystal structure of compstatin bound to C3c (reported under PDB code 2QKI). For 4MeW, we constructed in silico a mutant by performing methylation of the tryptophan at position 4 in 4W9A using UCSF Chimera [35]. For Cp10, we obtained the first model from the undeposited NMR ensembleFootnote 1 of this analog alone in solution [25]. For Cp20 and Cp40, we obtained the unpublished crystal structures1 of these analogs bound to C3b, and extracted the relevant chains. For all these analogs, hydrogen atoms were added to the structural model using UCSF Chimera [35].
Structural models of complexes involving compstatin
Instead of modeling the entire protein C3, we chose to model only its β-ring (see Background section). This is a reasonable choice in the context of this study, for several reasons. First, compstatin is known to bind domains MG4 and MG5 of the β-ring without yielding any significant structural change in C3 [29]. Second, the structure of the β-ring is known to be very stable, as we observed when simulating it alone in solution. Third, the β-ring is conserved in all C3-like proteins to which compstatin binds. Observations collected from simulating compstatin binding to the β-ring alone can thus be generalized to all these proteins.
Even when restricting ourselves to using only the β-ring (and not the whole C3 protein), MD simulations are still very computationally expensive. Therefore, instead of simulating complexes involving all the compstatin analogs presented in the Background section, we chose to simulate only two complexes involving compstatin analogs 4W9A and Cp40. The rationale for this choice is that we mostly wanted to try and pinpoint mechanistic aspects of the higher binding affinity of the most potent analog, Cp40, in comparison to an older analog. This choice was also driven by the availability of crystal structures of compstatin bound to C3-like proteins. More specifically, to create the model involving 4W9A and the β-ring, we used the crystal structure of compstatin bound to C3c (under PDB code 2QKI) [29]. To create the complex featuring Cp40, we used an unpublished crystal structure1 of compstatin bound to C3b. In both cases, we extracted chain A and residues 746–804 in chain B, to form the β-ring, as well as chain G, which is compstatin.
Molecular dynamics simulations
All simulations were run with the GROMACS v4.6.5 package [36] using the parameter set of the CHARMM27 force field [37], as well as the SPC water model. A cubic box was defined with 1.5 nm of liquid layer around the modeled structure, with periodic boundary conditions. Sodium (Na + ) and chloride (Cl−) counter-ions were added to neutralize each system, with a final concentration of 0.15 mol/L. Temperature and pressure coupling were controlled by the algorithms v-rescale (with tau-t = 0.1 ps) and parrinello-rahman (with tau-p = 2 ps), respectively. A cutoff value of 1.2 nm was used for both the van der Waals and Coulomb interactions, using fast particle-mesh Ewald electrostatics. Input MD files compatible with CHARMM27 were created with SwissParam [38] for compstatin analogs, and with pdb2gmx (from the GROMACS package) for the protein receptor (i.e., C3’s β-ring). For the two complexes, input files were created by merging the files of compstatin and the β-ring.
Simulations were conducted following a previously described protocol [39]. Briefly, the production stage of the simulation was preceded by three steps of energy minimization (EM) and eight steps of equilibration (EQ). The first EM step was conducted using the steepest-descent algorithm and position restraints on the heavy atoms of the substrate (5000 kJ− 1 mol− 1 nm− 1), allowing for relaxation of the solvent only. The second step used the same algorithm, without restraint; the third one used the conjugate-gradient algorithm without restraint. The EQ phase started at a temperature of 310 K, for 300 ps, with position restraints on the substrate’s heavy atoms (5000 kJ− 1 mol− 1 nm− 1), allowing for the formation of solvation layers. Then, temperature was reduced to 280 K, and position restraints were gradually reduced. Then, temperature was progressively increased again, up to 300 K. These EQ steps constituted the first 500 ps of each MD simulation. The production stage ran for 200 ns (for free compstatin) or 100 ns (for the two complexes) at constant temperature (300 K), without restraint.
After the simulation and post-processing steps, visual inspection of the trajectories was performed with VMD [40] and xmgrace.Footnote 2 Substrate conformations were extracted at regular steps using trjconv (from the GROMACS v4.6.5 package) [36]. The occupancy of each hydrogen bond during the MD simulation was calculated with the plot_hbmap.pl script.Footnote 3
Results
Free compstatin
We ran a 200 ns MD simulation for the six compstatin analogs, free in solution, starting from a minimized version of their all-atom models (see Methods section). Note that, for analogs 1A1P and Cp10, the simulation starts from a “real” free conformation, while for the other analogs, the simulation starts from a bound-like conformation because their structural models were obtained by extracting compstatin from co-crystallized complexes. From each MD trajectory, we extract 667 frames, using a 0.3 ns timestep. We align each conformation of compstatin with the closed α-shaped conformation of 4W9A (as reported under PDB code 2QKI) and calculate the Cα RMSD between these two conformations.
The evolution of this RMSD to the closed α-shaped state (measured in Å) along the MD trajectory is reported for each compstatin analog in Fig. 2. These plots show that, except for 1A1P, which always remains in its open υ-shaped conformation, free compstatin usually switches (sometimes even back and forth) between its open υ-shaped conformation and its closed α-shaped conformation, and that it can adopt intermediate conformations, as illustrated by 4MeW and Cp20. This information is schematically represented in Fig. 3, for easier comparison between analogs. It shows that, after excluding 1A1P, Cp20 is the analog that spent the least amount of simulation time in its closed α-shaped state, while all other analogs spent a similar amount of simulation time in their closed α-shaped states, albeit not necessarily in a continuous fashion.

Root mean square deviation (RMSD) to compstatin’s closed α-shaped conformation (defined by the crystal structure of 4W9A bound to C3c, as reported under PDB code 2QKI), for each compstatin analog. The RMSD (considering only Cα atoms) is reported in Å for 667 frames extracted from a 200 ns MD simulation of each analog alone in solution

Schematic representation of the time spent in each state (open υ-shaped state, closed α-shaped state, or intermediate state) during the 200 ns MD simulation of every compstatin analog, free in solution
Bound compstatin
We ran a 100 ns MD simulation for compstatin analogs 4W9A and Cp40 in complex with the β-ring of C3-like proteins, starting from minimized versions of all-atom models derived from co-crystal structures (see Methods section). From both MD trajectories, we extract 1000 frames, between 2 and 100 ns, using a 0.098-ns timestep. Then, we perform the following analyses: First, we remove compstatin from each frame, align the remaining β-ring with that in the first frame, and calculate the Cα RMSD between them. Plotting this RMSD over simulation time corroborates that the β-ring’s conformation did not change in either MD simulation (data not shown). Second, we align compstatin in each frame with that in the first frame and calculate the Cα RMSD between them. Plots of this RMSD over simulation time confirm that both compstatin analogs remained in their closed α-shaped state during the MD simulation (data not shown).
To assess differences in the binding of 4W9A and Cp40 to the β-ring, we monitor the inter-molecular hydrogen bonds that form during each MD simulation. To quantify the strength and importance of each hydrogen bond, we calculate its occupancy, i.e., the percentage of frames in which it is observed. Results are reported in Table 2 for hydrogen bonds whose occupancy is greater than 5%.
Overall, hydrogen bond networks formed by Cp40 and 4W9A are quite similar. Two of the strongest bonds, with occupancy close to 100%, are observed for both analogs: those between the tryptophan residues in compstatin, at positions 4 and 7, and residues Gly345 and Met457 in the β-ring, respectively. Two other strong bonds are common to both analogs: the one between C3’s Met457 and compstatin’s Gln5, with occupancy above 90%, and the one between C3’s Arg456 and compstatin’s Trp4, with occupancy above 80%. Six weaker bonds with an occupancy around 50% and among which a lot of “switching” is observed are also common: they form between compstatin’s Ala9 / His10 and C3’s Asp491.
We can also observe differences between the hydrogen bond networks associated with Cp40 and 4W9A. First, a bond with acceptor in C3’s Asn390 and that switches between compstatin’s Cys2 (occupancy: 77%) and Trp4 (occupancy: 15%) in 4W9A, is stabilized toward Cys2 in Cp40, with occupancy close to 100%. Second, a very weak bond with a donor in C3’s Asn390 and acceptor in compstatin’s Cys2, with an occupancy of 10% in 4W9A, is replaced by a stronger bond involving D-Tyr0, with an occupancy of 61% in Cp40. On the other hand, two weak bonds have seen their occupancy reduced when comparing 4W9A to Cp40: the one between compstatin’s Gln5 and C3’s Leu455, whose occupancy has decreased from 46 to 31%, and the one between C3’s Arg459 and compstatin’s Asp6, whose occupancy has decreased from 35 to 7%.
Discussion
In previous experimental studies on compstatin, it was suggested that the improved binding affinity of the most recent analogs was partially due to their ability to adopt in solution a conformation similar to their bound state (i.e., a closed α-shaped conformation), therefore leading to a faster complex formation with C3-like proteins [25]. Our MD simulations of free compstatin confirm this view, as all analogs except the original one, seem to spend a significant amount of time in a closed α-shaped state (cf. Fig. 3). At some point, this behavior had been attributed to the N-methylation of compstatin’s backbone (at position 8), which differentiates analogs Cp10, Cp20, and Cp40 from previous ones [25]. Our MD simulations do not support this theory, as the behavior of 4W9A and 4MeW does not differ from that of the later analogs (cf. Fig. 3). The tendency of free compstatin to adopt a closed α-shaped conformation seems to have appeared with the creation of 4W9A, most likely as a result of the amino acid substitutions it underwent. This is consistent with a previous MD study that had concluded that the higher affinity of 4W9A might be linked to its increased flexibility [32], although no comparison could be done with the bound conformation, which was released only later.
Our MD simulations of free compstatin also suggest that the improved binding affinity of the most recent analogs is not due to an increase in the tendency of free compstatin to spend time in its closed α-shaped state. Indeed, the amount of time spent in that state is absolutely not correlated with the binding affinity of an analog. This is best illustrated by Cp20, which spent little time in its closed α-shaped conformation (cf. Fig. 3), despite being the second most potent analog in this study.
Our MD simulations of bound 4W9A and Cp40 highlight some of the mechanisms underlying the higher binding affinity of Cp40. Despite forming relatively similar hydrogen bond networks with C3’s β-ring, 4W9A and Cp40 present several noteworthy differences. Most notably, Cp40 forms an additional strong hydrogen bond that replaces an equivalent yet weaker bond in 4W9A. Interestingly, the D-Tyr0 residue that was added to Cp40 is involved in a relatively stable hydrogen bond that replaces an equivalent but very weak bond involving Cys2 in 4W9A. The stronger hydrogen bond network formed by Cp40 when binding to C3-like proteins is certainly not sufficient to explain Cp40’s stronger binding affinity on its own, but it certainly has a significant impact.
Despite their benefits, MD simulations are mostly limited to short time-scale interactions. However, interactions between C3-like proteins and compstatin might present sharper differences at longer time-scales. To study this possibility, we will carry out coarse-grained simulations of complexes involving compstatin, such as those we performed on C3-like proteins alone [41, 42]. One of the challenges will be to select force fields that can correctly assess the energetic contributions of this small peptide, especially for analogs comprising unusual amino acids.
Notes
2015 communication from D Ricklin and JD Lambris
References
Gros P, Milder FJ, Janssen BJ (2008) Complement driven by conformational changes. Nat Rev Immunol 8(1):48–58
Morgan BP, Harris CL (2015) Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov 14:857–877
Ricklin D, Lambris JD (2016) New milestones ahead in complement-targeted therapy. Semin Immunol 28(3):208–222
Sahu A, Kay BK, Lambris JD (1996) Inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J Immunol 157(2):884–891
Kocsis A, Kékesi KA, Szász R, Végh BM, Balczer J, Dobó J, et al. (2010) Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and -2: significant contribution of MASP-1 to lectin pathway activation. J Immunol 185(7):4169–4178
Mohan RR, Wilson M, Gorham RD, Harrison RE, Morikis VA, Kieslich CA, et al. (2018) Virtual screening of chemical compounds for discovery of complement C3 ligands. ACS Omega 3(6):6427–6438
Alcon licenses complement pathway inhibitor for macular degeneration (2009) Nat Rev Drug Discov 8(12):922
Chi ZL, Yoshida T, Lambris JD, Iwata T (2010) Suppression of drusen formation by compstatin, a peptide inhibitor of complement C3 activation, on cynomolgus monkey with early-onset macular degeneration. In: Lambris J, Adamis A (eds) Inflammation and retinal disease: complement biology and pathology. vol. 703 of advances in experimental medicine and biology. Springer, New York, pp 127–135
Gorham RD, Forest DL, Tamamis P, López de Victoria A, Kraszni M, Kieslich CA, et al. (2013) Novel compstatin family peptides inhibit complement activation by drusen-like deposits in human retinal pigmented epithelial cell cultures. Exp Eye Res 116:96–108
Leung E, Landa G (2013) Update on current and future novel therapies for dry age-related macular degeneration. Expert Rev Clin Pharmacol 6(5):565–579
Silasi-Mansat R, Zhu H, Popescu NI, Peer G, Sfyroera G, Magotti P, et al. (2010) Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood 116(6):1002–1010
Silasi-Mansat R, Zhu H, Georgescu C, Popescu N, Keshari RS, Peer G, et al. (2015) Complement inhibition decreases early fibrogenic events in the lung of septic baboons. J Cell Mol Med. 19(11):2549–2563
Risitano AM, Ricklin D, Huang Y, Reis ES, Chen H, Ricci P, et al. (2014) Peptide inhibitors of C3 activation as a novel strategy of complement inhibition for the treatment of paroxysmal nocturnal hemoglobinuria. Blood 123(13):2094–2101
Zhang Y, Shao D, Ricklin D, Hilkin BM, Nester CM, Lambris JD, et al. (2015) Compstatin analog Cp40 inhibits complement dysregulation in vitro in C3 glomerulopathy. Immunobiology 220(8):993–998
Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, et al. (2014) Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J Immunol 192(12):6020–6027
Maekawa T, Briones RA, Resuello RR, Tuplano JV, Hajishengallis E, Kajikawa T, et al. (2016) Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J Clin Periodontol 43(3):238–249
Reis ES, DeAngelis RA, Chen H, Resuello RR, Ricklin D, Lambris JD (2015) Therapeutic C3 inhibitor Cp40 abrogates complement activation induced by modern hemodialysis filters. Immunobiology 220 (4):476–482
Ekdahl KN, Lambris JD, Elwing H, Ricklin D, Nilsson PH, Teramura Y, et al. (2011) Innate immunity activation on biomaterial surfaces: a mechanistic model and coping strategies. Adv Drug Deliv Rev 63(12):1042–1050
Mastaglio S, Ruggeri A, Risitano AM, Angelillo P, Yancopoulou D, Mastellos DC, et al. (2020) The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin Immunol 215:108450
Amyndas initiates phase 2 trial of complement inhibitor AMY-101 in COVID-19 (2020). www.amyndas.com/covid-19. Accessed 7 Jul 2020
Mallik B, Katragadda M, Spruce LA, Carafides C, Tsokos CG, Morikis D, et al. (2005) Design and NMR characterization of active analogues of compstatin containing non-natural amino acids. J Med Chem 48(1):274–286
Katragadda M, Magotti P, Sfyroera G, Lambris JD (2006) Hydrophobic effect and hydrogen bonds account for the improved activity of a complement inhibitor, compstatin. J Med Chem 49(15):4616–4622
Qu H, Magotti P, Ricklin D, Wu EL, Kourtzelis I, Wu YQ, et al. (2011) Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol Immunol 48(4):481–489
Tamamis P, López de Victoria A, Gorham RD, Bellows-Peterson ML, Pierou P, Floudas CA, et al. (2012) Molecular dynamics in drug design: new generations of compstatin analogs. Chem Biol Drug Des 79(5):703–718
Qu H, Ricklin D, Bai H, Chen H, Reis ES, Maciejewski M, et al. (2013) New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology 218(4):496–505
Gorham RD, Forest DL, Khoury GA, Smadbeck J, Beecher CN, Healy ED, et al. (2015) New compstatin peptides containing N-terminal extensions and non-natural amino acids exhibit potent complement inhibition and improved solubility characteristics. J Med Chem 58(2):814–826
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. (2000) The protein data bank. Nucleic Acids Res 28(1):235–242
Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson-Ekdahl K, et al. (2005) Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 437(7058):505–511
Janssen BJ, Halff EF, Lambris JD, Gros P (2007) Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J Biol Chem 282(40):29241–29247
Sfyroera G, Ricklin D, Reis ES, Chen H, Wu EL, Kaznessis YN, et al. (2015) Rare loss-of-function mutation in complement component C3 provides insight into molecular and pathophysiological determinants of complement activity. J Immunol 194(7):3305–3316
Morikis D, Assa-Munt N, Sahu A, Lambris JD (1998) Solution structure of Compstatin, a potent complement inhibitor. Protein Sci 7(3):619–627
Tamamis P, Skourtis SS, Morikis D, Lambris JD, Archontis G (2007) Conformational analysis of compstatin analogues with molecular dynamics simulations in explicit water. J Mol Graph 26(2):571–580
Reis ES, Mastellos DC, Yancopoulou D, Risitano AM, Ricklin D, Lambris JD (2015) Applying complement therapeutics to rare diseases. Clin Immunol 161(2):225–240
Press release: Amyndas’ lead candidate AMY-101 receives orphan drug status from the FDA and the EMA for the treatment of C3 glome- rulopathy (2016). http://c3g.today/?p=305. Accessed 5 Jul 2019)
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. (2004) UCSF chimera–a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Pronk S, Páll S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29 (7):845–854
Bjelkmar P, Larsson P, Cuendet MA, Hess B, Lindahl E (2010) Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J Chem Theory Comput 6(2):459–466
Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) Swissparam: a fast force field generation tool for small organic molecules. J Comput Chem 32(11):2359–2368
Devaurs D, Papanastasiou M, Antunes DA, Abella JR, Moll M, Ricklin D, et al. (2018) Native state of complement protein C3d analysed via hydrogen exchange and conformational sampling. Int J Comput Biol Drug Des 11(1-2):90–113
Humphrey W, Dalke A, Schulten K (1996) VMD Visual molecular dynamics. J Mol Graph 14(1):33–38
Devaurs D, Antunes DA, Papanastasiou M, Moll M, Ricklin D, Lambris JD (2017) Coarse-grained conformational sampling of protein structure improves the fit to experimental hydrogen-exchange data. Front Mol Biosci 4(13)
Devaurs D, Antunes DA, Kavraki LE (2018) Revealing unknown protein structures using computational conformational sampling guided by experimental hydrogen-exchange data. Int J Mol Sci 19(11):3406
Acknowledgments
The authors are extremely thankful to John Lambris and Daniel Ricklin for sharing data on the structural models of several compstatin analogs. All molecular structures are depicted by images produced with the PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC. PyMOL was also used to align molecular structures and calculate RMSD between them. Visual inspection and modeling of compstatin analogs also involved UCSF Chimera [35], developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311).
Author contributions
All authors contributed to the study conception and design. Computational modeling and simulations, as well as data collection and analysis were performed by Didier Devaurs and Dinler Antunes. The first draft of the manuscript was written by Didier Devaurs and all authors commented on subsequent versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported in part by the National Science Foundation (NSF) under Grant CCF 1423304, and by Rice University Funds. Computational simulations were run on equipment supported in part by the Data Analysis and Visualization Cyberinfrastructure funded by NSF under Grant OCI 0959097, on equipment supported by the Cyberinfrastructure for Computational Research funded by NSF under Grant CNS 0821727, as well as on equipment supported in part by the Big-Data Private-Cloud Research Cyberinfrastructure MRI-award funded by NSF under grant CNS 1338099 and by Rice University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
Crystal structures used in this study are available in the Protein Data Bank (PDB) [27]. Other structural models and ensembles produced by molecular dynamics simulations are available from the corresponding author, L.K., upon request.
Rights and permissions
About this article

Cite this article
Devaurs, D., Antunes, D.A. & Kavraki, L.E. Computational analysis of complement inhibitor compstatin using molecular dynamics. J Mol Model 26, 231 (2020). https://doi.org/10.1007/s00894-020-04472-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-020-04472-8