
Abstract
Purpose
High throughput sequencing analysis has facilitated the rapid analysis of the entire titin (TTN) coding sequence. This has resulted in the identification of a growing number of recessive titinopathy patients. The aim of this study was to (1) characterize the causative genetic variants and clinical features of the largest cohort of recessive titinopathy patients reported to date and (2) to evaluate genotype–phenotype correlations in this cohort.
Methods
We analyzed clinical and genetic data in a cohort of patients with biallelic pathogenic or likely pathogenic TTN variants. The cohort included both previously reported cases (100 patients from 81 unrelated families) and unreported cases (23 patients from 20 unrelated families).
Results
Overall, 132 causative variants were identified in cohort members. More than half of the cases had hypotonia at birth or muscle weakness and a delayed motor development within the first 12 months of life (congenital myopathy) with causative variants located along the entire gene. The remaining patients had a distal or proximal phenotype and a childhood or later (noncongenital) onset. All noncongenital cases had at least one pathogenic variant in one of the final three TTN exons (362–364).
Conclusion
Our findings suggest a novel association between the location of nonsense variants and the clinical severity of the disease.
Similar content being viewed by others
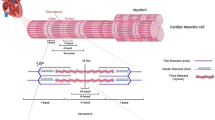

INTRODUCTION
Titin is the largest known human protein and it forms the third myofilament structure spanning the sarcomere from the Z-disk to the M-band.1 The titin I-band acts as a molecular spring generating the passive force needed to maintain the sarcomeric integrity.2 Titin is encoded by a large and complex gene (TTN) with 364 exons.1 Titin transcripts undergo extensive alternative splicing.1,3,4 The canonical skeletal muscle isoform N2A contains 312 exons.1 Five isoforms have a cardiac expression and the isoform N2BA, with its 311 exons, is the longest.1 The theoretical isoform that includes all putative TTN exons (363 exons with the exception of exon 48) is the inferred complete TTN metatranscript (NM_001267550.1).1,3 Exons not included in any of the cardiac and skeletal muscle isoforms are, thereby, defined metatranscript-only exons.3
Pathogenic TTN variants cause a wide range of skeletal muscle and cardiac disorders.5 Titin-related myopathies are a heterogeneous group of inherited muscle disorders that vary in terms of mode of inheritance (dominant versus recessive), age at onset, pattern of muscle involvement, severity, and rate of progression.5 The clinically relevant genotype–phenotype correlations identified to date are as follows:
-
1.
Patients with adult-onset, autosomal dominant tibial muscular dystrophy (TMD, 600334) carry heterozygous causative variants in the final exon (exon 364).6
-
2.
Patients with adult-onset hereditary myopathy with early respiratory failure (HMERF, 603689) carry missense variants in a specific exon (exon 344).7,8,9
-
3.
All remaining TTN-related skeletal myopathies are recessively inherited.5
-
4.
TTN variants affecting the inferred complete TTN metatranscript but not the well characterized adult skeletal muscle or cardiac isoforms (“metatranscript-only” pathogenic variants) identified to date1,3 have occurred in patients with arthrogryposis multiplex congenita and/or a congenital, usually slowly progressive, myopathy.10,11,12
-
5.
Heterozygous titin-truncating variants (TTNtv) in exons constitutively expressed in heart (i.e., TTNtv that impact exons included in the N2BA and N2B cardiac isoforms) are associated with an increased risk of adult-onset dilated cardiomyopathy (604145).13
-
6.
Patients harboring biallelic TTNtv that impact N2B and N2BA-encoding exons also appear to be at increased risk of congenital cardiac anomalies and/or early-onset cardiac involvement, e.g., dilated cardiomyopathy.11
In this study, we evaluate available clinical and genetic data of 23 previously unreported patients and 100 reported patients with convincingly pathogenic biallelic TTN variants (nonsense, frameshift, canonical splice site variants, and/or missense variants identified in more than one patient with additional functional evidence supporting pathogenicity).14 In addition, we undertook a comprehensive genotype–phenotype correlation analysis and identified an association between the onset and the severity of the clinical features and the type and location of the causative variants.
MATERIALS AND METHODS
Previously unreported patients
For this study, we recruited 23 patients from 20 unrelated families with a confirmed recessive titinopathy. The patients had been on regular follow-up over many years. Electrophysiological examination results (nerve conduction studies and needle electromyogram [EMG]), creatine kinase (CK) measurements, histological and histochemical muscle examination results, and cardiac function test results were available for most patients (Table 1).
Patients’ DNA was analyzed using gene panels or exome sequencing (ES).15,16,17 A bioinformatic pipeline aiming at the identification of single-nucleotide variants, small insertions or deletion (indels), and copy-number variants (CNV) was used.17,18,19 In all cases, segregation analysis (via Sanger or massively parallel sequencing [MPS]) was performed to confirm the phase of the variants.
The transcriptional impact of variants in canonical splice sites was evaluated by in silico tools (Human Splice Finder and SpliceAI20,21).
Complementary DNA (cDNA) analysis was performed in cases with a canonical splice variant and an available muscle biopsy sample. QIAGEN RNeasy Plus Universal Mini Kit (Qiagen, MD, USA) was used to extract RNA from the muscle biopsies of patients 8, 9, and 14 (patients 10a/10b have a previously reported and characterized splicing variant). cDNA synthesis was performed using RevertAid H Minus Reverse Transcriptase (Thermo Scientific, USA). Reverse transcription polymerase chain reactions (RT-PCR) was performed using primers designed with Primer3 software (available on request) and a DreamTaq™ DNA Polymerase (Thermo Scientific).
All variants were reported according to Human Genome Variation Society recommendations (http://varnomen.hgvs.org) using the inferred complete TTN metatranscript as reference (NM_001267550.1; LRG391_t1). Exons were numbered 1 to 364 according to the LRG scheme as adopted by the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/genes/TTN). Variants reported in this paper have been submitted to LOVD.
In cases with an available muscle biopsy sample (1, 8, 9, 10a, 11, 14, 15, 18, 19, 20), western blotting (WB) was performed according to standard methods.22 To study C‐terminal titin protein fragments of <200 kDa, two previously described in-house–generated antibodies (rabbit polyclonal antibody M10-16 and mouse monoclonal antibody 11-4-322 directed against the Ig-like domain M10 of titin) were used. HRP-conjugated secondary antibodies (Dako, Denmark), ECL detection (Pierce SuperSignal West Femto substrate, Thermo Fisher), and the ChemiDoc MP digital imager (Bio-Rad Laboratories) were used to visualize the protein bands.22
Ethics statement
All the patients or their legal guardians provided written informed consent. The study, approved by the ethics committee of the Helsinki University Hospital, was performed in accordance with the Declaration of Helsinki.
Literature search
To identify previously reported patients with recessive titinopathies, the medical literature was searched using PubMed (search terms: titin, TTN, titinopathy, congenital myopathy, muscular dystrophy; last search undertaken on 31 July 2019). Only previously reported patients with a recessive skeletal muscle titinopathy carrying variants that can be convincingly classified as pathogenic according to current guidelines23,24 were included (n = 100 cases) (Supplementary Fig. 1). A list of all previously reported patients included in this cohort and their causative TTN variants is provided in Supplementary Table 1.
RESULTS
Previously unreported patients
Clinical findings
Table 1 describes clinical data of newly identified (unreported) cases.
Patient 1, a 39-year-old Finnish woman, presented with a proximal myopathy (limb girdle muscular dystrophy, [LGMDR10, 608807]), mainly affecting the lower limbs with onset in early childhood (running difficulties first noted at age 3).
Twelve patients belonging to eight families (families 2–9) presented with distal weakness with the onset of overt symptoms ranging from infancy to early adulthood. Two siblings (family 10), aged 18 years and 5 years respectively, were examined because of hyperCKemia (up to 20× UNL).
The remaining ten patients presented with a congenital myopathy. Their features included arthrogryposis multiplex congenita, severe congenital or infant-onset axial hypotonia, respiratory failure, and/or delayed motor development (Table 1 and Fig. 1).

Patient 19 at neonatal age displaying multiple joint contractures in hyperextension involving hands and feet (a, b). Lower limb T1-weighted MRIs of patient 19 at age 7 (c, d). At thigh level (c), a prominent involvement of semitendinosus and vasti was detected. Adductor magnus and rectus femori were relatively spared with focal areas of complete intramuscular fat replacement. In the lower leg (d), soleus, gastrocnemius medialis, and peronei muscles were involved, while tibialis anterior showed only focal changes.
To date, five patients (1, 8, 9, 11, 18) have developed an overt (clinical rather than subclinical) dilated cardiomyopathy (DCM).
Molecular genetics
In total we identified 33 causative variants in our cohort of previously unreported patients. Twenty-five of the 33 variants were not previously described. Fourteen variants were in the M-band, 8 in the A-band, 9 in the I-band, and 2 in the Z-disk protein region.
Twelve causative variants were insertions or deletions resulting in frameshift, 7 were nonsense variants, and 11 were canonical splice site variants. One patient (1) is a heterozygous carrier of a FINmaj variant that changes four amino acids (EVTW > VKEK) in the final exon.6 A large intragenic deletion encompassing exons 34 to 41 was found in one family (family 4).18 A previously reported missense variant (c.102214T>C; p.Trp34072Arg in exon 359)25,26 was identified in patient 13.
The previously reported Iberian variant (c.107889delA:p.Lys35963fs in exon 364)22,27 was identified in six patients: patients 2a and 2b were homozygous for this variant, and the remaining patients (3, 4a, 4b, 8) carried a second causative variant in trans. Patients' parents and relatives who were heterozygous carriers of the Iberian variant did not have any evidence of weakness or other neuromuscular features, confirming that this variant causes a fully recessive muscular disease. In addition, the previously described variant c.107635C>T, p.Gln35879* (exon 363), which shows a founder effect in countries from the Balkan region,27,28 was identified in patients 6 and 7 (from Bosnia and Albania, respectively).
Metatranscript-only pathogenic variants were identified in three different patients (18, 19, and 20) presenting with hypotonia and arthrogryposis.
Impact of canonical splice site variants
Among the 11 variants affecting a canonical splicing site (Supplementary Table 2), one variant (c.107377+1G>A in intron 363—patients 10a and 10b) had been reported and characterized previously.14,27 Using RT-PCR, we characterized the splicing defect caused by four novel canonical splice variants (Fig. 2). We showed that the variant c.4646-1G>A (intron 26) in patient 8 causes the loss of the first 14 nucleotides of exon 27, resulting in frameshift and a premature stop codon (Fig. 2a). In patient 9, the variant c.107377+2dup (intron 363) results in retention of the first intronic nucleotide with a consequent frameshift and a premature stop codon. In the same patient, the variant c.54190+1G>A (intron 281) causes an intronic retention. The retained intron contains multiple in-frame stop codons resulting into a premature stop codon (Fig. 2b). Finally, the variant c.10115-1G>C (intron 43) in patient 14 causes the activation of a cryptic acceptor site causing the skipping of the first nine nucleotides of exon 44 and resulting in a near–full length protein, missing three amino acids (Fig. 2c). All remaining canonical splice site variants were predicted to affect splicing by in silico tools.20,21

In patient 8, the variant c.4646-1G>A in intron 26 activates a cryptic splice site causing the loss of the first 14 nucleotides of exon 27 predicted to result in a premature stop codon (a). In patient 9, the variant c.107377+2dup (intron 363) results in the retention of the first intronic nucleotide that is predicted to cause a frameshift and a premature stop codon (b). In the same patient, the variant c.54190+1G>A (intron 281) causes an intronic retention (Sanger sequencing performed after gel extraction). The retained intron contains multiple in-frame stop codons (b). In patient 14, the variant c.10115-1G>C (intron 43) causes the activation of a cryptic acceptor site causing the skipping of the first nine nucleotides of exon 44. This is predicted to result in a near–full length protein, missing three amino acids (c).
Muscle histopathology and western blotting
Seventeen patients underwent at least one muscle biopsy. As is detailed in Table 1, the histopathological and ultrastructural features were highly variable. Myopathic changes and rimmed vacuoles were common findings (seen in seven biopsies).
A loss of C‐terminal antigen recognition was seen in patients 1, 11, and 15 (Fig. 3). A severe reduction of C‐terminal fragments was seen in patients 8, 9, 10a, and a moderate reduction in patient 14 (Fig. 3). Additionally, patient 10a, carrying the previously reported splice site variant in intron 362, shows a truncated fragment probably resulting from the aberrant splicing in the C-terminal region (as already observed in a previously reported patient—patient IV in ref. 14—carrying the same splicing variant) (Fig. 3). Finally, a normal C-terminal pattern was observed in the three remaining patients 18, 19, and 20 (WB for patient 20 is not shown). Two different antibodies, M10-1 and 11-4-3, gave identical results; however, only blots detected with M10-1 are shown in Fig. 3.

Western blotting of skeletal muscle lysates using the M10-1 antibody shows the expression of the last M10 (ex364) domain of titin at protein level. The small C-terminal protein fragments (P13, p15 and p18, arrowheads in [a]) are present in pooled control sample (c), but show variable expression in patients with different TTN pathogenic variants (a–h). C100 = control 100% loading; C50 = 50% loading. Tibial muscular dystrophy (TMD) patients heterozygous (he) and limb-girdle muscular dystrophy 2J (LGMD2J) patients homozygous (ho) for the FINmaj variant serve as disease controls. When some M10 domain is present, even though the small fragments are absent or severely reduced, the M10-reactive high molecular weight band is visible in the upper compartment of the blots (asterisk). Myosin heavy chain (MyHC) band was used as loading control.
Spectrum of TTN variants and their related phenotypes
One hundred twenty-three cases with a recessive titinopathy were compared. These included the 23 aforementioned new cases and 100 previously reported cases.
One hundred thirty-two TTN causative variants had been identified in patients with a recessive titinopathy (Supplementary Table 1). Thirteen were metatranscript-only pathogenic variants.
Most of the identified pathogenic variants were indels resulting in frameshift and a premature stop codon (n = 59), nonsense variants (n = 36), and variants affecting a canonical splice site (n = 33) (Supplementary Table 3). Two were missense variants. In particular, the variant p.Trp34072Arg in exon 359 (gnomAD minor allele frequency [MAF] = 8.11e-6) within the kinase (TK) domain was identified in three different families. This variant causes the loss of function of TK and the loss of TK-Nbr1 binding.25 The second missense variant (p.Ile35947Asn in exon 364, gnomAD MAF = 4.01e-6), originally identified in a Belgian family with an autosomal dominant TMD,29 appears to reduce the titin-obscurin and titin-obscurin-like affinity (−0.17 and −0.949 Kcal/mol, respectively).30
The variants span the entire gene although an enrichment of variants in M-band and a reduction of Z-disk variants was evident (Supplementary Table 3). Ninety patients were compound heterozygous for two different causative variants and 33 had homozygous causative variants. Interestingly, besides the FINmaj variant showing a founder effect in the Finnish population, three variants (c.107889del, c.107635C>T and c.36122del) reoccurred in 36 cases (29.3% of the cases).
Patients showed four major clinical phenotypes: congenital (onset at birth or in early infancy, n = 72), proximal (11), and distal (37) myopathy with a childhood or later onset. Three patients were described with an Emery-Dreifuss–like phenotype without cardiomyopathy.31 Two siblings (10a and 10b in the current paper) presented with hyperCKemia, although lower limb MRI in patient 10a showed selective fatty replacement in the semitendinosus and degenerative atrophic loss of muscle tissue in medial gastrocnemius (Supplementary Fig. 2).
Patients with congenital myopathy (70) carried a wide spectrum of variants along the gene. Forty of them had biallelic variants outside of the M-band (Supplementary Table 4); of these, 23 had at least one metatranscript-only pathogenic variant and 25 had at least one allele with a variant affecting the splicing. Thirty patients with a congenital phenotype had at least one pathogenic variant in the M-band. Only 2 of the 70 congenital myopathy patients showed causative variants within the last three exons (patient 8 in ref. 26 and patient 1044-1/family 15 in ref. 32 and ref. 11).
Six patients with a congenital myopathy and a severe hypotonia at birth showed biallelic pathogenic variants (nonsense variants or indels) in exon 359 (the first exon of the M-band) resulting in a premature stop codon.
All the patients with a noncongenital (childhood or later onset) distal or proximal myopathy had a variant affecting the last two exons (i.e., variants in exons 363 and 364 or a splice variant in intron 362). All patients with a FINmaj variant, in homozygosity or in compound heterozygosity with a second causative variant, had a pre–adult-onset proximal myopathy.22 Conversely, patients with a monoallelic FINmaj variant develop TMD, an adult-onset dominant distal phenotype.7
The three patients previously reported with an Emery-Dreifuss–like myopathy (proximal weakness and multiple early-onset contractures with the first symptoms occurring during infancy or childhood) had at least one variant in exon 360 or 361.31
DISCUSSION
In the last few years, MPS has facilitated the sequencing of the entire TTN coding sequence allowing the identification of disease-causing variants in a large number of patients with undiagnosed myopathies.5,33 Our cohort of 23 previously unreported patients with a recessive skeletal titinopathy displays a wide spectrum of clinical phenotypes heterogeneous in terms of age of onset, disease course, and histopathological findings. However, the observed clinical features appear to represent a continuum of phenotypes that can be further dissected to improve our current understanding of genotype–phenotype correlations. For this study, we re-evaluated clinical and genetic data of previously reported patients satisfying the criteria for the diagnosis of a recessive titinopathy.14 Some correlations between the type of pathogenic variant and the phenotype clearly emerged.
Our analysis confirms that metatranscript-only pathogenic variants are only identified in patients with a congenital titinopathy.10,11,12 These patients develop prenatal-onset weakness and many experience only minimal progression of the skeletal muscle phenotype after birth. Metatranscript-only exons have a variable expression. Recent RNA sequencing–based studies have suggested that metatranscript-only exons 213–217 have a certain level of expression in adult muscles (e.g., in gastrocnemius muscle samples), although their expression is much higher in fetal muscles.3,4 Further studies are needed to clarify the effective usage of each titin exon in anatomically different muscles during development. So far, however, it has been hypothesized that correct fetal muscular development and assembly requires the presence of specific, still uncharacterized, fetal isoforms with the inclusion of metatranscript-only exons.
The term titin-truncating variant (TTNtv) can be misleading. The literal definition of “truncating variant” refers to any change shortening the size of the encoded protein. However, “TTNtv” has been used to describe nonsense, splicing, and frameshift variants, although their downstream effect on titin protein in postnatal muscles may vary dramatically. A large number of the identified TTNtv are splicing variants that may affect the splicing in only a limited number of transcripts, still resulting in a significant amount of normal protein. Moreover, most of the TTN exons are symmetric (i.e., exon length multiple of 3) and most splicing variants probably cause in-frame loss, resulting in a near–full length protein (as for the variant c.10115-1 in patient 14). A recent mouse model–based study demonstrated that, as a consequence of an in-frame intragenic deletion, a shorter titin (~75% shortened PEVK segment in I-band) is correctly located in the sarcomere.34 This causes an increased passive stiffness without any dramatic impact on the mouse phenotype.34 Downstream effect of splicing variants on the messenger RNA (mRNA) and protein level in postnatal muscles should be carefully evaluated whenever possible, to minimize the chance that a causal relationship between the titin variants and the clinical phenotype is misinterpreted.
Also, variants causing a premature stop codon have a different impact depending on nonsense-mediated decay (NMD). It is reasonable to hypothesize that premature stop codons in the I- and A-bands would mostly result in NMD (although exceptions are expected). This is supported by the nonidentification of a predicted truncated protein product in muscle patients carrying this kind of variant. On the contrary, premature stop codons in the M-band would reasonably result in a near–full length protein that is correctly integrated into the sarcomere. Interestingly, a clear inverted relationship between the position of truncation in the M-band and the clinical severity in patients is observed. Early M-band truncations impacting exon 359 result in a severe TTN-related phenotype, characterized by a severe neonatal hypotonia and respiratory distress. In a previously described mouse model, the selective deletion of the first two M-band exons results in embryonic lethality in midgestation since the produced truncated titin is not able to correctly integrate in the M-band,35,36 suggesting that these exons are required for normal embryonic development. The identification of patients with premature stop codons localized immediately downstream the TK domain (p.Ala35063Cysfs*6 in case 11 and p.Pro34879Glnfs*36 in case 12 as well as p.Leu34837Glufs*12 in pat 18 described in ref. 11) suggests that this domain is essential but most of the M-band protein is somehow dispensable for a normal prenatal developmental.
Although there appear to be occasional exceptions to the rule (e.g., cases 8 in ref. 26 and 1044-1 in ref. 32 and ref. 11) in general, it appears that C-terminal M-band nonsense variants impacting exons 362–364 result in a milder phenotype with a noncongenital onset (Fig. 4). However, we still lack a clear understanding of why some patients have proximal-predominant muscle involvement, whereas others have distal-predominant involvement. The most frequent pathogenic variant in this region is the aforementioned FINmaj variant able to disrupt the Ig fold of the M10 domain. Although FINmaj is not a truncating variant per se, it ultimately causes a loss of C-terminal titin because the mutated protein induces a secondary pathological cleavage of the M-band, as seen in patient 1 and in previously reported TMD and LGMD2J cases.6,37

The last six TTN exons (exons 359–364) encode for protein domains located in the sarcomeric M-band. Premature termination codons in early M-band exons (e.g., in exon 359) result in the loss of most titin M-band domains and, thereby, in severe congenital myopathies with a neonatal hypotonia and respiratory distress leading to a life-threatening risk for the affected baby. Moreover, parents may have experienced previous miscarragies. Premature termination codons in the last two exons result in near–full length protein, only lacking the most terminal part of the protein, and, consequently, in a milder phenotype with a later onset. Patients with the asterisk are homozygous for the reported variant. The remaining patients have a second allele with a nonsense variant in I- or A-band, most probably causing a nonsense-mediated decay.
Many heterozygous small indels, nonsense, or splicing variants in cardiac constitutive exons predispose to dominant DCM with an incomplete penetrance.11 In particular, variants in the A‑band, distal I‑band, or in the exon 49 have a higher odds ratio than variants at the Z‑disk, M‑band, or proximal I-band.38 Previous data suggest that patients with a recessive skeletal muscle titinopathy carrying two pathogenic variants impacting cardiac isoforms (N2BA and N2B) have a higher probability of a cardiac involvement than patients with other causative variants.11 On the contrary, patients with a pathogenic variant in metatranscript-only exons have less probability of cardiac abnormalities.11 So far, in our cohort of previously unreported titinopathy cases, only five patients show a cardiac involvement, although most of them undergo a yearly cardiac follow up. Four patients (1, 8, 9, and 11) with cardiomyopathy have variants affecting all the main isoforms (N2BA, N2B, N2A, Novex-1, and Novex-2). Only one patient (patient 18) among the 23 patients with metatranscript-only variants described so far has a cardiomyopathy. Of note is the fact that patient 18 is one of the oldest recesssive titinopathy patients with a metatranscript-only variant reported to date. The genotype–phenotype correlation for cardiac involvement remains unclear. However, several aspects should be considered. First, age should be considered as TTN variants causing a cardiomyopathy show an age-related penetrance11,38,39 and most young patients with a recessive skeletal muscle titinopathy may develop cardiac disease in later years. The second consideration is type of variant; as mentioned, some splicing variants can still result in a significant amount of full length or near–full length protein and, thereby, they should not be considered DCM actionable variants. Third, family history should be considered because we often lack a detailed family history that would allow us to correctly interpret the cardiac effect of the identified TTN variants. In a larger picture, the suggestions recently provided for the clinical interpretation of TTN variants for the diagnosis of DCM are however a very useful framework.11
The variable histological findings observed in our cohort as well as in the previously reported cases reflect the severity of the biopsied muscles, depending on the stage of the disease and the consequent muscle involvement (some muscles can show a total fatty replacement, others can be relatively spared).
Finally, the analysis of the C‐terminal titin protein fragments requires a cautious interpretation. The p13, p15, and p18 fragments are produced by proteolytic cleavages within the second last domain, is7 (exon 363), of titin.37 The amount of these fragments reflects the amount of a normal M-band titin and, presumably, of a full length or near–full length protein. Their total absence (or a minor amount of fragments) is due to the following:
-
Biallelic pathogenic variants in the last two exons;
-
Compound heterozygosity of a variant causing NMD (out of the M-band) and a pathogenic variant in the last two exons;
-
Two M-band variants resulting in premature stop codons and causing the aforementioned severe congenital titinopathy.
A possible mechanism of read-through of nonsense variants or possible minor splicing isoforms skipping the mutated exon (if symmetric) may explain a minor amount of fragments seen on western blot in the aforementioned condition.
A normal C-terminal pattern has different possible explanations and does not exclude a diagnosis of titinopathy. Normal fragments, proving the presence of a normal M-band titin, may still be produced by an allele with a metatranscript-only variant or with splice variants that do not cause a premature stop codon but result in a near–full length protein. It is noteworthy that (1) titin C-terminal pattern is expected to be normal in case of missense variants outside of the last two exons and (2) sample degradation may also affect the amount of fragments.
Conclusion
We performed a comprehensive study of recessive TTN pathogenic variants and their associated phenotypes. We confirmed the previously reported genotype–phenotype correlations and identified a novel association between the location of premature stop codons in the M-band and the clinical severity of the disease.
Based on the literature review, we would propose recessive skeletal titinopathies as a phenotypic continuum in terms of age of onset and severity, partly depending on the amount and the size of the protein expressed in skeletal muscle (Supplementary Fig. 3).
A proximal or distal phenotype, with childhood or later onset, is seen in patients with a near–full length protein (variants in exons 362–364 and, usually, a missing C-terminus).14,22 A clear mechanism to explain the different patterns of muscle involvement (distal versus proximal) in these patients has not yet been identified.
Similarly, the phenotypic variability observed among patients with other similar genotypes is not yet fully explained. Patients 10a and 10b, for example, have a milder clinical phenotype (without contractures) compared with the previously reported patients carrying the same splicing variant, suggesting a possible role of the second allele on the phenotype.
A congenital or infantile phenotype (onset at birth or in the first year) is observed in children with the following three possible genotypes (in line with previous studies):10,11,12
-
Patients with at least one allele carrying a variant in metatranscript-only exons (generally, showing a slowly progressive disease);
-
Patients with biallelic variants outside of the M-band and at least one allele still producing a near–full length protein with an intact C-terminal portion (e.g., an allele with a splice variant in A- or I-band causing an in-frame deletion);
-
Patients with at least one pathogenic variant in the M-band resulting in a shorter, tailless protein (with an inverted relationship between the position of truncation in the M-band and the clinical severity/age of onset of the disease).
Although we delineate an improved genotype–phenotype correlation, this is far from exhaustive. The identification and characterization of additional patients is needed to confirm or refine these correlations. In addition, due to the inherent difficulties in confirming the pathogenicity of TTN variants, a comprehensive understanding of the clinical features in recessive titinopathies involving missense variants remains elusive.23,40
References
Bang ML, Centner T, Fornoff F, et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89:1065–1072.
Linke WA, Kulke M, Li H, et al. PEVK domain of titin: an entropic spring with actin-binding properties. J Struct Biol. 2002;137:194–205.
Savarese M, Jonson PH, Huovinen S, et al. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet Muscle. 2018;8:11.
Bryen SJ, Ewans LJ, Pinner J, et al. Recurrent TTN metatranscript-only c.39974-11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat. 2020;41:403–411.
Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis. 2016;3:293–308.
Hackman P, Marchand S, Sarparanta J, et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord. 2008;18:922–928.
Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492–500.
Palmio J, Leonard-Louis S, Sacconi S, et al. Expanding the importance of HMERF titinopathy: new mutations and clinical aspects. J Neurol. 2019;266:680–690.
Tasca G, Udd B. Hereditary myopathy with early respiratory failure (HMERF): still rare, but common enough. Neuromuscul Disord. 2018;28:268–276.
Fernandez-Marmiesse A, Carrascosa-Romero MC, Alfaro Ponce B, et al. Homozygous truncating mutation in prenatally expressed skeletal isoform of TTN gene results in arthrogryposis multiplex congenita and myopathy without cardiac involvement. Neuromuscul Disord. 2017;27:188–192.
Oates EC, Jones KJ, Donkervoort S, et al. Congenital titinopathy: comprehensive characterization and pathogenic insights. Ann Neurol. 2018;83:1105–1124.
Chervinsky E, Khayat M, Soltsman S, Habiballa H, Elpeleg O, Shalev S. A homozygous TTN gene variant associated with lethal congenital contracture syndrome. Am J Med Genet A. 2018;176:1001–1005.
Ware JS, Cook SA. Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat Rev Cardiol. 2018;15:241–252.
Savarese M, Maggi L, Vihola A, et al. Interpreting genetic variants in titin in patients with muscle disorders. JAMA Neurol. 2018;75:557–565.
Savarese M, Di Fruscio G, Mutarelli M, et al. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol Commun. 2014;2:100.
Savarese M, Di Fruscio G, Torella A, et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology. 2016;87:71–76.
Evila A, Arumilli M, Udd B, Hackman P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord. 2016;26:7–15.
Välipakka S, Savarese M, Johari M, et al. Copy number variation analysis increases the diagnostic yield in muscle diseases. Neurology Genetics. 2017;3:e204.
Valipakka S, Savarese M, Sagath L, et al. Improving copy number variant detection from sequencing data with a combination of programs and a predictive model. J Mol Diagn. 2020;22:40–49.
Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–548.
Evila A, Vihola A, Sarparanta J, et al. Atypical phenotypes in titinopathies explained by second titin mutations. Ann Neurol. 2014;75:230–240.
Savarese M, Johari M, Johnson K, et al. Improved criteria for the classification of titin variants in inherited skeletal myopathies. J Neuromuscul Dis. 2020;7:153–166.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Chauveau C, Bonnemann CG, Julien C, et al. Recessive TTN truncating mutations define novel forms of core myopathy with heart disease. Hum Mol Genet. 2014;23:980–991.
Avila-Polo R, Malfatti E, Lornage X, et al. Loss of sarcomeric scaffolding as a common baseline histopathologic lesion in titin-related myopathies. J Neuropathol Exp Neurol. 2018;77:1101–1114.
Evila A, Palmio J, Vihola A, et al. Targeted next-generation sequencing reveals novel TTN mutations causing recessive distal titinopathy. Mol Neurobiol. 2017;54:7212–7223.
Peric S, Glumac JN, Topf A, et al. A novel recessive TTN founder variant is a common cause of distal myopathy in the Serbian population. Eur J Hum Genet. 2017;25:572–581.
Van den Bergh PY, Bouquiaux O, Verellen C, et al. Tibial muscular dystrophy in a Belgian family. Ann Neurol. 2003;54:248–251.
Laddach A, Gautel M, Fraternali F. TITINdb-a computational tool to assess titin’s role as a disease gene. Bioinformatics. 2017;33:3482–3485.
De Cid R, Ben Yaou R, Roudaut C, et al. A new titinopathy: childhood-juvenile onset Emery-Dreifuss-like phenotype without cardiomyopathy. Neurology. 2015;85:2126–2135.
Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, et al. Recessive truncating titin gene, TTN, mutations presenting as centronuclear myopathy. Neurology. 2013;81:1205–1214.
Hackman P, Udd B, Bonnemann CG, Ferreiro A, Titinopathy Database C. 219th ENMC International Workshop Titinopathies International database of titin mutations and phenotypes, Heemskerk, The Netherlands, 29 April–1 May 2016. Neuromuscul Disord. 2017;27:396–407.
Brynnel A, Hernandez Y, Kiss B. et al. Downsizing the molecular spring of the giant protein titin reveals that skeletal muscle titin determines passive stiffness and drives longitudinal hypertrophy. Elife. 2018;7:e40532.
Gotthardt M, Hammer RE, Hubner N, et al. Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J Biol Chem. 2003;278:6059–6065.
Weinert S, Bergmann N, Luo X, Erdmann B, Gotthardt M. M line-deficient titin causes cardiac lethality through impaired maturation of the sarcomere. J Cell Biol. 2006;173:559–570.
Charton K, Sarparanta J, Vihola A, et al. CAPN3-mediated processing of C-terminal titin replaced by pathological cleavage in titinopathy. Hum Mol Genet. 2015;24:3718–3731.
Schafer S, de Marvao A, Adami E, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49:46–53.
Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628.
Savarese M, Valipakka S, Johari M, Hackman P, Udd B. Is gene-size an issue for the diagnosis of skeletal muscle disorders?. J Neuromuscul Dis. 2020;7:203–216.
Acknowledgements
The authors thank all the patients and family members for their cooperation; all the clinicians for collecting patient data; Meharji Arumilli at Folkhälsan Research Center (FHR) and Francesco Musacchia at TIGEM for bioinformatic help; Merja Soininen and Anni Evilä at FHR, Sini Penttilä and Sara Lehtinen at Neuromuscular Research Center Tampere, Denise Cassandrini at Stella Maris, Claire Chauveau at Institut de Myologie, and Monica Traverso at Istituto Gaslini for sample and data acquisition; the Oxford Genomics Centre at the Wellcome Centre for Human Genetics (funded by Wellcome Trust grant reference 203141/Z/16/Z) for the generation and initial processing of the sequencing data. We also thank Magnus Ehrnrooth Foundation (M.S.), Päivikki ja Sakari Sohlbergin Säätiö (M.S. and Mridul Johari), Biomedicum Helsinki säätiö (Mridul Johari), Jane and Aatos Erkko Foundation (P.H.), Medicinska Understödsföreningen Liv och Hälsa rf (P.H.), Folkhälsan Research Foundation (B.U.), Erkko Foundation (B.U.), Juselius Foundation (B.U.), Finnish Academy (B.U.), Alfred Kordelin Foundation (S.V.), AFM-Telethon (grant number 22431 to CF), National Health and Medical Research Council (NHMRC) (GNT1090428 to ECO) for their support. N.M. and J.J.V. are funded by a project PI16/00316 supported by the Instituto de Salud Carlos III (ISCIII) Madrid: “Estudio de miopatías distales: diagnóstico mediante NGS, ampliación de estudios sobre su historia natural y exploración de factores patogénicos,” a grant by Fundación Isabel Gemio, and by the Generalitat Valenciana (grant PROMETEO/2019/075) to N.M. F.M. and Anna Sarkozy thank the European Community’s Seventh Framework Programme (FP7/2007–2013) funded grant “Integrated European –omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS)” (grant agreement number 2012–305121); the Muscular Dystrophy UK Grant on Gene Identification to F.M.; the Highly Specialised Services for Congenital Myopathies and Congenital dystrophies in England, and the support of the MRC and BRC Neuromuscular Centre Biobank at UCL is also gratefully acknowledged. Figure 4 and Supplementary Fig. 3 were created with BioRender (www.biorender.com).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Savarese, M., Vihola, A., Oates, E.C. et al. Genotype–phenotype correlations in recessive titinopathies. Genet Med 22, 2029–2040 (2020). https://doi.org/10.1038/s41436-020-0914-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0914-2
Keywords
This article is cited by
-
Exploring TTN variants as genetic insights into cardiomyopathy pathogenesis and potential emerging clues to molecular mechanisms in cardiomyopathies
Scientific Reports (2024)
-
Digenic inheritance involving a muscle-specific protein kinase and the giant titin protein causes a skeletal muscle myopathy
Nature Genetics (2024)
-
Clinical and functional characterization of a long survivor congenital titinopathy patient with a novel metatranscript-only titin variant
Acta Neuropathologica Communications (2023)
-
Tools to differentiate between Filamin C and Titin truncating variant carriers: value of MRI
European Journal of Human Genetics (2023)
-
Aberrant mRNA processing caused by splicing mutations in TTN-related neuromuscular disorders
Journal of Human Genetics (2023)
