Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin

1
Department of Dermatology, Faculty of Medicine and Medical Center, University of Freiburg, Hauptstraße 7, 79104 Freiburg, Germany
2
Faculty of Biology, University of Freiburg, Schänzlestraße 1, 79104 Freiburg, Germany
*
Authors to whom correspondence should be addressed.
Biomolecules 2020, 10(8), 1170; https://doi.org/10.3390/biom10081170
Submission received: 30 June 2020
/
Revised: 1 August 2020
/
Accepted: 5 August 2020
/
Published: 11 August 2020
(This article belongs to the Special Issue Desiphering the Network of Cell Receptors and Matrix in Health and Disease)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Skin consists of the epidermis and dermis, which are connected by a specialized basement membrane—the epidermal basement membrane. Both the epidermal basement membrane and the underlying interstitial extracellular matrix (ECM) created by dermal fibroblasts contain distinct network-forming macromolecules. These matrices play various roles in order to maintain skin homeostasis and integrity. Within this complex interplay of cells and matrices, cell surface receptors play essential roles not only for inside-out and outside-in signaling, but also for establishing mechanical and biochemical properties of skin. Already minor modulations of this multifactorial cross-talk can lead to severe and systemic diseases. In this review, major epidermal and dermal cell surface receptors will be addressed with respect to their interactions with matrix components as well as their roles in fibrotic, inflammatory or tumorigenic skin diseases.
1. Introduction
Skin consists of two distinct compartments: the superficial epidermis and the dermis below. The epidermis includes the innermost stratum basale, stratum spinosum, stratum granulosum and stratum corneum as the outer layer of skin, which is characterized by keratinocytes at progressing differentiation stages, but also involve non-epithelial cells, such as immune cells [1,2]. Additionally, hair follicles and sebaceous and sweat glands are associated appendages of the epidermis [3]. The dermis is subdivided into the upper, papillary dermis and the deep, reticular dermis, which is directly followed by the subcutaneous adipose layer [2]. The dermis also includes blood and lymphatic vessels, nerve endings, hair follicles and sweat glands [1,2]. Even though various immune cells are present in the dermis [1], the major cell type are fibroblasts [4], which produce an interstitial extracellular matrix (ECM) [5]. The ECM orchestrates skin homeostasis, functions as signaling platform and reservoir for soluble factors, and also serves as a structural scaffold that provides both mechanical resilience as well as elasticity [5]. This interstitial ECM consists of collagens, proteoglycans, laminins, fibrillin microfibrils, elastin and matricellular proteins, such as thrombospondins or tenascins [5]. Moreover, a specialized basement membrane—the epidermal basement membrane—exists at the epidermal–dermal junction and builds an anchoring sheet that firmly links the epidermis to the underlying interstitial ECM, while it at the same time functionally divides the epidermis and dermis [6,7,8]. Biochemically, this basement membrane is built in tandem by epidermal keratinocytes and dermal fibroblasts [5].
Both the epidermal basement membrane and the interstitial ECM interact with the cells they tangent or embed in a bidirectional manner in order to create a functioning homeostatic and integer tissue. In this context, a crucial role is assigned to cell surface receptors, since they coordinate cell-signaling events for ECM synthesis, degradation or remodeling. At the same time, they respond to external physical or chemical stimuli and communicate these into cellular responses, such as proliferation, differentiation or migration.
On the basis of these bidirectional cell–matrix interactions in skin, this review will provide a general overview of cell surface receptors in healthy, injured or diseased skin, with a focus on malignancies that involve unremitting inflammation, fibrosis and cancer.
2. Integrins
Integrins are the main adhesion proteins that bridge the cellular cytoskeleton with the extracellular matrix (ECM) and thereby serve as bidirectional signal transducers regulating cell proliferation, homeostasis, differentiation, adhesion, migration and apoptosis [9,10,11,12]. The integrin family consists of genetically-distinct alpha and beta subunits that heterodimerize to form functioning transmembrane receptors [10]. In humans, 18 α- and eight β-subunits associate non-covalently into 24 heterodimer pairs [13,14,15]. Each subunit generally comprises a short cytoplasmic region, a single transmembrane segment and a larger ectodomain [14]. After synthesis, both integrin subunits heterodimerize in the endoplasmic reticulum and are subsequently exported to the plasma membrane [16]. Integrins interact with various ECM components, which are recognized either by a specific region on the α-subunit or by motifs on both subunits [17]. Common motifs in the ligands are recognized by groups of integrins such as, e.g., the arginine-glycine-aspartic acid (RGD) in many proteins including thrombospondin-1, tenascin-C, vitronectin, and fibronectin [10,18,19], the leucine-aspartate-valine (LDV) motif in fibronectin [14,20], a triple-helical GFOGER sequence in collagens [14,20,21] and isoleucine-aspartic acid-glycine (IDG) motif of tenascin-C [19]. Importantly, proteolytic cleavage products of ECM components, such as endostatin (from collagen XVIII), endorepellin (from perlecan) or tumstatin (from collagen IV) are ligands and may have similar or different preferences for integrins than the proteins they derive from [20,22,23,24].
At the plasma membrane, integrins undergo conformational changes to transform from an inactive form with low ligand affinity, sometimes over an intermediate form, to a high affinity form to a fully activated ligand-bound integrin [25]. Important for these changes are, in a selection of nine α-subunits, the in-the-ectodomain-inserted von Willebrand factor A domain (I-domain) [10,14,26]. Depending on the ligand bound, the integrin heterodimers regulate different cellular events and literature has presented evidence that at least some integrins select their binding partner in a force- and conformation-dependent manner when they are embedded in a complex ECM that offers various ligands [27,28]. Conversely, the force exerted by the ECM modifies integrin conformation and thus modulates integrin activation, clustering, trafficking and endocytosis as well as various cellular reactions such as proliferation, migration or invasion [17]. Therefore, integrins are widely recognized as mechanoreceptors that translate intra- and extracellular forces into signaling events, which has been reviewed in detail [17,29,30,31,32].
In the classical model of integrin signaling, adaptor and signaling proteins intracellularly cluster around transmembrane integrins to generate focal adhesions, dynamic multi-protein structures that connect the ECM with the actin cytoskeleton [12,33,34]. The focal adhesion proteins including talin, tensin, kindlins and vinculin structurally link the intracellular domain of integrins to the cytoskeletal acto-myosin complexes [35]. Moreover, these proteins are involved in the subsequent recruitment of the focal adhesion kinase (FAK), which is not only a central regulator of focal adhesion (dis-)assembly [36], but also a key player in signaling, since it complexes with Src kinase to be phosphorylated by the latter [37]. This can, in turn, activate various downstream targets and pathways [33,38], for example the PI3K/AKT [34], NFκB [34] or JNK pathway [39] but also cytoskeleton re-organization via Rac1 can be initiated [34].
The main integrins expressed in skin are: α2β1 [40], α3β1 [40] α5β1 [40], α6β4 [40] and αV integrins associating with β3 [41], β5 [40] and β6 subunits [40]. These integrins will be the focus of the review and we will highlight their roles in skin homeostasis as well as selected skin anomalies.
2.1. Integrin α6β4
Integrin α6β4 is an integrin rather specialized to epithelial cells. In skin, high abundances of integrin α6β4 subunits are found in basal keratinocytes at their basal plasma membrane adjacent to the basement membrane [40,42,43,44]. The major ligands of integrin α6β4 in skin are the epidermal basement membrane core components laminin-332 and laminin-511 [45].
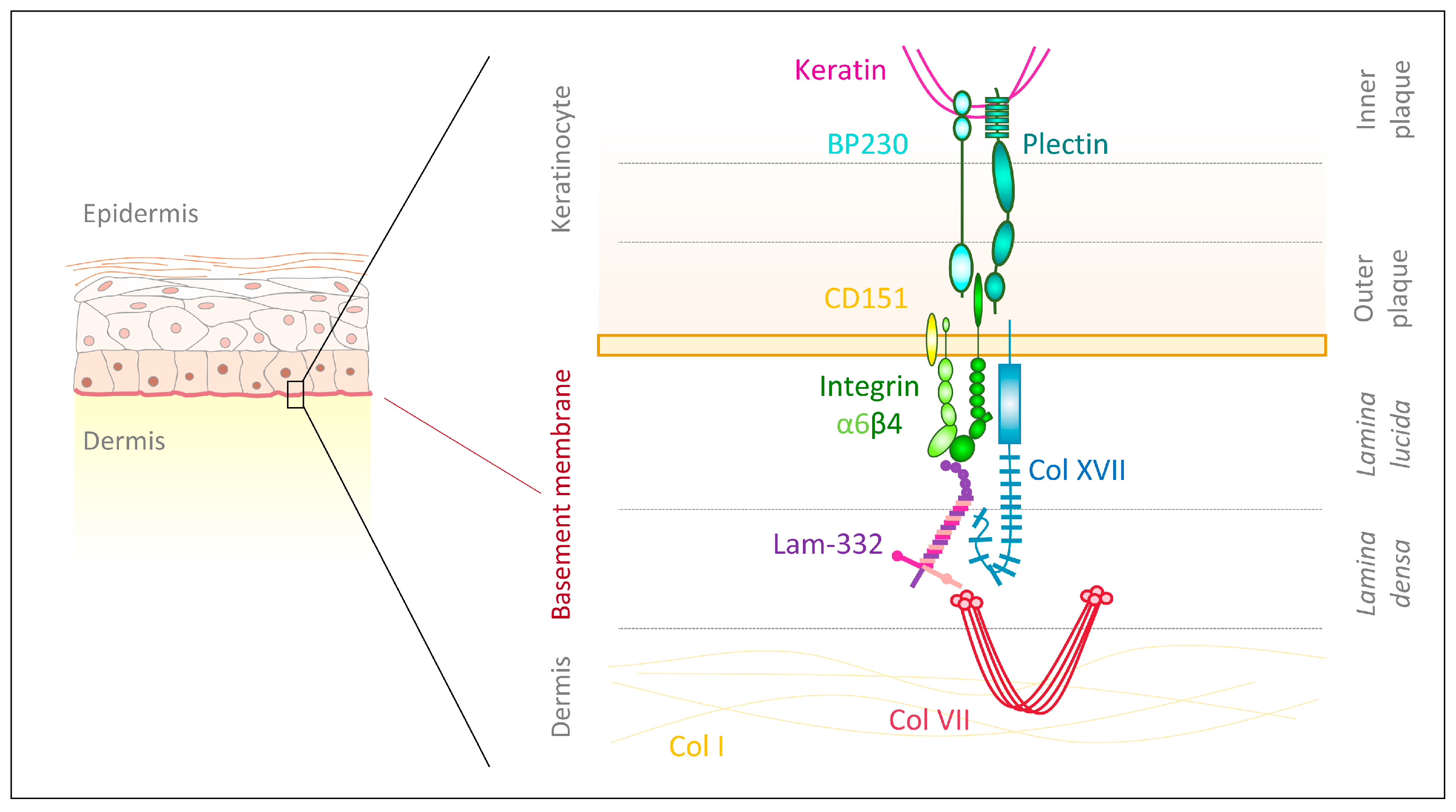
The integrin β4 differs from other subunits, due to its atypically large cytoplasmic domain containing four type III fibronectin-like repeats [46] and five potential N-glycosylation motifs [47]. The β4 subunit can be phosphorylated through integrin-associated Src family kinase and it subsequently interacts with the adaptor protein Shc and Ras to control ERK and JNK signaling [48]. Additionally, integrin β4 phosphorylation causes activation of MAPK and NF-κB, which foster wound healing and epidermal growth [49,50]. This extended intracellular domain allows for the formation of anchoring complexes—hemidesomsomes—which maintain skin integrity and homeostasis [51]. Type I hemidesmosomes (Figure 1), as they are found in the epidermis, are complexes of integrin α6β4 with intracellular plaques and extracellular anchoring filaments [51]. Within the inner plaque, plectin isoform 1a (P1a) and bullous pemphigoid antigen 1 isoform e (BPAG1e/PB230) link intracellular keratin filaments to the cytoplasmic part of integrin β4, which is located in the outer plaque close to the plasma membrane [51]. Extracellularly, integrin α6 offers binding sites for the tetraspanin CD151 to stabilize hemidesmosomes [51]. Moreover, the transmembrane collagen XVII (BPAG2/BP180) strengthens hemidesmosomes by providing intracellular binding sites for integrin β4, plectin and BPAG1e as well as extracellular docking sites for integrin α6 and laminin-332, though the latter is not sufficient to maintain cellular adhesion [51,52].
Mutations in the ITGA6 or ITGB4 gene, encoding for the integrin α6β4 subunits result in the skin blistering disease junctional epidermolysis bullosa (JEB) associated with pyloric atresia (JEB-PA) [53]. Integrin α6β4 deficiency causes aberrant hemidesomsome formation [44,54,55], in the case of ITGB4 mutations in part through formation of integrin α6β1 [44], which is normally very lowly expressed in basal keratinocytes.
Integrin α6β4 also associates with the receptor tyrosine kinases EGFR family receptors [56,57] through galectin-3-mediated connection of N-glycans [47]. Integrin α6β4-mediated cell adhesion and cell motility are regulated by phosphorylation of the integrin β4 cytoplasmic domain. Serine, tyrosine and threonine phosphorylation of it promotes hemidesmosome disruption [48,57]; conversely, dephosphorylation allows the β4 intracellular domain to associate with the keratin filaments, resulting in hemidesmosome assembly [46,48,58]. It has been reported that laminin-332, which is essential for cell adhesion, inhibits with its short arm of the γ2 chain (γ2sa) EGF-induced phosphorylation of integrin β4 and thereby stabilizes hemidesmosomes [59]. This is regulated by the specific binding of γ2sa to the proteoglycan syndecan-1, which acts as γ2sa receptor on the cell surface and possibly induces signaling cascades that negatively regulate integrin β4 phosphorylation and thus promote stable cell adhesion [59]. On the other hand, the activation of EGFR induces tyrosine phosphorylation of the cytoplasmic integrin β4 subunit through the Src family kinase Fyn [57], further downstream phosphoinositol-3-kinase (PI3K) and ERK are activated to foster cell migration and tumor invasion [57,60,61,62]. In addition, integrin α6β4-mediated PI3K signaling impacts gene expression, for example, the transcription of the integrin α2 subunit or translation of the α3 integrin subunit, which, inter alia, determine migration velocity [63]. Moreover, this signaling controls the translation of genes relevant for (carcinoma) cell survival [64].
Despite being essential for firm, stable cell adhesion evidence supports integrin α6β4 to promote tumor invasion and progression [62,65,66,67,68]. This duality is, in part, enabled through phosphorylation of the integrin β4 subunit’s cytoplasmic domain [69]. Elevated and suprabasal expression of integrin α6β4 is seen in all stages of squamous cell carcinoma (SCC) progression and it has been reported that high suprabasal expression primes SCCs for early relapse [68,70,71,72]. Multiple mechanisms and molecular pathways, including glycan modifications and modulation of the immune microenvironment, underlie integrin α6β4-mediated tumor progression and are extensively reviewed elsewhere [73].
2.2. Integrins Containing the β1 Subunit
The integrin subunit β1 pairs with the α1, α2, α3, α4, α5, α6, α7, α8, α9, α10, α11 and αV subunits [10]. The cytoplasmic domain of integrin β1 directly binds various proteins to anchor itself to actin filaments, such as kindlin and talin [74,75,76]. Both proteins keep the integrin in its active form and promote the interaction with the actin cytoskeleton [77,78]. Src family kinases are able to phosphorylate the integrin β1 cytoplasmic tail at two tyrosine residues in the region crucial for talin and kindlin recruitment, which prevents talin and kindlin binding and thereby controls integrin activity [77]. It has been speculated that phosphorylation may modulate integrin signaling such that it initiates transformation and adhesion-independent growth [79,80,81,82].
The major constitutive integrin β1 integrins in skin are in the epidermis α2β1 and α3β1 [40]. After wounding α5β1 and α9β1 can be increased [40]. β1 integrins are also part of specialized niches including integrin α6β1 in hair follicle stem cells [83] and α8β1 in mesenchymal cells in the hair follicle buldge [84].
Integrin α2β1 is in skin found along the lateral and apical surface of basal keratinocytes [85]. It is commonly considered a collagen receptor; however, its ability to bind intact collagen fibrils has been challenged and may rely on fibril-associated proteins [86]. Transmembrane collagen XXIII has been proposed to be an epidermal integrin α2β1 ligand [87]. Many additional proteins bind integrin α2β1 including endorepellin/perlecan [23], which should be considered a major interaction partner at the epidermal basement membrane. Upon collagen interactions, integrin α2β1 lowers cell proliferation but enhances degradation by matrix metalloproteinases (MMP-1, MMP-13) [88,89,90,91,92]. This fosters ECM remodeling and is thought to support the migration of keratinocytes during human wound healing [93]. In contrast, during murine skin wound healing, re-epithelialization, granulation tissue formation and wound contraction by myofibroblasts appear to be independent from integrin α2β1 [94].
Integrin α3β1 localizes at basolateral sites of basal keratinocytes [85], where it is recruited to focal contacts [95] and thereby links keratinocytes to the underlying basement membrane [76]. Integrin α3β1 binds laminin-332 [96] and laminin-511 [97]. Mutations in ITGA3 encoding the integrin α3 subunit cause congenital nephrotic syndrome, interstitial lung disease, and skin fragility [98], which is classified as a form of junctional epidermolysis bullosa [53]. Both humans and mice with integrin α3β1 deficiency present microblisters at the dermal–epidermal junction with laminin-332 present at both the blister roof and floor [95,98,99].
As integrin α6β4, integrin α3β1 regulates keratinocyte migration. Keratinocytes isolated from integrin α3-deficient mice migrate faster and with increased directional persistence [99], they show elevated stress fiber formation and an accumulation of actin-associated proteins to focal contacts [100]. Additionally, integrin α5β1 and α2β1 activities are enhanced in these cells [100], indicating the β1 subunit to increasingly pair with other α-subunits.
In the epidermis minor integrin, integrin α9β1 promotes re-epithelialization [101].
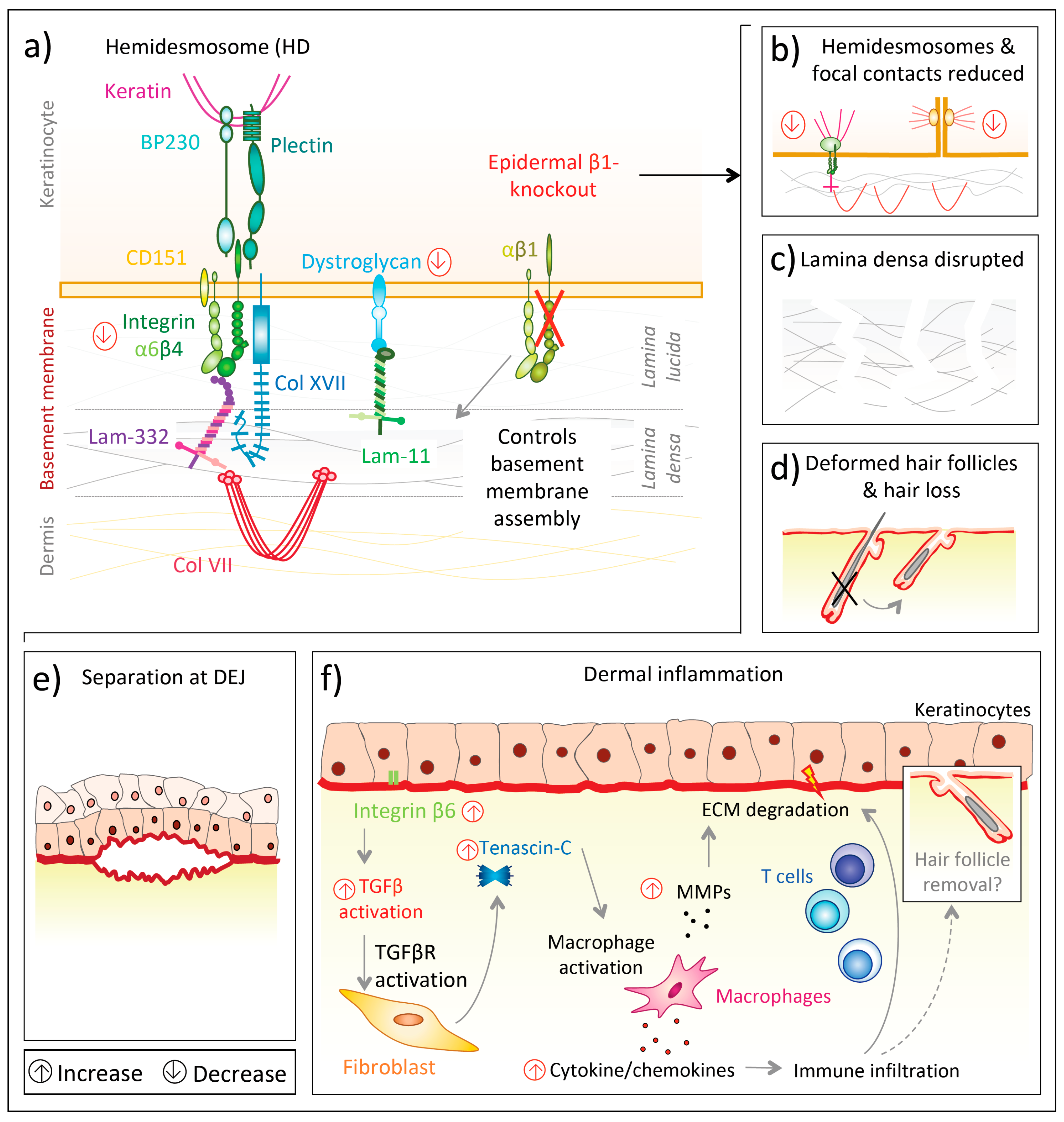
Deficiency of all epidermal β1 integrins is much more severe (Figure 2a) than lack of individual β1 integrins, indicating cooperation and additive effects of them. Keratinocyte-specific integrin β1 deletion in mice under the keratin 5 promoter resulted in severe hair loss as well as mechanically induced skin wounds, though the epidermal barrier function remained stable [102]. In this knockout model, separation at the dermal–epidermal junction was observed, hemidesmosomes were rare (Figure 2b) and the basement membrane was altered, with diminished lamina densa [102] (Figure 2c). Additionally, integrin β1-deficient basal keratinocytes proliferated only weakly and their level of integrin α6β4 was reduced, as was the laminin receptor dystroglycan [102,103] (Figure 2a). Mutant mice displayed thickened epidermis and the authors hypothesized this to be caused by delayed terminal differentiation of suprabasal cells [102]. Moreover, Brakebusch et al. [102] found multiple signs of dermal inflammation. Subsequently, enhanced dermal deposition of collagen I, fibronectin, tenascin-C and perlecan as well as skin stiffening was observed, indicating the presence of dermal fibrogenic processes in mice lacking epidermal integrin β1 [102]. In another mouse model with conditional epidermal integrin β1 deficiency under the keratin 14 promoter [104] newborn mice had a flattened basal epidermal layer and only a thin suprabasal layer before the stratum corneum. Additionally, hair follicle development was absent (Figure 2d). Basal keratinocytes from knockout mice proliferated less, though did not prematurely undergo terminal differentiation [104]. Moreover, the basement membrane assembly was compromised, with laminin-332 scattering into the upper dermis and also the abundance of other integrins, such as α6β4, was disturbed or deficient [104]. Accordingly, the back skin of knockout mice was highly fragile and separated at the dermal–epidermal junction upon mechanical challenges [104] (Figure 2e). This separation was possibly a consequence of scarce and morphologically altered hemidesmosomes at the dermal–epidermal junction, as well as a discontinuous lamina densa [104]. Thus, the authors suggested that integrin α6β4, to establish firm hemidesmosomes, requires integrin β1 to control the assembly of an intact basement membrane [104].
The effects on ECM organization and the dermal immune microenvironment upon integrin β1 deficiency have been reinforced by subsequent studies. Kurbet et al. [105] showed that the loss of epidermal integrin β1 disorganizes the basement membrane in early (day E16.5) mouse embryos and progressively causes a sterile inflammation despite an otherwise intact epidermal barrier. (Figure 2f).
β1-containing integrins are in skin not only essential for keratinocytes but also for dermal fibroblasts. Liu et al. [106] found that mice with a fibroblast-specific knockout of integrin β1 had reduced collagen I and αSMA expression and presented a thinned dermis [106,107,108]. This phenotype was in part caused by a reduced Rac1 activation and lowered abundance of reactive oxygen species (ROS) in integrin β1 knockout mice [106]. Moreover, these mice were resistant to bleomycin-evoked dermal fibrosis [108]. The lowered ability of knockout fibroblasts to produce collagen I and αSMA and to differentiate into myofibroblasts also delayed closure of dermal punch wounds and impaired granulation tissue formation and wound contraction in integrin β1-deficient mice [107].
Additionally, dermal fibroblasts from explants of integrin β1 knockout mice showed reduced proliferation as well as migration on collagen I-coated surfaces and also impaired contraction of collagen matrices [107]. Since contractile forces are necessary to activate latent transforming growth factor β (TGFβ), integrin β1-null fibroblasts have a lower ability to activate latent TGFβ [107].
Dermal fibroblasts also express integrin α11β1 as a collagen receptor [109] and upregulate this integrin upon mechanical challenges of the ECM [110]. Integrin α11β1 crucially regulates pro-fibrotic signaling events and also is involved during tissue repair [110]. Accordingly, Schulz et al. [109,111] found that collagen remodeling during skin wound healing is regulated in tandem by integrin α11β1 and non-canonical TGFβ1 signaling. Indeed, wound contraction and granulation tissue formation were diminished in integrin α11β1-deficient mice independent of integrin α2β1. Moreover, these mice presented scar tissue with reduced tensile strength, due to the impaired conversion of dermal fibroblasts into myofibroblasts [109].
In squamous cell carcinomas (SCCs), integrin β1 is required for cell adhesion, spreading and dermal invasion, but, in contrast to normal keratinocytes, not for proliferation [112]. Both of the major epidermal β1 integrins, α2β1 and α3β1, have been investigated in the context of non-melanoma skin cancers. In high-risk cSCCs, arising in the genetic skin blistering disease recessive dystrophic epidermolysis bullosa (RDEB), which is caused by collagen VII deficiency, Martins et al. [113] found that neutralization of integrin α2 with an antibody reduced adhesion of SCC keratinocytes to recombinant human collagen VII, which in turn increased the expression of integrin αVβ6 and TGFβ1 as well as the phosphorylation of Smad2. Thus, they concluded that in cSCC, keratinocyte–collagen VII interaction via integrin α2β1 restrain TGFβ1 signaling [113]. Two-stage chemical carcinogenesis on epidermal integrin α3β1-deficient mice yielded fewer and smaller SCCs compared to wild-type mice [114]. This was explained by enhanced terminal differentiation of α3β1-deficient keratinocytes leading to lower accumulation of mutations in living keratinocytes [114]. Similarly, Meves et al. [79] found the cytoplasmic domain of integrin β1 to endorse skin tumorigenesis independent from its tyrosine phosphorylation status in a Src/FAK-dependent manner that inhibits keratinocyte differentiation. Subsequent to tumor initiation, integrin β1 supports skin tumor invasion and dissemination [115,116,117,118]. Growth factor receptors including EGFR co-operate to facilitate these processes [119,120,121]. Interestingly, the expression levels of EGFR have been shown to depend on matrix attached integrin β1 [122]. EGFR inhibition downregulates integrin β1; vice versa, EGFR activation may stimulate expression of integrin β1 [123]. Combined targeting of EGFR and the integrin β1 subunit has shown promise in preclinical studies to sensitize radioresistant head and neck SCCs to conservative radiotherapy [124].
2.3. Integrins Containing the αV Subunit
Integrin αV belongs to the non-I-domain group of α subunits [10] and heterodimerizes with the β1, β3, β5, β6 or β8 subunit [10,12], though it is likely that a hierarchy exists on which β-subunit is preferred for heterodimerization [125].
In healthy adult skin, integrin αV is present in the epidermis and dermis, where it reaches its maximal expression levels in the plasma membrane of proliferative basal keratinocytes [33]. It is not restricted to the basal side of keratinocytes but distributed throughout the cell membrane [33]. In the epidermis integrin β5 is the primary heterodimerization partner of integrin αV [126,127]; however, although constitutively present, its abundance is modest [40]. Integrin αVβ6 is found in hair follicle stem cells [40,128] and integrin αVβ8 is expressed in suprabasal epidermal layers in normal skin [40]. Integrin αVβ3 is weakly expressed in healthy skin [41]. Given their low abundance in healthy adult skin, it is reasonable that αV integrins are not required for skin maintenance [33]. After injury, their expression is heavily increased, and they are required for skin regeneration [33]. αV subunit-containing integrins interact with ligands containing an RGD tripeptide motif. Nevertheless, within the integrin αV family, there are differences in the ligand preferences as determined by the heterodimerization partner of integrin αV. Here, integrin αVβ3 displays most promiscuous ligand binding [28] (Figure 3).
To generalize, αV integrins can be viewed as regenerative integrins, and are involved in multiple physiological and pathophysiological regenerative processes. In this context, their ability to activate latent TGFβ1 and 3 is an important trait. This activation occurs through the RGD sequence within the latency associated peptide LAP of TGFβ1 and TGFβ3 [129,130,131]. To be activated by integrin αVβ3, αVβ5, αVβ6 or αVβ8, latent TGFβ has to be also associated with LTBP1 [131,132] or with glycoprotein-A repetitions predominant protein (GARP) [133,134], which is predominantly expressed on T-regulatory cells. LTBP1 localizes and anchors latent TGFβ to the ECM [131,132] while GARP links it to cell surfaces [133]. On the other side, the integrin β-subunits are associated with the cytoskeleton and thereby transmit traction forces from the latter onto the LAP-TGFβ-complex, which ultimately liberates active TGFβ [130,131,132,134]. Additionally, upon binding to latent TGFβ1, integrin αVβ8 is able to simultaneously recruit and bind the membrane type 1 matrix metalloprotease (MT1-MMP), to proteolytically release active TGFβ1 [132,135]. Similarly, integrin αVβ3 is suggested to interact with MMP2 and MMP9 to proteolytically liberate active TGFβ1 [132]. Once TGFβ1 is activated, the β-subunit of integrin αVβ3 complexes with the TGFβRII to control the bioactivity of TGFβ1 as well as to modulate TGFβ1-induced signaling and downstream processes, such as proliferation, ECM deposition or invasion [132,136,137,138].
The integrin-mediated release of active TGFβ is facilitated by increased tissue stiffness [139], which lowers the force needed by the cell to evoke a conformational shift in the ECM-anchored LAP. As TGFβ1 is a pleiotropic fibrotic factor and tissue stiffness is a consequence of fibrosis, a self-perpetuating TGFβ activating loop is created. αVβ3 integrins are the main players of cellular rigidity sensing and cooperate tightly with α5β1 integrins to perceive and react to ECM stiffness [13]. In fibroblasts, integrin α5β1 adheres to fibronectin and creates tension via myosin II activation, while integrin αVβ3 regulates structural adaptations in response to force [13]. In fact, αV integrins cluster at adhesion sites susceptible to high traction forces, but cellular tension due to substrate stiffness is needed to increase the lifetime of fibronectin-αVβ3 integrin complexes. This, in turn, strengthens focal adhesions and induces stress-fiber formation to calibrate cell contractility according to substrate stiffness [13]. Interestingly, data suggest that the mechanical load on the integrin αV-integrin may regulate ligand-binding preferences [28]. Moreover, integrin αVβ3 associates with EGFR, this complex is activated by EGF or fibronectin and subsequently generates paxillin-dependent adhesion and survival signals to prevent anoikis [140].
In terms of its role in cell adhesion, Duperret et al. [33] found integrin αV to associate in large paxillin-containing focal adhesions in fibroblasts, while integrin β1 interacts with smaller focal adhesions. On the other hand, in keratinocytes, integrin β1 is tightly localized with focal adhesions, while integrin αV cannot be detected in a specific subcellular location but spreads throughout the plasma membrane [33]. In this distribution, outside of focal adhesions, integrin αV heterodimerizes with integrins β5 or β6 and together they signal via the focal adhesion kinase (FAK) and the transcription factor c-Myc to control the transition from G1 to S phase in cell cycle, as well as cell proliferation, especially during epidermal tissue generation [33]. Additionally, integrin αV regulates FAK expression, activity and directs it to focal adhesions in keratinocytes [33].
αV integrins are players in wound healing and their roles appear contextual as both overexpression and loss of the same integrin can cause delay of healing [141,142,143]. The various roles of integrin αV during wound healing are reviewed extensively elsewhere [128,144,145]. These integrins are also implicated in the promotion of non-melanoma skin cancer cell migration and invasion. In particular, integrin αVβ6 appears to enhance migration and invasion of SCC cells [146,147], in addition it hampers fibronectin matrix assembly [147] and promotes tumor growth [148].
3. Proteoglycans
Proteoglycans are macromolecules containing a core protein with one or more covalently bound glycosaminoglycan (GAG) chains [149]. Either via the protein core or their GAGs, they are able to interact with growth factors and other ECM components to modulate signal transduction, ECM organization and skin architecture [5]. According to their location, they can be classified into intracellular, cell-surface, pericellular and extracellular proteoglycans [150]. In this review, syndecans and CD44 are discussed as cell-surface proteoglycans that function as essential receptors for components of the basement membrane as well as the dermal ECM.
3.1. Syndecans
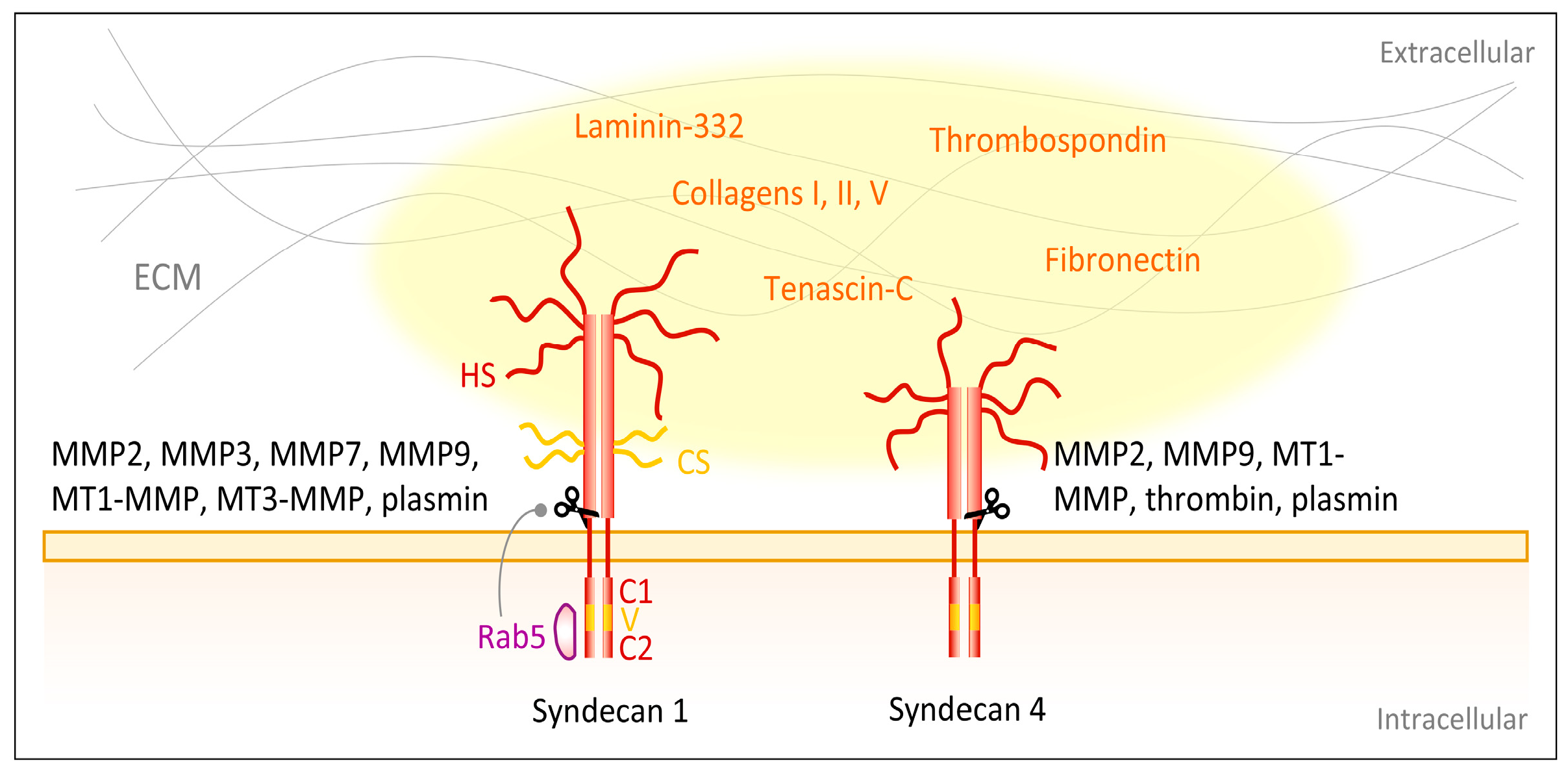
Syndecans are a family of transmembrane proteoglycans and in mammals they comprise four members (syndecan-1, -2, -3, -4) [151] that are present on various epithelial, stromal, endothelial and hematopoietic cells during certain phases of development [152,153,154]. All syndecans consist of an N-terminal extracellular signaling peptide, followed by a transmembrane domain, which also facilitates the dimerization of the protein via a conserved GXXXG motif [155] and thereby assists outside-in-signaling [156,157]. C-terminally, two conserved regions C1 and C2 on the cytoplasmic tail are separated by a variable (v) region that also exerts distinct intracellular roles, such as actin assembly [156,158]. All three intracellular domains carry several serine and tyrosine residues and their phosphorylation status regulates syndecan downstream signaling [159,160,161], which is mediated through binding of intracellular adaptor or scaffold proteins [162,163,164]. For example, the C2 region ends in an EFYA sequence that is able to interacts with PDZ domain proteins [163]. A PDZ domain-containing protein that interacts with syndecans is syntenin-1, which negatively regulates syndecan-4 function [165] and supports syndecan recycling through endosomal compartments [166,167]. The extracellular domain of syndecans harbors several sites for covalent attachment of glycosaminoglycans, which mainly are heparan sulfate (HS) chains. However, syndecan-1 [168,169] and syndecan-4 [170] have been identified as hybrid-type proteoglycans able to carry both HS and chondroitin sulfate (CS) chains [152]. These negatively charged GAGs foster and regulate the binding of cationic extracellular ligands [156,169], among other functions promoting attachment of cells to their surrounding extracellular matrix [171].
Syndecan ectodomains can undergo protease-mediated shedding. Syndecan-1 and syndecan-4 are cleaved by the matrix metalloproteinases MMP-2 [172,173], MMP3 [173], MMP7 [173], MMP-9 [172,173], MT1-MMP [173,174] and MT3-MMP [174], as well as by the serine proteinases thrombin [173] and plasmin [173] (Figure 4). The cleavage sites on the ectodomains of these syndecans are located in close proximity to the plasma membrane [173,175,176,177] and most proteinases recognize and cleave several sites of the core protein [173]. The syndecan protein cores are released together with their GAGs as an entire unit and may be pericellularly retained to compete with plasma membrane-linked syndecans [173]. The shed ectodomains are involved in multiple pathophysiological processes including wound healing [178,179], bacterial and viral pathogenesis [180,181,182] as well as tumor progression [183,184]. Various regulators of syndecan shedding have been identified, such as the HS chains [185]. Reduction of the HS chains increases syndecan-1 shedding [185]. The small GTPase Rab5 has been described to control syndecan-1 shedding, since it specifically binds the cytoplasmic domain of syndecan-1 and dissociation of Rab5 leads to increased shedding [176]. The dissociation can be evoked by binding of the GTPase Rab5 to the cytoplasmic tails of closely interacting integrin β1-subunit-containing integrins [176].
In skin, syndecan-1 and -4 are expressed with high abundances in the epidermis [171,186]. Syndecan-1 is mainly found in the stratum spinosium and granulosum, and only weakly in basal keratinocytes; it is absent in the stratum corneum of intact skin [187,188,189]. On a cellular level, syndecan-1 locates polarized to the basolateral surface of epithelial cells [190,191,192] and is present with high abundance at cell-cell contacts [193]. Human dermal fibroblasts do not constitutively express syndecan-1 [186] and produce only low levels of syndecan-4 [171]. Furthermore, syndecan-2 is not part of healthy adult human skin [194] but is elevated under certain pathological conditions. Its levels are raised in fibrotic dermis, due to the induction of TGFβ and the insulin-like growth factor binding protein-3 (IGFBP-3) [195]. Syndecan-1 is also overexpressed in keloid scars compared to normal or hypertrophic scars [196].
Loss of syndecan-1 expression has also been linked to decreased intercellular adhesion in acantholytic and spongiotic processes, which may foster blister formation in acantholytic or spongiotic dermatosis and also in pemphigus vulgaris or foliaceus [197] and thereby emphasizes an involvement of syndecans in cellular adhesion, skin homeostasis and integrity. In line with this, syndecan-1 and syndecan-4 double knockout mice show increased P-cadherin levels in the epidermal stratum spinosum and stratum basale, and a disturbed organization of lower epidermal layers, while the suprabasal cells keep their cytoplasmic extensions reaching to the basement membrane [198]. Mechanistically, syndecans interact with transient receptor potential canonical (TRPC) calcium channels and may therefore be involved in regulating actin cytoskeleton, adhesion, junction assembly and cell migration via calcium homeostasis [198,199]. The interaction with TRPCs could also be important in the context of fibrosis. Gopal et al. [198] found syndecan-4 to mediate a myofibroblastic phenotype in primary mouse embryonic fibroblasts via TRPC7. The authors hypothesized that syndecan-4 indirectly interacts with TRPC7, for example via α-actinin, which is known to co-localize with both [198].
Syndecans have been reported to have both pro- and anti-inflammatory effects, most likely depending on the underlying model, the tissue of focus or the stage of inflammation, as well as characteristics of their ectodomains [199]. In the skin disease psoriasiform dermatitis, syndecan-1 has been alluded to an anti-inflammatory function, since it regulates the homeostasis of an interleukin-17-producing subset of γδ-T-cells (Tγδ17) [200].
In wound healing, syndecans may regulate inflammation and cell proliferation. After injury, syndecan-1 becomes highly abundant in keratinocytes at wound margins [187,188,201]. Most likely, TGFβ signaling via protein kinase A (PKA) is responsible for this elevation [202]. A functional role of syndecan-1 in wound healing was established from mouse studies. Syndecan-1-deficient mice showed defective keratinocyte proliferation and differentiation upon wounding [193], as well as decreased keratinocyte migration speed [203]. This may be because syndecan-1 deficiency alters the deposition and assembly as ECM proteins, including laminin-332 and fibrillar collagens, in addition to the cellular interactions with them [203]. It appears as if these events are partially driven by TGFβ1 signaling being constitutively active in syndecan-1-deficient keratinocytes leading to elevated surface abundance of αVβ6, αVβ8 and α6β4 integrins [203]. The altered collagen deposition was speculated to be dependent on the shed ectodomain of syndecan-1 protecting collagen molecules from degradation [203]. Syndecans also support dermal healing, as exemplified by syndecan-4 null mice, presenting with delayed dermal wound healing and diminished angiogenesis [204].
Of specific importance for skin are interactions between syndecan-1 and -4 and laminin-332 [59,205,206]. In a study using normal human keratinocytes, Carulli et al. [205] reported the binding region for both syndecans to lie within the C-terminal globular domains 4 and 5 (LG4-5) domain of the α3 chain of laminin-332; however, the two receptors specifically recognize overlapping but distinct sites and apply discrete binding mechanisms. While binding of syndecan-1 to LG4-5 has been shown to entirely depend on its GAGs [205,206], syndecan-4 also employs its protein core [205]. Upon its secretion and deposition into the basement membrane, laminin-332 rapidly undergoes specific maturation processes, including the cleavage of its LG4-5 domain [207]. The major integrin binding sites are located within the LG1-3 domains but with dependence on the laminin β and γ chain C-termini [208]. Non-processed laminin-332 has been reported to primarily interact with integrin α3β1 and removal of the LG4-5 domain enhances interactions with integrin α6β4 [209]. Syndecan-1 recruitment influences binding and distribution of integrin α3β1, pointing towards an interaction of both receptors [209].
Syndecan-1-mediated cellular adhesion to non-processed laminin-332 retaining LG4-5 has been shown to induce the formation of fascin-containing protrusions via the Rho GTPases Rac1 and cell division control protein Cdc42 [209]. This involves the rapid de-phosphorylation of tyrosine residues in the cytoplasmic regions of syndecan-1 [164] and is proposed to be modulated by the subsequent recruitment and binding of syntenin-1 [164], which in turn has been shown to regulate Cdc42 [210]. Additionally, syndecan-1 or syndecan-4 can complex with the hemidesmosomal integrin α6β4 and ErbB2 or EGFR, respectively, to stimulate migration [211,212,213]. The two syndecans recognize independent sites at the very C-terminus of the β4 integrin and binding critically relies on arginine and glutamic acid, for syndecan-1 and syndecan-4, respectively [212].
Apart from laminin-332, collagen I, III and V [214] and proteins abundant in the transitional ECM of wounded skin such as thrombospondin, fibronectin and tenascin-C [215,216,217] interact with syndecans (Figure 4). Fibronectin binds to the HS GAGs of syndecan-4 with its high affinity heparin-binding domain (HepII) [218] and has been described in several studies to foster cellular adhesion, especially via formation of focal adhesion and actin stress fibers [219,220]. Syndecan-4 is a main receptor for focal adhesion-formation on fibronectin [221,222,223] and has been shown to be recruited into these areas by protein kinase C (PKC) activity [218,223,224]. Additionally, syndecan-4 connects fibronectin with the cytoskeletal component α-actinin [217], providing another link between syndecans and cytoskeletal organization and cellular adhesion. The same binding site on fibronectin that is targeted by syndecan-4, is also recognized by tenascin-C, which therefore interferes with syndecan-4 binding and hampers cellular adhesion [216]. In agreement with this, Midwood et al. [225] showed that syndecan-4-deficient fibroblasts no longer respond to tenascin-C and are therefore able to spread on a fibrin–fibronectin matrix containing tenascin-C, whereas overexpression of syndecan-4 bypasses these inhibitory effects of tenascin-C and normalizes the changes caused by the latter.
Figure 4.
Syndecans interact with ECM components. Major skin syndecans are syndecan-1 and -4. They consist of extracellular signaling peptide containing glycosaminoglycans (GAGs), i.e., heparan sulfate (HS) and chondroitin sulfate (CS) chains, a transmembrane domain and intracellularly two conserved regions C1 and C2 separated by a variable (v) region [155,156,157,158]. Via the GAGs the syndecans interact with ECM ligands, such as laminin-332 [205,206], collagen I, III and V [214]. Additionally, proteins expressed in the transitional ECM of wounds, such as thrombospondin, fibronectin and tenascin-C are ligands [215,216,217,218]. Several proteinases shed the extracellular domain of syndecans, for example matrix metalloproteinases MMP2 [172,173], MMP3 [173], MMP7 [173], MMP-9 [172,173], MT1-MMP [173,174] and MT3-MMP [174] as well as the serine proteinases thrombin and plasmin [173]. The small GTPase Rab5 controls syndecan-1 shedding [176].
Figure 4.
Syndecans interact with ECM components. Major skin syndecans are syndecan-1 and -4. They consist of extracellular signaling peptide containing glycosaminoglycans (GAGs), i.e., heparan sulfate (HS) and chondroitin sulfate (CS) chains, a transmembrane domain and intracellularly two conserved regions C1 and C2 separated by a variable (v) region [155,156,157,158]. Via the GAGs the syndecans interact with ECM ligands, such as laminin-332 [205,206], collagen I, III and V [214]. Additionally, proteins expressed in the transitional ECM of wounds, such as thrombospondin, fibronectin and tenascin-C are ligands [215,216,217,218]. Several proteinases shed the extracellular domain of syndecans, for example matrix metalloproteinases MMP2 [172,173], MMP3 [173], MMP7 [173], MMP-9 [172,173], MT1-MMP [173,174] and MT3-MMP [174] as well as the serine proteinases thrombin and plasmin [173]. The small GTPase Rab5 controls syndecan-1 shedding [176].

Studies indicate syndecan-4 to be involved in mechanotransduction and mechanosignaling [226,227,228]. In fact, mechanical strain increases syndecan-4 expression [229] and during the early stages of mechanical loading, syndean-4 via protein kinase PKCα, activates myosin light chain 2, FAK and ERK [227,228]. In the later stages of mechanical stress, however, these downstream modulators are de-phosphorylated and thus downregulated [227]. Via these cascades, syndecan-4 is indicated to impact actin cytoskeleton assembly, contractility and spreading of epithelial cells [227]. Chronopoulos et al. [226] investigated the role of syndecan-4 as mechanotransducer by locally applying tension force to the receptor itself or its HS chains. This caused EGFR-mediated activation of the phosphoinositide 3-kinase (PI3K) [226]. The latter activated the focal adhesion proteins talin-1 and kindlin-2 and recruited them to focal adhesions, causing the formation of larger and more frequent talin-1 and kindin-2 comprising focal adhesions throughout the cell [226]. Moreover, PI3K activation generated diffusive phosphatidylinositol-3,4,5-triphosphate (PIP3), which interacted with kindlin-2 at focal adhesions and thereby activated integrin β1 [226]. The activated integrin β1-containing integrins, in turn, established new connections to fibronectin and subsequently triggered the activation of the small GTPase RhoA, which finally induced acto-myosin contraction to generate cellular stiffness [226]. Additionally, the application of force onto syndecan-4 strengthened the association of syndecan-4 with α-actinin and F-actin creating a “molecular scaffold” that, through YAP, a well-known mechanosensitive transcription co-activator involved in ECM remodeling [230,231], augmented mechanotransduction [226].
Syndecan-1 in skin seems to protect against cancer initiation and progression. Its loss is associated with transformation of epithelia into anchorage-independent mesenchyme-like cells [232] and also with epithelial malignancies like carcinoma [233]. Accordingly, mice deficient in syndecan-1 present higher conversion of benign papillomas into squamous cell carcinomas than their wild-type peers [189]. Correspondingly, in a study on sporadic and RDEB-associated cutaneous SCCs syndecan-1 was associated with invasion suppression [234]. In this context, MMP-7 expression has been inversely correlated to syndecan-1 abundance, indicating a role of MMP-7 in shedding the protein [234], a function which is beneficial for wound healing [235] but harmful with respect to tumor progression [234]. It should be mentioned that the outcome of syndecan-1 activity on tumor progression could be contextual. Its ectodoamin can regulate activation of integrin αVβ3, which could promote invasion and migration on ECM proteins such as vitronectin [236].
3.2. CD44
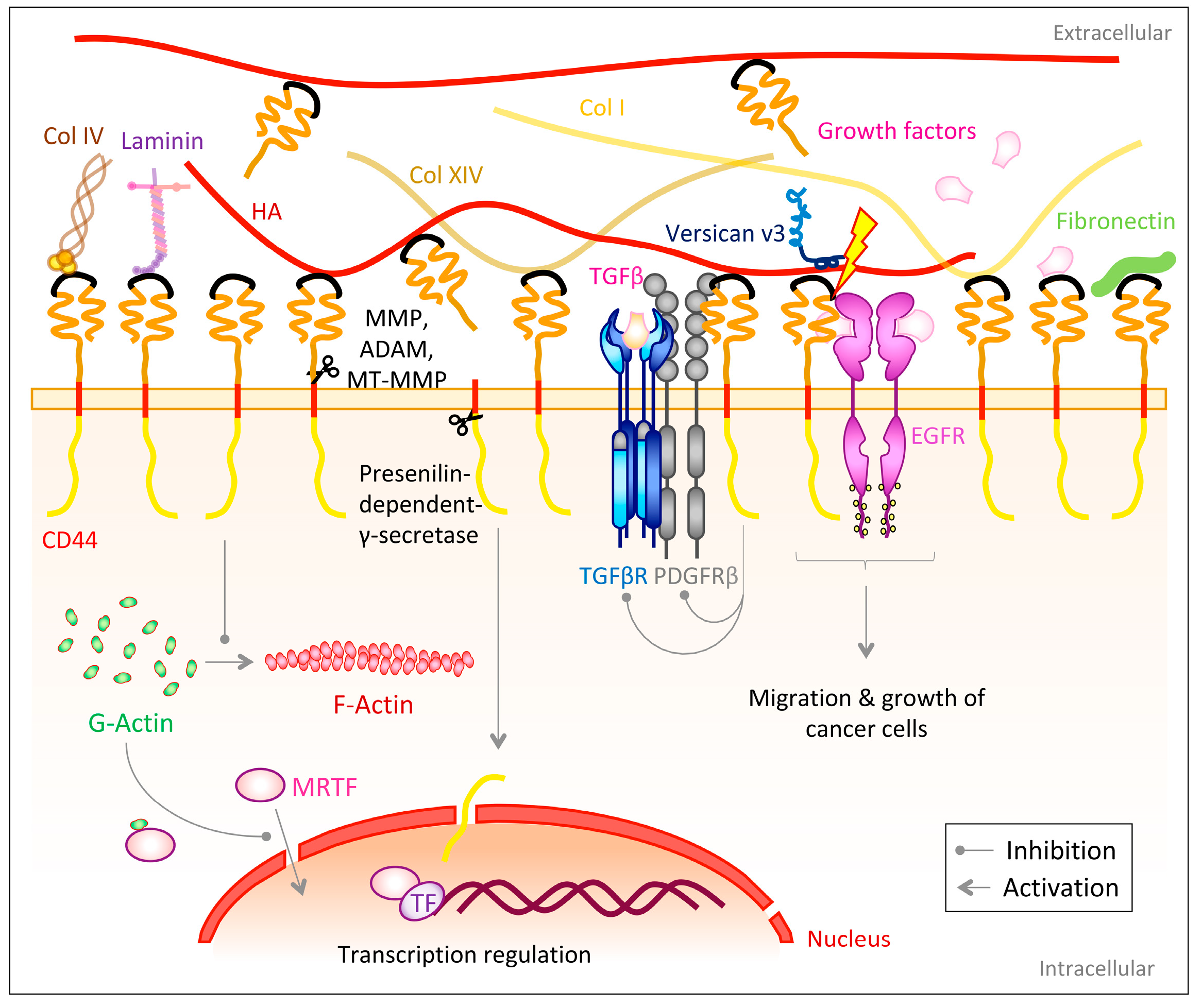
The transmembrane glycoprotein family CD44 belongs to the group of cell adhesion molecules [237] and is most commonly known as a receptor for hyaluronan (HA) [238,239]. HA is one of the most abundant ECM components in adult human skin and exists both membrane bound as a pericellular coat and freely in the extracellular space [240]. CD44 also serves as binding partner for other ECM components (Figure 5). However, these interactions are only rarely addressed in literature. The ECM proteins fibronectin [241], laminins [241], proteoglycans [242,243,244], heparin-binding growth factors [245] as well as collagen I [241], IV [246] and XIV have been shown to associate with CD44 [247]. Furthermore, CD44 interacts and cross-talks with other cell surface receptors, such as the transforming growth factor β receptor (TGFβR) and the platelet-derived growth factor receptor β (PDGFRβ) by forming a ternary complex, though CD44 is not critical for the interaction of the latter two receptors [248]. Nevertheless, HA-activated CD44 acts as negative regulator on TGFβR and PDGFRβ signaling, probably by recruiting a phosphatase to these growth receptors and/or by destabilizing them [248,249]. Additionally, CD44 has been shown in several studies to interact with the ErbB family of receptor tyrosine kinases and this has been associated with the modulation of tumor cell growth and motility [250,251,252] (Figure 5).
In humans, a single gene on the short arm of chromosome 11 with 19 exons encodes for various isoforms of CD44 [237,239,261]. Of these, the ubiquitously expressed protein, CD44s, represents the smallest isoform and consist of a globularly structured N-terminal extracellular domain with binding sites for its ligands, a transmembrane domain and a C-terminal cytoplasmic domain [237,239,261,262]. However, in the variant isoforms CD44v, the alternative splicing of exons 6–15 creates a variable part, the so-called stem region, which separates the extracellular domain from the transmembrane region [237,239,263]. Furthermore, CD44 can be post-translationally modified by phosphorylation [261] or glycosylation [264] and additionally, its extracellular domain is able to carry heparan sulfate (HS) [245] or chondroitin sulfate (CS) glycosaminoglycan (GAGs) side chains [265], which broadens its repertoire of forms and functions. However, there is limited knowledge about CD44′s specific functions as a proteoglycan [150].
The extracellular domain of CD44 can be proteolytically cleaved by matrix metalloproteinases (MMPs) [266,267], a disintegrin and metalloproteinases (ADAMs) [253] as well as membrane-type-MMPs (MT1-MMP and MT3-MMP) [254,255] (Figure 5), liberating the CD44 extracellular domain as a soluble NH2-terminal fragment [257] and thereby allowing the controlled release of cell-surface bound HA [263]. Ectodomain cleavage is regulated by extracellular calcium influx and activation of protein kinase C (PKC) and the small GTPase Rac [253]. On the opposite end after cleavage a membrane-bound COOH-terminal product (CD44EXT), containing the transmembrane and intracellular domain [257] is generated. This remaining domain is subsequently cleaved by the presenilin-dependent-γ-secretase [257,258], which in turn releases the CD44 intracellular domain (CD44ICD) into the cytoplasm. This intramembranous cleavage requires the previous removal of the ectodomain [268]. CD44ICD subsequently translocates to the nucleus to modulate transcriptions dependent on the 12-O-tetradecanoylphorbol 13-acetate (TPA)-responsive element (TRE) [257,268]. Interestingly, one target of this CD44ICD regulated transcription might be the CD44 gene itself, which comprises TRE elements in its promoter region [268].
In skin, the expression patterns of CD44 are independent from gender, age or ethnicity of donors, as well as from the anatomical origin of the sample [269]. However, while dermal fibroblasts mainly express CD44s [269] and only minimal amounts of variable CD44 transcripts [270], 18 distinct and unique transcripts have been identified in epidermal keratinocytes [269]. Moreover, adult keratinocytes display distinct CD44 expression patterns dependent on their differentiation level, with the strongest intensities in the stratum spinosum and stratum basale [271,272]. An increased expression of CD44 is found in inflamed or neoplastic skin [273,274], both on keratinocytes as well as on infiltrated lymphocytes close to the lesion [273] and is also reported for allergic and irritant contact dermatitis [275].
Although, complete CD44 deficiency in mice does not significantly alter the speed of macroscopic healing of punch biopsy wounds, such mice display alterations of the regeneration of the dermal collagen matrix [238]. During the early phases of wound healing, CD44-deficient mice presented increased inflammatory and reduced fibrogenic responses, such as enhanced leukocyte infiltration but delayed and altered accumulation and spatial distribution of fibroblasts positive for the fibroblast activation protein (FAP) as well as lowered levels of fibrillar collagens [238]. Upon wound closure, however, an accumulation of fibrillar collagens was observed due to a decreased collagen degradation, which promoted severe scarring as well as a lowered tensile strength of the tissue [238].
CD44 knockout mice have been reported to have thinned epidermis with altered differentiation, diminished apical localization of lamellar bodies as well as a delayed recovery of the skin barrier function upon acute perturbation of the stratum corneum [276,277], which has been associated with changed expression of tight junction proteins, allocating CD44 a role in tight junction assembly [277]. To specifically address the role of epidermal CD44, Shatirishvili et al. [278] employed a mouse model to, under the control of the keratin 14 promoter, delete CD44 in the epidermis (CD44Δker mice). They observed delayed wound healing, a compromised proliferation and differentiation of keratinocytes as well as a decreased keratinocyte adhesion to and migration on HA coated surfaces [278]. Moreover, atomic force microscopy on skin samples from these CD44Δker mice revealed a reduction in epidermal stiffness, whereas dermal stiffness remained unaffected when compared to wild-type mice [278]. The authors hypothesized that the decrease in epidermal stiffness caused the delayed wound healing properties of these mice and may itself be initiated by a lowered HA production as well as a lack of CD44-dependent HA adhesion [278].
Following the influence of CD44 on dermal healing and ECM deposition, CD44 would also be expected to have effects on fibrosis. Indeed, there is a large volume of studies describing the role of CD44 and its ligand HA in TGFβ-mediated pro-fibrotic signaling; however, findings are contradictory indicating contextuality of CD44 in regulation of fibrogenic processes [259]. While some studies describe CD44 as a supporting and stimulating factor of myofibroblast differentiation and fibrosis [259,279,280], others identify it as an inhibitor of fibrosis and TGFβ signaling [248,259,281,282]. In a murine in vitro model of dermal fibroblasts, Wang et al. [259] identified CD44 as inhibitor of α-SMA gene expression, independent from both the extracellular HA coat as well as HA biosynthesis. Instead, CD44 has been described to, in a yet unknown manner, prevent the conversion of G- to F-actin (actin polymerization) and thereby causing accumulation of G-actin in the cytoplasm. The latter binds cytoplasmatic myocardin-related transcription factor (MRTF) and hinders it from translocating to the nucleus, where it could co-activate the α-SMA transcription factor serum response factor (SRF) (Figure 5) [259].
4. Growth Factor Receptors
Growth factor receptors are transmembrane receptors employing protein kinase activity to activate intracellular signaling cascades and thereby modulate, inter alia, cell proliferation, differentiation, metabolism or migration. Prominent players in skin development, homeostasis as well as inflammatory and fibrotic skin malignancies are the transforming growth factor β receptors (TGFβR) and the epidermal growth factor receptor (EGFR). Therefore, and due to their interaction with various ECM components, they are highlighted in this review.
4.1. TGFβR
In mammals, there are three transforming growth factor β (TGFβ) isoforms (TGFβ1,-2, -3) [283]; however, the whole family of cytokine genes consists of 33 members [284]. The TGFβ family members signal through receptors (TGFβR) [285], which can be classified into type I (activin receptor-like kinase, ALK) and type II receptors and in humans, seven type I (ALK1–ALK7) and five type II receptors (TGFβRII, ActRII, ActRIIB, AMHRII, BMPRII) exist [285].
Here, with a skin-centric focus, we concentrate on the receptors themselves rather than their ligands. We will refer to them as type I (TGFβRI) or type II receptors (TGFβRII).
The TGFβRs consist N-terminally of small extracellular cysteine-rich domains for ligand binding followed by the transmembrane region and the cytoplasmic kinase domain [286]. TGFβRI additionally holds a regulatory juxtamembrane domain [286]. The receptors assemble into a heterotetrameric complex consisting of two type I and two type II receptors upon TGFβ binding to receptor type II [285,287,288,289,290,291,292]. Subsequently, TGFβRI is recruited and phosphorylated by TGFβRII [285,291,293,294]. This fosters the binding of receptor-regulated Smads (R-Smads) as well as their phosphorylation (i.e., Smad2 and 3 for ALK5 and Smad1, 5 and 8 for ALK1) by TGFβRI [285,295,296,297]. Canonically, ALK5-activated Smad2/3 associate with Smad4 and translocate to the nucleus, where they regulate gene transcription [285,291,293].
Apart from utilizing the canonical Smad signaling pathway, TGFβ is able to regulate signaling and gene transcription via other pathways, such as Ras-ERK-MAPK [293,298,299,300], p38MAPK [301,302], JNK [303,304], PI3K/AKT [305,306], NF-κB [307,308], RhoA [309,310], Rac [311] and Cdc42 [310,311]. Some of these pathways are additionally modulated by betaglycan, which is also referred to as type III TGFβ receptor (TGFβRIII) [312,313]. This transmembrane proteoglycan functions as co-receptor by binding TGFβ and presenting it to TGFβRII, thereby activating downstream signaling cascade [314,315]. Additionally, other transmembrane proteins or ECM components interact and cooperate with TGFβRs to modulate their signaling activities. These include: TGFβRII interaction with the fibronectin receptor, integrin α5β1, at the cell surface, which promotes fibronectin internalization, recycling and incorporation into fibrils in a Smad- and transcription-independent mechanism [316]; TGFβRII clustering with integrin αVβ3 enhancing TGFβ1-induced proliferative effects in the presence of tenascin-C or vitronectin [136]; and direct interaction of integrin α2β1 with TGFβRI and II [317].
In normal human skin, TGFβRI and TGFβRII are expressed in the strata basale, spinosum and granulosum of the epidermis, but can also be found in the dermis, though to a lesser extent [318,319,320]. Additionally, they are highly expressed in sweat and sebaceous glands as well as in hair follicles [320,321,322]. TGFβRII expression is strongly downregulated in aged human dermis [323,324,325].
A broad body of literature on the effects of TGFβ on fibrosis and wound healing exists [326,327,328,329,330,331,332,333,334] and several mouse models [335,336,337,338,339,340] have been developed to study them. However, many of these models present skin anomalies and/or decreased viability [341,342,343], which make them challenging to employ for wound healing studies. Nevertheless, they highlight the role of TGFβ in restricting cell growth and inducing apoptosis in wound healing processes and promoting ECM deposition and remodeling. For example, Liu et al. [335] examined mice with a point mutation in TGFβRI that caused partial receptor activation and found accelerated wound closure and cartilage formation in an ear-punch wound experiment. Mice expressing a dominant-negative TGFβRII mutant exclusively in the basal and suprabasal epidermis displayed thickened, wrinkled skin with a hyperplastic and hyperkeratotic epidermis [336]. Similarly, full-thickness excisional wounds re-epithelialized faster in transgenic mice that express a dominant negative TGFβRII only in keratinocytes [337]. Others created an inducible, fibroblastic TGFβRII knockout model and also found enhanced wound closure, faster re-epithelialization and increased macrophage infiltration [338]. Additionally, collagen deposition and remodeling, wound contraction as well as expression of integrin subunits α1, α2 and β1 were decreased [338]. Similarly, in mice where TGFβRII was deleted postnatally in dermal fibroblasts, excisional wound formation resulted in reduced wound contraction and scarring, while epidermal proliferation was increased [339]. Another fibroblast-selective expression of a kinase-deficient TGFβRII in transgenic mice led to TGFβ hyperactivity with increased fibroblast proliferation, increased ECM production as well as dermal fibrosis [340].
Martins et al. [113] found TGFβRI to be highly expressed in RDEB-associated high-risk SCCs, which occur in a heavily fibrotic microenvironment [344]. Interestingly, this marker was absent or expressed at low levels in non-EB-SCCs [113]. The group also established a stable knockdown of collagen VII in squamous cell carcinoma cells and xenografted 3D cultures onto nude mice. In their system, increased TGFβRI expression in invasive cells within the stroma of xenografts was observed and they suggested a role of TGFβ signaling in RDEB tumorigenesis [113]. Accordingly, Knaup et al. [345] characterized TGFβ1 signaling as architect of RDEB-associated SCC development in a comprehensive gene expression study comparing a non-malignant RDEB keratinocyte cell line to a RDEB SCC keratinocyte cell line.
Cammareri et al. [346] reported mutations in both TGFβRI and II to occur in human sporadic cutaneous SCC samples, while no mutations were detected in distant or perilesional skin. Many of these mutations resulted in a loss of function of canonical Smad signaling, provoking a loss of the TGFβ mediated tumor suppression and the authors grade this as driving event in sporadic cutaneous SCC development [346].
4.2. EGFR
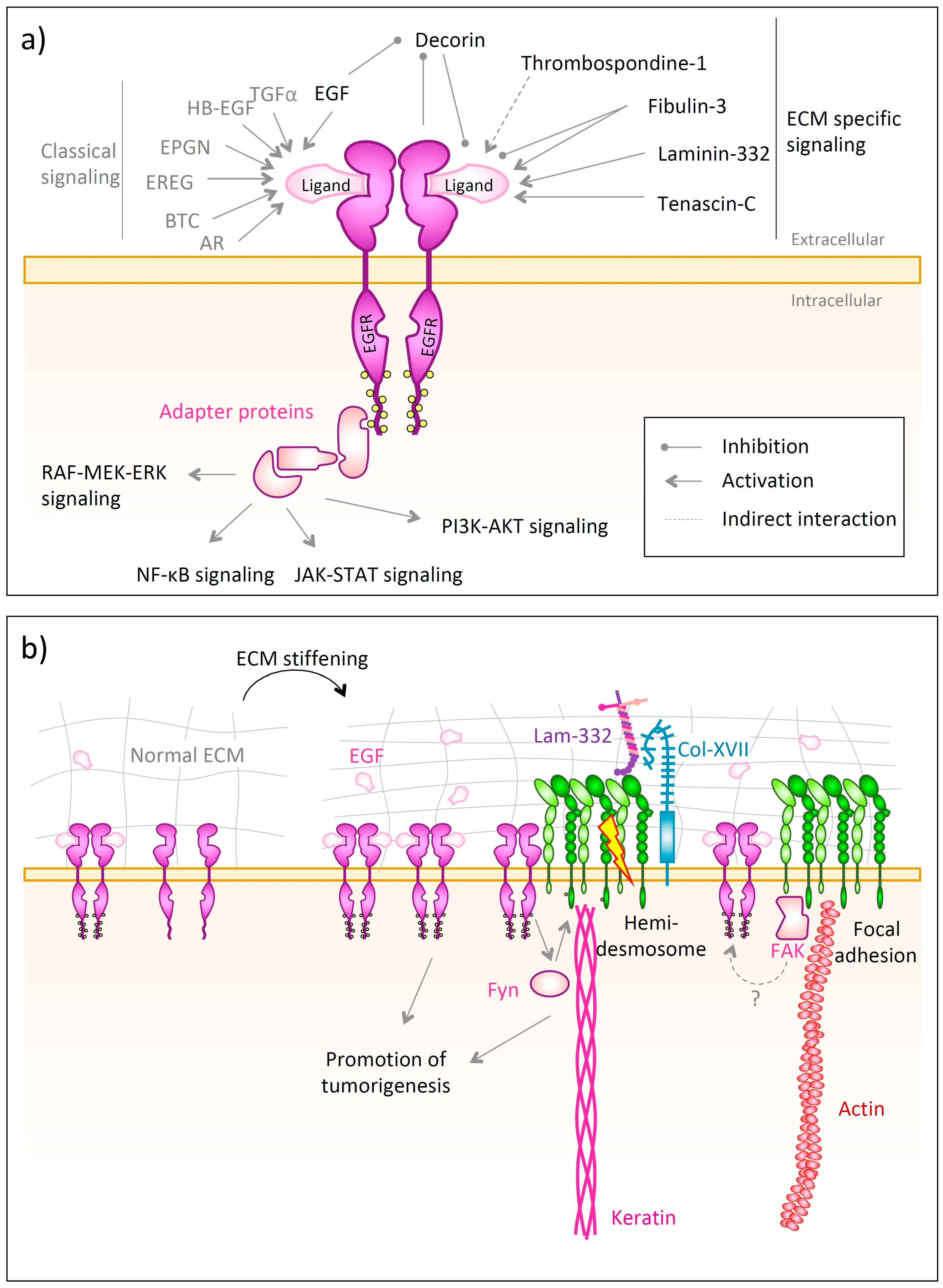
The epidermal growth factor (EGF) receptor (EGFR) is a transmembrane protein that belongs to the EGFR family of receptor tyrosine kinases along with that also includes ErbB2, ErbB3, and ErbB4 [347]. In skin, it is expressed both in epidermal keratinocytes [348] as well as dermal fibroblasts [349], though is most highly abundant in proliferative basal epidermal layers [348].
EGFR consists of an extracellular ligand-binding domain, a transmembrane helix followed intracellularly by a kinase domain and tyrosine-rich C-terminal phosphorylation sites [350,351]. The binding of a ligand, such as EGF, TGF-α, amphiregulin (AR), betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF), epigen (EPGN) or epiregulin (EREG) [352,353] to the monomeric extracellular domain causes receptor dimerization and auto-phosphorylation on the intracellular tyrosine residues [351]. This, in turn, allows the docking of adaptor proteins containing Src homology 2 (SH2) or phosphotyrosine binding (PTB) domains [352]. These activate various signaling pathways, such as the Ras-RAF-MEK-ERK pathway [352], the NF-κB cascade [354], the JAK-STAT [355] or the phosphatidylinositol 3-kinase (PI3K)/AKT pathway [352] (Figure 6a). Apart from forming homodimers, EGFR can also heterodimerize with other members of the ErbB family [356]. In order to downregulate EGFR signaling, the receptor undergoes endocytosis and is then sorted to be either recycled to the plasma membrane or to be degraded in the lysosome [357]. One prominent regulator involved in lyososomal degradation of EGFR is the E3 ubiquitin ligase c-Cbl, which binds to active EGFR and then facilitates its poly-ubiquitination and lysosomal degradation [357,358].
Cell–ECM interactions are crucial to maintain cellular metabolic activity by modulating signaling pathways and thereby regulating gene expression or growth factor availability. Detachment of epithelial cells from an ECM causes, inter alia, downregulation of EGFR and integrin β1, both on protein and mRNA level [363]. However, when embedded in an ECM, cells interact with their components utilizing EGFR in a bidirectional manner. For example, ECM composition impacts gene expression in response to EGF stimulation [364], but on the other hand, EGF has been reported to induce the expression of fibronectin in a dose-dependent manner via EGFR signaling [365,366].
Apart from the classical ligands, many ECM proteins influence EGFR signaling and interact with the receptor directly or indirectly. These are decorin, which binds with high affinity to the EGFR [367], causing downregulation of kinase activity and blocking intracellular calcium mobilization and thereby acting as tumor suppressor [367,368,369], tenascin C [370], the γ2 chain of laminin-332 [371], thrombospondin-1 [372] and fibulin-3 [373].
The differentiation and proliferation of epithelial cells is dependent on EGFR signaling. Various studies emphasize the role of EGFR in epidermal development, proliferation and differentiation [374,375,376,377], as well as epithelial motility [378,379,380] and adhesion [381]. This is highlighted by the manifestations of EGFR deficiency in humans. Campbell et al. [382] found an EGFR missense mutation being associated with fragile and highly inflamed skin in a 12 months old infant. While the child was first clinically diagnosed with a subtype of epidermolysis bullosa, it was later specified to carry a mutation located in the extracellular domain of EGFR in a region involved in receptor dimerization. On one hand, this mutation caused unstable EGFR to be rendered at the plasma membrane making it more prone to endocytosis. On the other hand, the mutation also suppressed EGFR phosphorylation and activation of downstream targets, such as ERK or AKT. Clinically, this led to inflamed, frequently infected skin with a reduction in desmosomal proteins as well as alterations in epidermal differentiation. Contrastingly, others have linked EGFR activity with desmosome disassembly and reduced cellular adhesion, whereas the inhibition of EGFR stabilizes desmosomes [383,384,385,386]. In this context, a crucial role is assigned to a disintegrin and metalloproteinase 17 (ADAM17) due to its capability to shed various EGFR ligands [387,388], which fuels EGFR signaling in differentiated keratinocytes. This, in turn, induces protein kinase C and phospholipase C γ1 pathways that terminate in transglutaminase 1 expression [374], which is crucial for crosslinking insoluble proteins of the cornified envelope at the outermost layer of the epidermis [374,389]. Correspondingly, human ADAM17 deficiency due to a loss-of-function mutation within exon 5 of ADAM17 has been associated with epithelial barrier defects resulting in neonatal inflammatory skin and bowel disease [390]. The observed lack of ADAM17 activity possibly prevents various EGFR ligands to be released from the plasma membrane via ectodomain shedding, therefore impeding EGFR activation [391] and corresponding downstream effects.
EGFR relies on the interplay with other receptors for its functions in skin. This is exemplified by interactions with epidermal integrins. It has been shown that a fraction of EGFR directly associates with the hemidesmosomal integrin α6β4 in keratinocytes [57,362] (Figure 6b). Activated EGFR, in turn, activates the Src family kinase Fyn, which phosphorylates the β4 cytoplasmic domain of the integrin. However, this requires the engagement of integrin α6β4 with laminin-332 or its clustering due to specific antibodies [362]. This phosphorylation of the β4 cytoplasmic domain promotes hemidesmosome disruption, which is a requirement for normal keratinocyte migration during wound healing, but also paves the way for squamous carcinoma invasion, proliferation and survival [57,362].
The mechanical properties of tissues are emerging as essential regulators of EGFR activity. Kenny et al. [359] investigated the proliferative response of keratinocytes on tissue stiffness, which was strongly mediated by EGF signaling through EGFR (Figure 6b). In their study, normal human keratinocytes were seeded on model silicone substrates with different elastic moduli coated with either collagen I or fibronectin. The authors revealed the EGFR phosphorylation and activation to be dependent on focal adhesion assembly and cytoskeletal tension. Furthermore, they could correlate EGFR phosphorylation with dermal stiffening in keloid scars, underlining the convolution of biochemistry and biomechanics in scar formation. This is in line with data from Saxena et al. [360], who found EGFR activity to be necessary for rigidity sensing in a model of fibronectin-coated silicone substrates of different stiffness cultured with mouse fibroblasts.
From a pathological perspective, it is known that even moderate stiffening of the ECM sensitizes epithelial cells to EGF, allowing them to proliferate independently of their contact to neighboring cells—a mechanism which is a hallmark of cancer cells [361]. Accordingly, stiffening of the ECM is associated with tumor aggression, metastases and poor clinical outcome in various cancers [392,393]. Likewise, EGFR is frequently overexpressed or strongly activated in several tumor types and fuels tumorigenesis [394,395]. EGFR is also expressed in RDEB-associated SCCs, though levels vary strongly [396,397,398]. Rationally, EGFR inhibition using the monoclonal antibody cetuximab [365] has been tested in single patients with RDEB cSCC and shown some positive but transient effects on lymph node metastases with only mild adverse events [396].
5. Outlook
Intensive studies on the receptor-mediated interplay of cells with their surrounding ECM have been carried out during the last few decades, and they have revealed the presence of complex and highly intertwined communication networks that go far beyond simply anchoring cells and matrices together. In fact, these complex systems impact diverse cellular processes, and vice versa, modifications in single cellular receptors may alter the integrity or composition of the entire ECM, promoting systemic diseases. Examples highlighted in this review are mutations of single integrin subunits that result in junctional epidermolysis bullosa, mutations in EGFR that cause skin fragility, or mutations in TGFβRs that impact cutaneous SCCs development. These effects are, in part, also mediated by the tight interplay of cell-surface receptors in skin, such as the direct or indirect interaction of EGFR with syndecans and integrins, which impact each other in a bidirectional manner.
The research work reviewed in this article highlights that malignancies, even though they symptomatically or clinically appear similar, may actually hold distinct underlying mechanisms and should therefore be approached in different ways.
However, an exciting and promising feature of the reviewed diseases is that—despite disease-specific triggers—shared characteristics can be observed not only symptomatically, but also on a biochemical level. These common hallmarks, in turn, may be targeted for therapeutic purposes. A more comprehensive understanding of the bidirectional cell–matrix crosstalks would foster a move from descriptive studies towards the identification and validation of specific therapeutic targets that are common in several conditions. This may pave the way for the development of single therapeutic agents applicable to a broad range of conditions.
Author Contributions
S.K. wrote the first draft of the manuscript and prepared all figures, S.K. and A.N. edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The authors’ research on these topics are supported by the German Research Foundation (DFG) SFB850 project B11 (AN) and NY90-5/1 (AN).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nestle, F.O.; Di Meglio, P.; Qin, J.-Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Yadav, N.; Parveen, S.; Chakravarty, S.; Banerjee, M. Skin anatomy and morphology. In Skin Aging & Cancer: Ambient UV-R Exposure; Dwivedi, A., Agarwal, N., Ray, L., Tripathi, A.K., Eds.; Springer: Singapore, 2019; pp. 1–10. ISBN 978-981-13-2541-0. [Google Scholar]
- Abdo, J.M.; Sopko, N.A.; Milner, S.M. The applied anatomy of human skin: A model for regeneration. Wound Med. 2020, 28, 100179. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular matrix and dermal fibroblast function in the healing wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Nyström, A.; Bruckner-Tuderman, L. Matrix molecules and skin biology. Semin. Cell Dev. Biol. 2019, 89, 136–146. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Theocharis, A.D.; Neill, T.; Karamanos, N.K. Complexity of matrix phenotypes. Matrix Biol. Plus 2020, 6–7, 100038. [Google Scholar] [CrossRef]
- Uitto, J.; Has, C.; Vahidnezhad, H.; Youssefian, L.; Bruckner-Tuderman, L. Molecular pathology of the basement membrane zone in heritable blistering diseases: The paradigm of epidermolysis bullosa. Matrix Biol. 2017, 57–58, 76–85. [Google Scholar] [CrossRef]
- Has, C.; Nyström, A. Chapter four—Epidermal basement membrane in health and disease. In Current Topics in Membranes: Basement Membranes; Miner, J.H., Ed.; Academic Press: Cambridge, MA, USA, 2015; pp. 117–170. ISBN 1063-5823. [Google Scholar]
- Kerr, B.A.; Byzova, T.V. Integrin alpha v (ITGAV). In Encyclopedia of Signaling Molecules; Choi, S., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 2634–2645. ISBN 978-3-319-67199-4. [Google Scholar]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [Green Version]
- Hegde, S.; Raghavan, S. A skin-depth analysis of integrins: Role of the integrin network in health and disease. Cell Commun. Adhes. 2013, 20, 155–169. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Schiller, H.B.; Hermann, M.-R.; Polleux, J.; Vignaud, T.; Zanivan, S.; Friedel, C.C.; Sun, Z.; Raducanu, A.; Gottschalk, K.-E.; Théry, M.; et al. β1- and αv-class integrins cooperate to regulate myosin II during rigidity sensing of fibronectin-based microenvironments. Nat. Cell Biol. 2013, 15, 625–636. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [Green Version]
- Goodman, S.R. Chapter 6—Cell adhesion and the extracellular matrix. In Medical Cell Biology, 3rd ed.; Goodman, S.R., Ed.; Academic Press: Cambridge, MA, USA, 2008; pp. 191–225. ISBN 978-0-12-370458-0. [Google Scholar]
- Tiwari, S.; Askari, J.A.; Humphries, M.J.; Bulleid, N.J. Divalent cations regulate the folding and activation status of integrins during their intracellular trafficking. J. Cell Sci. 2011, 124, 1672–1680. [Google Scholar] [CrossRef] [Green Version]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Danen, E.H.J. Integrins: An overview of structural and functional aspects. In Madame Curie Bioscience Database; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Tucker, R.P.; Chiquet-Ehrismann, R. Tenascin-C: Its functions as an integrin ligand. Int. J. Biochem. Cell Biol. 2015, 65, 165–168. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2010, 339, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Knight, C.G.; Morton, L.F.; Onley, D.J.; Peachey, A.R.; Messent, A.J.; Smethurst, P.A.; Tuckwell, D.S.; Farndale, R.W.; Barnes, M.J. Identification in collagen type I of an integrin alpha2 beta1-binding site containing an essential GER sequence. J. Biol. Chem. 1998, 273, 33287–33294. [Google Scholar] [CrossRef] [Green Version]
- Rehn, M.; Veikkola, T.; Kukk-Valdre, E.; Nakamura, H.; Ilmonen, M.; Lombardo, C.; Pihlajaniemi, T.; Alitalo, K.; Vuori, K. Interaction of endostatin with integrins implicated in angiogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 1024–1029. [Google Scholar] [CrossRef]
- Woodall, B.P.; Nyström, A.; Iozzo, R.A.; Eble, J.A.; Niland, S.; Krieg, T.; Eckes, B.; Pozzi, A.; Iozzo, R.V. Integrin alpha2beta1 is the required receptor for endorepellin angiostatic activity. J. Biol. Chem. 2008, 283, 2335–2343. [Google Scholar] [CrossRef] [Green Version]
- Sudhakar, A.; Sugimoto, H.; Yang, C.; Lively, J.; Zeisberg, M.; Kalluri, R. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. USA 2003, 100, 4766–4771. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.-H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Shimaoka, M.; Xiao, T.; Liu, J.-H.; Yang, Y.; Dong, Y.; Jun, C.-D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Costell, M.; Fässler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Schäfer, M.; Mykuliak, V.V.; Ripamonti, M.; Heiser, L.; Weißenbruch, K.; Krübel, S.; Franz, C.M.; Hytönen, V.P.; Wehrle-Haller, B.; et al. Induction of ligand promiscuity of αVβ3 integrin by mechanical force. J. Cell Sci. 2020. [Google Scholar] [CrossRef]
- Baker, E.L.; Zaman, M.H. The biomechanical integrin. J. Biomech. 2010, 43, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Moore, S.W.; Roca-Cusachs, P.; Sheetz, M.P. Stretchy proteins on stretchy substrates: The important elements of integrin-mediated rigidity sensing. Dev. Cell 2010, 19, 194–206. [Google Scholar] [CrossRef] [Green Version]
- Puklin-Faucher, E.; Sheetz, M.P. The mechanical integrin cycle. J. Cell Sci. 2009, 122, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.A. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2010, 2, a005066. [Google Scholar] [CrossRef]
- Duperret, E.K.; Dahal, A.; Ridky, T.W. Focal-adhesion-independent integrin-alpha v regulation of FAK and c-Myc is necessary for 3D skin formation and tumor invasion. J. Cell Sci. 2015, 128, 3997–4013. [Google Scholar] [CrossRef] [Green Version]
- Duperret, E.K.; Ridky, T.W. Focal adhesion complex proteins in epidermis and squamous cell carcinoma. Cell Cycle 2013, 12, 3272–3285. [Google Scholar] [CrossRef]
- Stutchbury, B.; Atherton, P.; Tsang, R.; Wang, D.-Y.; Ballestrem, C. Distinct focal adhesion protein modules control different aspects of mechanotransduction. J. Cell Sci. 2017, 130, 1612–1624. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.-L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Guan, J.-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, D.A.; Mitra, S.K.; Hauck, C.R.; Streblow, D.N.; Nelson, J.A.; Ilic, D.; Huang, S.; Li, E.; Nemerow, G.R.; Leng, J.; et al. Differential regulation of cell motility and invasion by FAK. J. Cell Biol. 2003, 160, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Watt, F.M. Role of integrins in regulating epidermal adhesion, growth and differentiation. EMBO J. 2002, 21, 3919–3926. [Google Scholar] [CrossRef] [Green Version]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Increased expression of integrin αvβ3 contributes to the establishment of autocrine TGF-β signaling in scleroderma fibroblasts. J. Immunol. 2005, 175, 7708–7718. [Google Scholar] [CrossRef] [Green Version]
- Phillips, R.J.; Aplin, J.D.; Lake, B.D. Antigenic expression of integrin alpha 6 beta 4 in junctional epidermolysis bullosa. Histopathology 1994, 24, 571–576. [Google Scholar] [CrossRef]
- Brown, T.A.; Gil, S.G.; Sybert, V.P.; Lestringant, G.G.; Tadini, G.; Caputo, R.; Carter, W.G. Defective integrin alpha 6 beta 4 expression in the skin of patients with junctional epidermolysis bullosa and pyloric atresia. J. Investig. Dermatol. 1996, 107, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Niessen, C.M.; van der Raaij-Helmer, M.H.; Hulsman, E.H.; van der Neut, R.; Jonkman, M.F.; Sonnenberg, A. Deficiency of the integrin beta 4 subunit in junctional epidermolysis bullosa with pyloric atresia: Consequences for hemidesmosome formation and adhesion properties. J. Cell Sci. 1996, 109 Pt 7, 1695–1706. [Google Scholar]
- Nishiuchi, R.; Takagi, J.; Hayashi, M.; Ido, H.; Yagi, Y.; Sanzen, N.; Tsuji, T.; Yamada, M.; Sekiguchi, K. Ligand-binding specificities of laminin-binding integrins: A comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol. 2006, 25, 189–197. [Google Scholar] [CrossRef]
- Spinardi, L.; Ren, Y.L.; Sanders, R.; Giancotti, F.G. The beta 4 subunit cytoplasmic domain mediates the interaction of alpha 6 beta 4 integrin with the cytoskeleton of hemidesmosomes. Mol. Biol. Cell 1993, 4, 871–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariya, Y.; Gu, J. N-glycosylation of β4 integrin controls the adhesion and motility of keratinocytes. PLoS ONE 2011, 6, e27084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dans, M.; Gagnoux-Palacios, L.; Blaikie, P.; Klein, S.; Mariotti, A.; Giancotti, F.G. Tyrosine phosphorylation of the beta 4 integrin cytoplasmic domain mediates Shc signaling to extracellular signal-regulated kinase and antagonizes formation of hemidesmosomes. J. Biol. Chem. 2001, 276, 1494–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolopoulos, S.N.; Blaikie, P.; Yoshioka, T.; Guo, W.; Puri, C.; Tacchetti, C.; Giancotti, F.G. Targeted deletion of the integrin beta4 signaling domain suppresses laminin-5-dependent nuclear entry of mitogen-activated protein kinases and NF-kappaB, causing defects in epidermal growth and migration. Mol. Cell Biol. 2005, 25, 6090–6102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Dong, Z.; Zhang, Y.; Miao, J. The roles of integrin β4 in vascular endothelial cells. J. Cell Physiol. 2012, 227, 474–478. [Google Scholar] [CrossRef]
- Walko, G.; Castanon, M.J.; Wiche, G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015, 360, 529–544. [Google Scholar] [CrossRef] [Green Version]
- Aho, S.; Uitto, J. Direct interaction between the intracellular domains of bullous pemphigoid antigen 2 (BP180) and beta 4 integrin, hemidesmosomal components of basal keratinocytes. Biochem. Biophys. Res. Commun. 1998, 243, 694–699. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.-D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Tidman, M.J.; Eady, R.A. Hemidesmosome heterogeneity in junctional epidermolysis bullosa revealed by morphometric analysis. J. Investig. Dermatol. 1986, 86, 51–56. [Google Scholar] [CrossRef]
- Eady, R.A.; McGrath, J.A.; McMillan, J.R. Ultrastructural clues to genetic disorders of skin: The dermal-epidermal junction. J. Investig. Dermatol. 1994, 103, 13S–18S. [Google Scholar] [CrossRef] [Green Version]
- Falcioni, R.; Antonini, A.; Nistico, P.; Di Stefano, S.; Crescenzi, M.; Natali, P.G.; Sacchi, A. Alpha 6 beta 4 and alpha 6 beta 1 integrins associate with ErbB-2 in human carcinoma cell lines. Exp. Cell Res. 1997, 236, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, A.; Kedeshian, P.A.; Dans, M.; Curatola, A.M.; Gagnoux-Palacios, L.; Giancotti, F.G. EGF-R signaling through Fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes: Role in epithelial cell migration and carcinoma invasion. J. Cell Biol. 2001, 155, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Gagnoux-Palacios, L.; Dans, M.; van’t Hof, W.; Mariotti, A.; Pepe, A.; Meneguzzi, G.; Resh, M.D.; Giancotti, F.G. Compartmentalization of integrin alpha6beta4 signaling in lipid rafts. J. Cell Biol. 2003, 162, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Tsubota, Y.; Hashimoto, J.; Kariya, Y.; Miyazaki, K. The short arm of laminin gamma2 chain of laminin-5 (laminin-332) binds syndecan-1 and regulates cellular adhesion and migration by suppressing phosphorylation of integrin beta4 chain. Mol. Biol. Cell 2007, 18, 1621–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainiero, F.; Murgia, C.; Wary, K.K.; Curatola, A.M.; Pepe, A.; Blumemberg, M.; Westwick, J.K.; Der, C.J.; Giancotti, F.G. The coupling of alpha6beta4 integrin to Ras-MAP kinase pathways mediated by Shc controls keratinocyte proliferation. EMBO J. 1997, 16, 2365–2375. [Google Scholar] [CrossRef] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. A signaling adapter function for alpha6beta4 integrin in the control of HGF-dependent invasive growth. Cell 2001, 107, 643–654. [Google Scholar] [CrossRef] [Green Version]
- Shaw, L.M.; Rabinovitz, I.; Wang, H.H.; Toker, A.; Mercurio, A.M. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell 1997, 91, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Kligys, K.R.; Wu, Y.; Hopkinson, S.B.; Kaur, S.; Platanias, L.C.; Jones, J.C.R. α6β4 integrin, a master regulator of expression of integrins in human keratinocytes. J. Biol. Chem. 2012, 287, 17975–17984. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Bachelder, R.E.; Lipscomb, E.A.; Shaw, L.M.; Mercurio, A.M. Integrin (alpha 6 beta 4) regulation of eIF-4E activity and VEGF translation: A survival mechanism for carcinoma cells. J. Cell Biol. 2002, 158, 165–174. [Google Scholar] [CrossRef]
- Tani, T.; Karttunen, T.; Kiviluoto, T.; Kivilaakso, E.; Burgeson, R.E.; Sipponen, P.; Virtanen, I. Alpha 6 beta 4 integrin and newly deposited laminin-1 and laminin-5 form the adhesion mechanism of gastric carcinoma. Continuous expression of laminins but not that of collagen VII is preserved in invasive parts of the carcinomas: Implications for acquisition of the invading phenotype. Am. J. Pathol. 1996, 149, 781–793. [Google Scholar]
- Kariya, Y.; Oyama, M.; Hashimoto, Y.; Gu, J.; Kariya, Y. beta4-integrin/PI3K signaling promotes tumor progression through the galectin-3-N-glycan complex. Mol. Cancer Res. 2018, 16, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitz, I.; Mercurio, A.M. The integrin alpha 6 beta 4 and the biology of carcinoma. Biochem. Cell Biol. 1996, 74, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, S.W.; Theivakumar, S.; Owens, D.M. Epidermal alpha6beta4 integrin stimulates the influx of immunosuppressive cells during skin tumor promotion. J. Dermatol. Sci. 2012, 66, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, R.L.; O’Connor, K.L. Clinical significance of the integrin α6β4 in human malignancies. Lab. Investig. 2015, 95, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, K.A.; Carey, T.E. Altered expression in squamous carcinoma cells of an orientation restricted epithelial antigen detected by monoclonal antibody A9. Cancer Res. 1986, 46, 3614–3623. [Google Scholar]
- Van Waes, C.; Surh, D.M.; Chen, Z.; Kirby, M.; Rhim, J.S.; Brager, R.; Sessions, R.B.; Poore, J.; Wolf, G.T.; Carey, T.E. Increase in suprabasilar integrin adhesion molecule expression in human epidermal neoplasms accompanies increased proliferation occurring with immortalization and tumor progression. Cancer Res. 1995, 55, 5434–5444. [Google Scholar]
- Wolf, G.T.; Carey, T.E.; Schmaltz, S.P.; McClatchey, K.D.; Poore, J.; Glaser, L.; Hayashida, D.J.; Hsu, S. Altered antigen expression predicts outcome in squamous cell carcinoma of the head and neck. J. Natl. Cancer Inst. 1990, 82, 1566–1572. [Google Scholar] [CrossRef]
- Kariya, Y.; Kariya, Y.; Gu, J. Roles of integrin α6β4 glycosylation in cancer. Cancers 2017, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Michael, M.; Begum, R.; Chan, G.K.; Whitewood, A.J.; Matthews, D.R.; Goult, B.T.; McGrath, J.A.; Parsons, M. Kindlin-1 regulates epidermal growth factor receptor signaling. J. Investig. Dermatol. 2019, 139, 369–379. [Google Scholar] [CrossRef]
- Brakebusch, C.; Fässler, R. The integrin-actin connection, an eternal love affair. EMBO J. 2003, 22, 2324–2333. [Google Scholar] [CrossRef]
- Rippa, A.L.; Vorotelyak, E.A.; Vasiliev, A.V.; Terskikh, V.V. The role of integrins in the development and homeostasis of the epidermis and skin appendages. Acta Nat. 2013, 5, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Moser, M.; Legate, K.R.; Zent, R.; Fässler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895. [Google Scholar] [CrossRef] [PubMed]
- Bouaouina, M.; Lad, Y.; Calderwood, D.A. The N-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate beta1 and beta3 integrins. J. Biol. Chem. 2008, 283, 6118–6125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meves, A.; Geiger, T.; Zanivan, S.; DiGiovanni, J.; Mann, M.; Faessler, R. Beta 1 integrin cytoplasmic tyrosines promote skin tumorigenesis independent of their phosphorylation. Proc. Natl. Acad. Sci. USA 2011, 108, 15213–15218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, T.; Jove, R.; Fässler, R.; Mosher, D.F. Role of the cytoplasmic tyrosines of β1A integrins in transformation by v-src. Proc. Natl. Acad. Sci. USA 2001, 98, 3808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pylayeva, Y.; Giancotti, F.G. Development requires activation but not phosphorylation of β1 integrins. Genes Dev. 2006, 20, 1057–1060. [Google Scholar] [CrossRef] [Green Version]
- Plantefaber, L.C.; Hynes, R.O. Changes in integrin receptors on oncogenically transformed cells. Cell 1989, 56, 281–290. [Google Scholar] [CrossRef]
- Morgner, J.; Ghatak, S.; Jakobi, T.; Dieterich, C.; Aumailley, M.; Wickström, S.A. Integrin-linked kinase regulates the niche of quiescent epidermal stem cells. Nat. Commun. 2015, 6, 8198. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, H.; Ferreira, M.; Donati, G.; Marciano, D.K.; Linton, J.M.; Sato, Y.; Hartner, A.; Sekiguchi, K.; Reichardt, L.F.; Watt, F.M. The basement membrane of hair follicle stem cells is a muscle cell niche. Cell 2011, 144, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Symington, B.; Takada, Y.; Carter, W. Interaction of integrins α3β1 and α2β1: Potential role in keratinocyte intercellular adhesion. J. Cell Biol. 1993, 120, 523–535. [Google Scholar] [CrossRef]
- Woltersdorf, C.; Bonk, M.; Leitinger, B.; Huhtala, M.; Käpylä, J.; Heino, J.; Gil Girol, C.; Niland, S.; Eble, J.A.; Bruckner, P.; et al. The binding capacity of α1β1-, α2β1- and α10β1-integrins depends on non-collagenous surface macromolecules rather than the collagens in cartilage fibrils. Matrix Biol. 2017, 63, 91–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, G.; Zwolanek, D.; Eckes, B.; Niland, S.; Käpylä, J.; Zweers, M.C.; Ishada-Yamamoto, A.; Krieg, T.; Heino, J.; Eble, J.A.; et al. Collagen XXIII, novel ligand for integrin alpha2beta1 in the epidermis. J. Biol. Chem. 2011, 286, 27804–27813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riikonen, T.; Westermarck, J.; Koivisto, L.; Broberg, A.; Kähäri, V.-M.; Heino, J. Integrin α2β1 is a positive regulator of collagenase (MMP-1) and collagen α1(I) gene expression. J. Biol. Chem. 1995, 270, 13548–13552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langholz, O.; Röckel, D.; Mauch, C.; Kozlowska, E.; Bank, I.; Krieg, T.; Eckes, B. Collagen and collagenase gene expression in three-dimensional collagen lattices are differentially regulated by alpha 1 beta 1 and alpha 2 beta 1 integrins. J. Cell Biol. 1995, 131, 1903–1915. [Google Scholar] [CrossRef] [PubMed]
- Ravanti, L.; Heino, J.; Lopez-Otin, C.; Kahari, V.M. Induction of collagenase-3 (MMP-13) expression in human skin fibroblasts by three-dimensional collagen is mediated by p38 mitogen-activated protein kinase. J. Biol. Chem. 1999, 274, 2446–2455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivaska, J.; Reunanen, H.; Westermarck, J.; Koivisto, L.; Kähäri, V.-M.; Heino, J. Integrin α2β1 mediates isoform-specific activation of p38 and upregulation of collagen gene transcription by a mechanism involving the α2 cytoplasmic tail. J. Cell Biol. 1999, 147, 401–416. [Google Scholar] [CrossRef]
- Ojalill, M.; Parikainen, M.; Rappu, P.; Aalto, E.; Jokinen, J.; Virtanen, N.; Siljamäki, E.; Heino, J. Integrin α2β1 decelerates proliferation, but promotes survival and invasion of prostate cancer cells. Oncotarget 2018, 9, 32435–32447. [Google Scholar] [CrossRef]
- Parks, W.C. What is the α2β1 integrin doing in the epidermis? J. Investig. Dermatol. 2007, 127, 264–266. [Google Scholar] [CrossRef] [Green Version]
- Zweers, M.C.; Davidson, J.M.; Pozzi, A.; Hallinger, R.; Janz, K.; Quondamatteo, F.; Leutgeb, B.; Krieg, T.; Eckes, B. Integrin alpha2beta1 is required for regulation of murine wound angiogenesis but is dispensable for reepithelialization. J. Investig. Dermatol. 2007, 127, 467–478. [Google Scholar] [CrossRef] [Green Version]
- DiPersio, C.M.; Hodivala-Dilke, K.M.; Jaenisch, R.; Kreidberg, J.A.; Hynes, R.O. Alpha3beta1 integrin is required for normal development of the epidermal basement membrane. J. Cell Biol. 1997, 137, 729–742. [Google Scholar] [CrossRef]
- Delwel, G.O.; de Melker, A.A.; Hogervorst, F.; Jaspars, L.H.; Fles, D.L.; Kuikman, I.; Lindblom, A.; Paulsson, M.; Timpl, R.; Sonnenberg, A. Distinct and overlapping ligand specificities of the alpha 3A beta 1 and alpha 6A beta 1 integrins: Recognition of laminin isoforms. Mol. Biol. Cell 1994, 5, 203–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruegel, J.; Miosge, N. Basement membrane components are key players in specialized extracellular matrices. Cell Mol. Life Sci. 2010, 67, 2879–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin α3 mutations with kidney, lung, and skin disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margadant, C.; Raymond, K.; Kreft, M.; Sachs, N.; Janssen, H.; Sonnenberg, A. Integrin α3β1 inhibits directional migration and wound re-epithelialization in the skin. J. Cell Sci. 2009, 122, 278. [Google Scholar] [CrossRef] [Green Version]
- Hodivala-Dilke, K.M.; DiPersio, C.M.; Kreidberg, J.A.; Hynes, R.O. Novel roles for alpha3beta1 integrin as a regulator of cytoskeletal assembly and as a trans-dominant inhibitor of integrin receptor function in mouse keratinocytes. J. Cell Biol. 1998, 142, 1357–1369. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Chen, C.; Pal-Ghosh, S.; Stepp, M.A.; Sheppard, D.; van de Water, L. Loss of integrin alpha9beta1 results in defects in proliferation, causing poor re-epithelialization during cutaneous wound healing. J. Investig. Dermatol. 2009, 129, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Brakebusch, C.; Grose, R.; Quondamatteo, F.; Ramirez, A.; Jorcano, J.L.; Pirro, A.; Svensson, M.; Herken, R.; Sasaki, T.; Timpl, R.; et al. Skin and hair follicle integrity is crucially dependent on beta 1 integrin expression on keratinocytes. EMBO J. 2000, 19, 3990–4003. [Google Scholar] [CrossRef] [Green Version]
- Henry, M.D.; Campbell, K.P. A role for dystroglycan in basement membrane assembly. Cell 1998, 95, 859–870. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Bauer, C.; Mundschau, G.; Li, Q.; Fuchs, E. Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J. Cell Biol. 2000, 150, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Kurbet, A.S.; Hegde, S.; Bhattacharjee, O.; Marepally, S.; Vemula, P.K.; Raghavan, S. Sterile inflammation enhances ECM degradation in integrin beta1 KO embryonic skin. Cell Rep. 2016, 16, 3334–3347. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Leask, A. Integrin β1 is required for dermal homeostasis. J. Investig. Dermatol. 2013, 133, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Xu, S.-w.; Blumbach, K.; Eastwood, M.; Denton, C.P.; Eckes, B.; Krieg, T.; Abraham, D.J.; Leask, A. Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo. J. Cell Sci. 2010, 123, 3674–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Kapoor, M.; Denton, C.P.; Abraham, D.J.; Leask, A. Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis Rheum. 2009, 60, 2817–2821. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.-N.; Zeltz, C.; Sørensen, I.W.; Barczyk, M.; Carracedo, S.; Hallinger, R.; Niehoff, A.; Eckes, B.; Gullberg, D. Reduced granulation tissue and wound strength in the absence of α11β1 integrin. J. Investig. Dermatol. 2015, 135, 1435–1444. [Google Scholar] [CrossRef] [Green Version]
- Schulz, J.-N.; Plomann, M.; Sengle, G.; Gullberg, D.; Krieg, T.; Eckes, B. New developments on skin fibrosis—Essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 2018, 68–69, 522–532. [Google Scholar] [CrossRef]
- Schulz, J.-N.; Blumbach, K.; Brunner, G.; Niehoff, A.; Gullberg, D.; Krieg, T.; Eckes, B. Disruption of the b1-integrin-ILK axis attenuates fibrotic reactions. Wound Repair Regen 2014, 22, A96. [Google Scholar]
- Brockbank, E.C.; Bridges, J.; Marshall, C.J.; Sahai, E. Integrin β1 is required for the invasive behaviour but not proliferation of squamous cell carcinoma cells in vivo. Br. J. Cancer 2005, 92, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Martins, V.L.; Caley, M.P.; Moore, K.; Szentpetery, Z.; Marsh, S.T.; Murrell, D.F.; Kim, M.H.; Avari, M.; McGrath, J.A.; Cerio, R.; et al. Suppression of TGFbeta and angiogenesis by type VII collagen in cutaneous SCC. J. Natl. Cancer Inst. 2016. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; Secades, P.; van Hulst, L.; Kreft, M.; Song, J.-Y.; Sonnenberg, A. Loss of integrin α3 prevents skin tumor formation by promoting epidermal turnover and depletion of slow-cycling cells. Proc. Natl. Acad. Sci. USA 2012, 109, 21468. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Zou, L.; Ma, G.; Wu, X.; Huang, F.; Feng, T.; Li, S.; Lin, Q.; He, X.; Liu, Z.; et al. Integrin β1 is a critical effector in promoting metastasis and chemo-resistance of esophageal squamous cell carcinoma. Am. J. Cancer Res. 2017, 7, 531–542. [Google Scholar]
- Wang, D.; Muller, S.; Amin, A.R.M.; Huang, D.; Su, L.; Hu, Z.; Rahman, M.A.; Nannapaneni, S.; Koenig, L.; Chen, Z.; et al. The pivotal role of integrin beta1 in metastasis of head and neck squamous cell carcinoma. Clin. Cancer Res. 2012, 18, 4589–4599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, M.; Nakamura, S.; Sasaki, M.; Kurahara, S.; Ikebe, T.; Harada, T.; Shirasuna, K. Expression of integrins in squamous cell carcinoma of the oral cavity. Correlations with tumor invasion and metastasis. Am. J. Clin. Pathol. 1999, 111, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, H.; Mueller, S.C.; Nomizu, M.; Yamada, Y.; Yeh, Y.; Chen, W.-T. Activation of β1 integrin signaling stimulates tyrosine phosphorylation of p190 RhoGAP and membrane-protrusive activities at invadopodia. J. Biol. Chem. 1998, 273, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.C.; Coppolino, M.G. SNARE-dependent interaction of Src, EGFR and β1 integrin regulates invadopodia formation and tumor cell invasion. J. Cell Sci. 2014, 127, 1712. [Google Scholar] [CrossRef] [Green Version]
- Kean, M.J.; Williams, K.C.; Skalski, M.; Myers, D.; Burtnik, A.; Foster, D.; Coppolino, M.G. VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J. Cell Sci. 2009, 122, 4089. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Miyamoto, S.; Mekada, E. Integrin alpha 2 beta 1-dependent EGF receptor activation at cell-cell contact sites. J. Cell Sci. 2000, 113 Pt 12, 2139–2147. [Google Scholar]
- Reginato, M.J.; Mills, K.R.; Paulus, J.K.; Lynch, D.K.; Sgroi, D.C.; Debnath, J.; Muthuswamy, S.K.; Brugge, J.S. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 2003, 5, 733–740. [Google Scholar] [CrossRef]
- Wang, F.; Weaver, V.M.; Petersen, O.W.; Larabell, C.A.; Dedhar, S.; Briand, P.; Lupu, R.; Bissell, M.J. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: A different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA 1998, 95, 14821–14826. [Google Scholar] [CrossRef]
- Eke, I.; Zscheppang, K.; Dickreuter, E.; Hickmann, L.; Mazzeo, E.; Unger, K.; Krause, M.; Cordes, N. Simultaneous β1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. J. Natl. Cancer Inst. 2015. [Google Scholar] [CrossRef]
- Koistinen, P.; Heino, J. The selective regulation of alpha v beta 1 integrin expression is based on the hierarchical formation of alpha v-containing heterodimers. J. Biol. Chem. 2002, 277, 24835–24841. [Google Scholar] [CrossRef] [Green Version]
- Janes, S.M.; Watt, F.M. Switch from alpha v beta 5 to alpha v beta 6 integrin expression protects squamous cell carcinomas from anoikis. J. Cell Biol. 2004, 166, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualini, R.; Bodorova, J.; Ye, S.; Hemler, M.E. A study of the structure, function and distribution of beta 5 integrins using novel anti-beta 5 monoclonal antibodies. J. Cell Sci. 1993, 105 Pt 1, 101–111. [Google Scholar]
- Longmate, W.M.; DiPersio, C.M. Integrin regulation of epidermal functions in wounds. Adv. Wound Care 2014, 3, 229–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef]
- Wipff, P.-J.; Hinz, B. Integrins and the activation of latent transforming growth factor beta1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, J.; Dong, X.; Shi, M.; Lu, C.; Springer, T.A. GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 2012, 23, 1129–1139. [Google Scholar] [CrossRef]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Scaffidi, A.K.; Petrovic, N.; Moodley, Y.P.; Fogel-Petrovic, M.; Kroeger, K.M.; Seeber, R.M.; Eidne, K.A.; Thompson, P.J.; Knight, D.A. Alpha(v)beta(3) integrin interacts with the transforming growth factor beta (TGFbeta) type II receptor to potentiate the proliferative effects of TGFbeta1 in living human lung fibroblasts. J. Biol. Chem. 2004, 279, 37726–37733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galliher, A.J.; Schiemann, W.P. Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Tamaki, K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am. J. Pathol. 2006, 168, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Zoppi, N.; Barlati, S.; Colombi, M. FAK-independent alphavbeta3 integrin-EGFR complexes rescue from anoikis matrix-defective fibroblasts. Biochim. Biophys. Acta 2008, 1783, 1177–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häkkinen, L.; Koivisto, L.; Gardner, H.; Saarialho-Kere, U.; Carroll, J.M.; Lakso, M.; Rauvala, H.; Laato, M.; Heino, J.; Larjava, H. Increased expression of beta6-integrin in skin leads to spontaneous development of chronic wounds. Am. J. Pathol. 2004, 164, 229–242. [Google Scholar] [CrossRef]
- AlDahlawi, S.; Eslami, A.; Hakkinen, L.; Larjava, H.S. The alphavbeta6 integrin plays a role in compromised epidermal wound healing. Wound Repair Regen 2006, 14, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Duperret, E.K.; Natale, C.A.; Monteleon, C.; Dahal, A.; Ridky, T.W. The integrin αv-TGFβ signaling axis is necessary for epidermal proliferation during cutaneous wound healing. Cell Cycle 2016, 15, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Schnittert, J.; Bansal, R.; Storm, G.; Prakash, J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 2018, 129, 37–53. [Google Scholar] [CrossRef]
- Jakhu, H.; Gill, G.; Singh, A. Role of integrins in wound repair and its periodontal implications. J. Oral Biol. Craniofac. Res. 2018, 8, 122–125. [Google Scholar] [CrossRef]
- Thomas, G.J.; Lewis, M.P.; Whawell, S.A.; Russell, A.; Sheppard, D.; Hart, I.R.; Speight, P.M.; Marshall, J.F. Expression of the αvβ6 integrin promotes migration and invasion in squamous carcinoma cells. J. Investig. Dermatol. 2001, 117, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, D.M.; But, M.; Regezi, J.; Schmidt, B.L.; Atakilit, A.; Dang, D.; Ellis, D.; Jordan, R.; Li, X. Expression of integrin beta 6 enhances invasive behavior in oral squamous cell carcinoma. Matrix Biol. 2002, 21, 297–307. [Google Scholar] [CrossRef]
- Ahmed, N.; Niu, J.; Dorahy, D.J.; Gu, X.; Andrews, S.; Meldrum, C.J.; Scott, R.J.; Baker, M.S.; Macreadie, I.G.; Agrez, M.V. Direct integrin alphavbeta6-ERK binding: Implications for tumour growth. Oncogene 2002, 21, 1370–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomin, V.H.; Mulloy, B. Glycosaminoglycans and proteoglycans. Pharmaceuticals 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Bartlett, A.H.; Hayashida, K.; Park, P.W. Molecular and cellular mechanisms of syndecans in tissue injury and inflammation. Mol. Cells 2007, 24, 153–166. [Google Scholar]
- Deepa, S.S.; Yamada, S.; Zako, M.; Goldberger, O.; Sugahara, K. Chondroitin sulfate chains on syndecan-1 and syndecan-4 from normal murine mammary gland epithelial cells are structurally and functionally distinct and cooperate with heparan sulfate chains to bind growth factors. A novel function to control binding of midkine, pleiotrophin, and basic fibroblast growth factor. J. Biol. Chem. 2004, 279, 37368–37376. [Google Scholar] [CrossRef] [Green Version]
- Rapraeger, A.C. Molecular interactions of syndecans during development. Semin. Cell Dev. Biol. 2001, 12, 107–116. [Google Scholar] [CrossRef]
- Kasza, I.; Suh, Y.; Wollny, D.; Clark, R.J.; Roopra, A.; Colman, R.J.; MacDougald, O.A.; Shedd, T.A.; Nelson, D.W.; Yen, M.-I.; et al. Syndecan-1 is required to maintain intradermal fat and prevent cold stress. PLoS Genet. 2014, 10, e1004514. [Google Scholar] [CrossRef] [Green Version]
- Dews, I.C.; Mackenzie, K.R. Transmembrane domains of the syndecan family of growth factor coreceptors display a hierarchy of homotypic and heterotypic interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20782–20787. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chung, H.; Jung, H.; Couchman, J.R.; Oh, E.-S. Syndecans as cell surface receptors: Unique structure equates with functional diversity. Matrix Biol. 2011, 30, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lee, E.; Kwon, S.; Park, H.; Yi, J.Y.; Kim, S.; Han, I.-O.; Yun, Y.; Oh, E.-S. Transmembrane domain-induced oligomerization is crucial for the functions of syndecan-2 and syndecan-4. J. Biol. Chem. 2005, 280, 42573–42579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, D.J.; Bendt, K.M.; Stahl, R.C. The cytoplasmic domain of syndecan-1 is required for cytoskeleton association but not detergent insolubility. Identification of essential cytoplasmic domain residues. J. Biol. Chem. 1996, 271, 15253–15260. [Google Scholar] [CrossRef] [Green Version]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, V.L.; Rapraeger, A.C. Tyrosine phosphorylation of syndecan-1 and -4 cytoplasmic domains in adherent B82 fibroblasts. J. Biol. Chem. 1998, 273, 35291–35298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhuri, P.; Colles, S.M.; Fox, P.L.; Graham, L.M. Protein kinase Cδ—Dependent phosphorylation of syndecan-4 regulates cell migration. Circ. Res. 2005, 97, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Woods, A. Syndecans: Transmembrane modulators of adhesion and matrix assembly. J. Clin. Investig. 2001, 107, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Couchman, J.R. Syndecans: Proteoglycan regulators of cell-surface microdomains? Nat. Rev. Mol. Cell Biol. 2003, 4, 926–937. [Google Scholar] [CrossRef]
- Sulka, B.; Lortat-Jacob, H.; Terreux, R.; Letourneur, F.; Rousselle, P. Tyrosine dephosphorylation of the syndecan-1 PDZ binding domain regulates syntenin-1 recruitment. J. Biol. Chem. 2009, 284, 10659–10671. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Yun, J.-H.; Yoo, J.; Lee, I.; Kim, H.; Son, H.-N.; Kim, I.-S.; Yoon, H.S.; Zimmermann, P.; Couchman, J.R.; et al. New structural insight of C-terminal region of syntenin-1, enhancing the molecular dimerization and inhibitory function related on syndecan-4 signaling. Sci. Rep. 2016, 6, 36818. [Google Scholar] [CrossRef]
- Friand, V.; David, G.; Zimmermann, P. Syntenin and syndecan in the biogenesis of exosomes. Biol. Cell 2015, 107, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Zhang, Z.; Degeest, G.; Mortier, E.; Leenaerts, I.; Coomans, C.; Schulz, J.; N’Kuli, F.; Courtoy, P.J.; David, G. Syndecan recyling is controlled by syntenin-PIP2 interaction and Arf6. Dev. Cell 2005, 9, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapraeger, A.; Jalkanen, M.; Endo, E.; Koda, J.; Bernfield, M. The cell surface proteoglycan from mouse mammary epithelial cells bears chondroitin sulfate and heparan sulfate glycosaminoglycans. J. Biol. Chem. 1985, 260, 11046–11052. [Google Scholar] [PubMed]
- Zhang, Y.; McKown, R.L.; Raab, R.W.; Rapraeger, A.C.; Laurie, G.W. Focus on molecules: Syndecan-1. Exp. Eye Res. 2011, 93, 329–330. [Google Scholar] [CrossRef] [Green Version]
- Shworak, N.W.; Shirakawa, M.; Mulligan, R.C.; Rosenberg, R.D. Characterization of ryudocan glycosaminoglycan acceptor sites. J. Biol. Chem. 1994, 269, 21204–21214. [Google Scholar]
- Gallo, R.L. Proteoglycans and cutaneous vascular defense and repair. J. Investig. Dermatol. Symp. Proc. 2000, 5, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Fears, C.Y.; Gladson, C.L.; Woods, A. Syndecan-2 is expressed in the microvasculature of gliomas and regulates angiogenic processes in microvascular endothelial cells. J. Biol. Chem. 2006, 281, 14533–14536. [Google Scholar] [CrossRef] [Green Version]
- Manon-Jensen, T.; Multhaupt, H.A.B.; Couchman, J.R. Mapping of matrix metalloproteinase cleavage sites on syndecan-1 and syndecan-4 ectodomains. FEBS J. 2013, 280, 2320–2331. [Google Scholar] [CrossRef]
- Endo, K.; Takino, T.; Miyamori, H.; Kinsen, H.; Yoshizaki, T.; Furukawa, M.; Sato, H. Cleavage of syndecan-1 by membrane type matrix metalloproteinase-1 stimulates cell migration. J. Biol. Chem. 2003, 278, 40764–40770. [Google Scholar] [CrossRef] [Green Version]
- Bernfield, M.; Götte, M.; Park, P.W.; Reizes, O.; Fitzgerald, M.L.; Lincecum, J.; Zako, M. Functions of cell surface heparan sulfate proteoglycans. Annu. Rev. Biochem. 1999, 68, 729–777. [Google Scholar] [CrossRef]
- Hayashida, K.; Stahl, P.D.; Park, P.W. Syndecan-1 ectodomain shedding is regulated by the small GTPase Rab5. J. Biol. Chem. 2008, 283, 35435–35444. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, M.L.; Wang, Z.; Park, P.W.; Murphy, G.; Bernfield, M. Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a TIMP-3-sensitive metalloproteinase. J. Cell Biol. 2000, 148, 811–824. [Google Scholar] [CrossRef] [PubMed]
- Elenius, V.; Gotte, M.; Reizes, O.; Elenius, K.; Bernfield, M. Inhibition by the soluble syndecan-1 ectodomains delays wound repair in mice overexpressing syndecan-1. J. Biol. Chem. 2004, 279, 41928–41935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, S.V.; Fitzgerald, M.L.; Bernfield, M. Regulated shedding of syndecan-1 and -4 ectodomains by thrombin and growth factor receptor activation. J. Biol. Chem. 1997, 272, 14713–14720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, P.W.; Pier, G.B.; Preston, M.J.; Goldberger, O.; Fitzgerald, M.L.; Bernfield, M. Syndecan-1 shedding is enhanced by LasA, a secreted virulence factor of Pseudomonas aeruginosa. J. Biol. Chem. 2000, 275, 3057–3064. [Google Scholar] [CrossRef] [Green Version]
- Park, P.W.; Pier, G.B.; Hinkes, M.T.; Bernfield, M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature 2001, 411, 98–102. [Google Scholar] [CrossRef]
- Hadigal, S.; Koganti, R.; Yadavalli, T.; Agelidis, A.; Suryawanshi, R.; Shukla, D. Heparanase-regulated syndecan-1 shedding facilitates herpes simplex virus 1 egress. J. Virol. 2020. [Google Scholar] [CrossRef]
- Yang, Y.; Yaccoby, S.; Liu, W.; Langford, J.K.; Pumphrey, C.Y.; Theus, A.; Epstein, J.; Sanderson, R.D. Soluble syndecan-1 promotes growth of myeloma tumors in vivo. Blood 2002, 100, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Mahtouk, K.; Hose, D.; Raynaud, P.; Hundemer, M.; Jourdan, M.; Jourdan, E.; Pantesco, V.; Baudard, M.; de Vos, J.; Larroque, M.; et al. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood 2007, 109, 4914–4923. [Google Scholar] [CrossRef] [Green Version]
- Ramani, V.C.; Pruett, P.S.; Thompson, C.A.; DeLucas, L.D.; Sanderson, R.D. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J. Biol. Chem. 2012, 287, 9952–9961. [Google Scholar] [CrossRef] [Green Version]
- Gallo, R.; Kim, C.; Kokenyesi, R.; Scott Adzick, N.; Bernfield, M. Syndecans-1 and -4 are induced during wound repair of neonatal but not fetal skin. J. Investig. Dermatol. 1996, 107, 676–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksala, O.; Salo, T.; Tammi, R.; Hakkinen, L.; Jalkanen, M.; Inki, P.; Larjava, H. Expression of proteoglycans and hyaluronan during wound healing. J. Histochem. Cytochem. 1995, 43, 125–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousselle, P.; Beck, K. Laminin 332 processing impacts cellular behavior. Cell Adh. Migr. 2013, 7, 122–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepp, M.A.; Pal-Ghosh, S.; Tadvalkar, G.; Rajjoub, L.; Jurjus, R.A.; Gerdes, M.; Ryscavage, A.; Cataisson, C.; Shukla, A.; Yuspa, S.H. Loss of syndecan-1 is associated with malignant conversion in skin carcinogenesis. Mol. Carcinog. 2010, 49, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, H.M.; Edwards, S.N.; Jalkanen, M. Analysis of transport and targeting of syndecan-1: Effect of cytoplasmic tail deletions. Mol. Biol. Cell 1994, 5, 1325–1339. [Google Scholar] [CrossRef] [Green Version]
- Haubeck, H.-D. Syndecane. In Lexikon der Medizinischen Laboratoriumsdiagnostik; Gressner, A.M., Arndt, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2019; pp. 2246–2247. ISBN 978-3-662-48986-4. [Google Scholar]
- Jalkanen, M.; Rapraeger, A.; Bernfield, M. Mouse mammary epithelial cells produce basement membrane and cell surface heparan sulfate proteoglycans containing distinct core proteins. J. Cell Biol. 1988, 106, 953–962. [Google Scholar] [CrossRef]
- Stepp, M.A.; Gibson, H.E.; Gala, P.H.; Iglesia, D.D.S.; Pajoohesh-Ganji, A.; Pal-Ghosh, S.; Brown, M.; Aquino, C.; Schwartz, A.M.; Goldberger, O.; et al. Defects in keratinocyte activation during wound healing in the syndecan-1-deficient mouse. J. Cell Sci. 2002, 115, 4517–4531. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-h.; Park, H.; Chung, H.; Choi, S.; Kim, Y.; Yoo, H.; Kim, T.-Y.; Hann, H.-J.; Seong, I.; Kim, J.; et al. Syndecan-2 regulates the migratory potential of melanoma cells. J. Biol. Chem. 2009, 284, 27167–27175. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, X.D.; Mlakar, L.R.; Yamaguchi, Y.; Su, Y.; Larregina, A.T.; Pilewski, J.M.; Feghali-Bostwick, C.A. Syndecan-2 is a novel target of insulin-like growth factor binding protein-3 and is over-expressed in fibrosis. PLoS ONE 2012, 7, e43049. [Google Scholar] [CrossRef] [Green Version]
- Bagabir, R.A.; Syed, F.; Shenjere, P.; Paus, R.; Bayat, A. Identification of a potential molecular diagnostic biomarker in keloid disease: Syndecan-1 (CD138) is overexpressed in keloid scar tissue. J. Investig. Dermatol. 2016, 136, 2319–2323. [Google Scholar] [CrossRef] [Green Version]
- Bayer-Garner, I.; Dilday, B.; Sanderson, R.; Smoller, B. Acantholysis and spongiosis are associated with loss of syndecan-1 expression. J. Cutan. Pathol. 2001, 28, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Søgaard, P.; Multhaupt, H.A.B.; Pataki, C.; Okina, E.; Xian, X.; Pedersen, M.E.; Stevens, T.; Griesbeck, O.; Park, P.W.; et al. Transmembrane proteoglycans control stretch-activated channels to set cytosolic calcium levels. J. Cell Biol. 2015, 210, 1199–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, S. Syndecans in inflammation at a glance. Front. Immunol. 2020, 11, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, A.K.; Sadasivam, M.; Archer, N.K.; Miller, R.J.; Dillen, C.A.; Ravipati, A.; Park, P.W.; Chakravarti, S.; Miller, L.S.; Hamad, A.R.A. Syndecan-1 regulates psoriasiform dermatitis by controlling homeostasis of IL-17-producing γδ T cells. J. Immunol. 2018, 201, 1651–1661. [Google Scholar] [CrossRef]
- Elenius, K.; Vainio, S.; Laato, M.; Salmivirta, M.; Thesleff, I.; Jalkanen, M. Induced expression of syndecan in healing wounds. J. Cell Biol. 1991, 114, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, K.; Johnston, D.R.; Goldberger, O.; Park, P.W. Syndecan-1 expression in epithelial cells is induced by transforming growth factor beta through a PKA-dependent pathway. J. Biol. Chem. 2006, 281, 24365–24374. [Google Scholar] [CrossRef] [Green Version]
- Stepp, M.A.; Liu, Y.; Pal-Ghosh, S.; Jurjus, R.A.; Tadvalkar, G.; Sekaran, A.; Losicco, K.; Jiang, L.; Larsen, M.; Li, L.; et al. Reduced migration, altered matrix and enhanced TGFbeta1 signaling are signatures of mouse keratinocytes lacking Sdc1. J. Cell Sci. 2007, 120, 2851–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echtermeyer, F.; Streit, M.; Wilcox-Adelman, S.; Saoncella, S.; Denhez, F.; Detmar, M.; Goetinck, P. Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J. Clin. Investig. 2001, 107, R9–R14. [Google Scholar] [CrossRef] [Green Version]
- Carulli, S.; Beck, K.; Dayan, G.; Boulesteix, S.; Lortat-Jacob, H.; Rousselle, P. Cell surface proteoglycans syndecan-1 and -4 bind overlapping but distinct sites in laminin α3 LG45 protein domain. J. Biol. Chem. 2012, 287, 12204–12216. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, O.; Bachy, S.; Odenthal, U.; Bernaud, J.; Rigal, D.; Lortat-Jacob, H.; Smyth, N.; Rousselle, P. Normal human keratinocytes bind to the alpha3LG4/5 domain of unprocessed laminin-5 through the receptor syndecan-1. J. Biol. Chem. 2003, 278, 44168–44177. [Google Scholar] [CrossRef] [Green Version]
- Tsubota, Y.; Yasuda, C.; Kariya, Y.; Ogawa, T.; Hirosaki, T.; Mizushima, H.; Miyazaki, K. Regulation of biological activity and matrix assembly of laminin-5 by COOH-terminal, LG4-5 domain of alpha3 chain. J. Biol. Chem. 2005, 280, 14370–14377. [Google Scholar] [CrossRef] [Green Version]
- Pulido, D.; Hussain, S.-A.; Hohenester, E. Crystal structure of the heterotrimeric integrin-binding region of laminin-111. Structure 2017, 25, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachy, S.; Letourneur, F.; Rousselle, P. Syndecan-1 interaction with the LG4/5 domain in laminin-332 is essential for keratinocyte migration. J. Cell. Physiol. 2008, 214, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.E.; Shen, X.-N.; Das, S.K.; Emdad, L.; Sarkar, D.; Fisher, P.B. MDA-9/Syntenin (SDCBP) modulates small GTPases RhoA and Cdc42 via transforming growth factor β1 to enhance epithelial-mesenchymal transition in breast cancer. Oncotarget 2016, 7, 80175–80189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Leavitt, L.; Ramaswamy, R.; Rapraeger, A.C. Interaction of syndecan and alpha6beta4 integrin cytoplasmic domains: Regulation of ErbB2-mediated integrin activation. J. Biol. Chem. 2010, 285, 13569–13579. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jin, H.; Beauvais, D.M.; Rapraeger, A.C. Cytoplasmic domain interactions of syndecan-1 and syndecan-4 with α6β4 integrin mediate human epidermal growth factor receptor (HER1 and HER2)-dependent motility and survival. J. Biol. Chem. 2014, 289, 30318–30332. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jin, H.; Rapraeger, A.C. Syndecan-1 and syndecan-4 capture epidermal growth factor receptor family members and the alpha3beta1 integrin via binding sites in their ectodomains: Novel synstatins prevent kinase capture and inhibit alpha6beta4-integrin-dependent epithelial cell motility. J. Biol. Chem. 2015, 290, 26103–26113. [Google Scholar] [CrossRef] [Green Version]
- Koda, J.E.; Rapraeger, A.; Bernfield, M. Heparan sulfate proteoglycans from mouse mammary epithelial cells. Cell surface proteoglycan as a receptor for interstitial collagens. J. Biol. Chem. 1985, 260, 8157–8162. [Google Scholar]
- Sun, X.; Mosher, D.F.; Rapraeger, A. Heparan sulfate-mediated binding of epithelial cell surface proteoglycan to thrombospondin. J. Biol. Chem. 1989, 264, 2885–2889. [Google Scholar]
- Huang, W.; Chiquet-Ehrismann, R.; Moyano, J.V.; Garcia-Pardo, A.; Orend, G. Interference of tenascin-C with syndecan-4 binding to fibronectin blocks cell adhesion and stimulates tumor cell proliferation. Cancer Res. 2001, 61, 8586–8594. [Google Scholar]
- Greene, D.K.; Tumova, S.; Couchman, J.R.; Woods, A. Syndecan-4 associates with alpha-actinin. J. Biol. Chem. 2003, 278, 7617–7623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Longley, R.L.; Tumova, S.; Couchman, J.R. Syndecan-4 binding to the high affinity heparin-binding domain of fibronectin drives focal adhesion formation in fibroblasts. Arch. Biochem. Biophys. 2000, 374, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Wilcox-Adelman, S.A.; Denhez, F.; Goetinck, P.F. Syndecan-4 modulates focal adhesion kinase phosphorylation. J. Biol. Chem. 2002, 277, 32970–32977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saoncella, S.; Echtermeyer, F.; Denhez, F.; Nowlen, J.K.; Mosher, D.F.; Robinson, S.D.; Hynes, R.O.; Goetinck, P.F. Syndecan-4 signals cooperatively with integrins in a Rhodependent manner in the assembly of focal adhesions and actin stress fibers. Proc. Natl. Acad. Sci. USA 1999, 96, 2805. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, K.; Kadomatsu, K.; Kojima, T.; Muramatsu, H.; Tsuzuki, S.; Nakamura, E.; Kusugami, K.; Saito, H.; Muramatsu, T. Syndecan-4 deficiency impairs focal adhesion formation only under restricted conditions. J. Biol. Chem. 2000, 275, 5249–5252. [Google Scholar] [CrossRef] [Green Version]
- Echtermeyer, F.; Baciu, P.C.; Saoncella, S.; Ge, Y.; Goetinck, P.F. Syndecan-4 core protein is sufficient for the assembly of focal adhesions and actin stress fibers. J. Cell Sci. 1999, 112 Pt 20, 3433–3441. [Google Scholar]
- Mostafavi-Pour, Z.; Askari, J.A.; Parkinson, S.J.; Parker, P.J.; Ng, T.T.C.; Humphries, M.J. Integrin-specific signaling pathways controlling focal adhesion formation and cell migration. J. Cell Biol. 2003, 161, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Baciu, P.C.; Goetinck, P.F. Protein kinase C regulates the recruitment of syndecan-4 into focal contacts. Mol. Biol. Cell 1995, 6, 1503–1513. [Google Scholar] [CrossRef] [Green Version]
- Midwood, K.S.; Valenick, L.V.; Hsia, H.C.; Schwarzbauer, J.E. Coregulation of fibronectin signaling and matrix contraction by tenascin-C and syndecan-4. Mol. Biol. Cell 2004, 15, 5670–5677. [Google Scholar] [CrossRef] [Green Version]
- Chronopoulos, A.; Thorpe, S.D.; Cortes, E.; Lachowski, D.; Rice, A.J.; Mykuliak, V.V.; Róg, T.; Lee, D.A.; Hytönen, V.P.; del Río Hernández, A.E. Syndecan-4 tunes cell mechanics by activating the kindlin-integrin-RhoA pathway. Nat. Mater. 2020, 19, 669–678. [Google Scholar] [CrossRef]
- Huang, C.-P.; Cheng, C.-M.; Su, H.-L.; Lin, Y.-W. Syndecan-4 promotes epithelial tumor cells spreading and regulates the turnover of PKCα activity under mechanical stimulation on the elastomeric substrates. Cell Physiol. Biochem. 2015, 36, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Bellin, R.M.; Kubicek, J.D.; Frigault, M.J.; Kamien, A.J.; Steward, R.L.; Barnes, H.M.; DiGiacomo, M.B.; Duncan, L.J.; Edgerly, C.K.; Morse, E.M.; et al. Defining the role of syndecan-4 in mechanotransduction using surface-modification approaches. Proc. Natl. Acad. Sci. USA 2009, 106, 22102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Chaikof, E.L. Mechanical stress regulates syndecan-4 expression and redistribution in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Nardone, G.; La Oliver-De Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skladal, P.; Pesl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Saunders, S.; Nguyen, H.; Bernfield, M. Loss of cell surface syndecan-1 causes epithelia to transform into anchorage-independent mesenchyme-like cells. Mol. Biol. Cell 1995, 6, 559–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szatmári, T.; Ötvös, R.; Hjerpe, A.; Dobra, K. Syndecan-1 in cancer: Implications for cell signaling, differentiation, and prognostication. Dis. Markers 2015. [Google Scholar] [CrossRef] [Green Version]
- Kivisaari, A.K.; Kallajoki, M.; Mirtti, T.; McGrath, J.A.; Bauer, J.W.; Weber, F.; Konigova, R.; Sawamura, D.; Sato-Matsumura, K.C.; Shimizu, H.; et al. Transformation-specific matrix metalloproteinases (MMP)-7 and MMP-13 are expressed by tumour cells in epidermolysis bullosa-associated squamous cell carcinomas. Br. J. Dermatol. 2008, 158, 778–785. [Google Scholar] [CrossRef]
- Chen, P.; Abacherli, L.E.; Nadler, S.T.; Wang, Y.; Li, Q.; Parks, W.C. MMP7 shedding of syndecan-1 facilitates re-epithelialization by affecting alpha(2)beta(1) integrin activation. PLoS ONE 2009, 4, e6565. [Google Scholar] [CrossRef] [Green Version]
- Beauvais, D.M.; Burbach, B.J.; Rapraeger, A.C. The syndecan-1 ectodomain regulates alphavbeta3 integrin activity in human mammary carcinoma cells. J. Cell Biol. 2004, 167, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Orian-Rousseau, V. CD44, a therapeutic target for metastasising tumours. Eur. J. Cancer 2010, 46, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Govindaraju, P.; Todd, L.; Shetye, S.; Monslow, J.; Pure, E. CD44-dependent inflammation, fibrogenesis, and collagenolysis regulates extracellular matrix remodeling and tensile strength during cutaneous wound healing. Matrix Biol. 2019, 75–76, 314–330. [Google Scholar] [CrossRef] [PubMed]
- Basakran, N.S. CD44 as a potential diagnostic tumor marker. Saudi. Med. J. 2015, 36, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Anderegg, U.; Simon, J.C.; Averbeck, M. More than just a filler—The role of hyaluronan for skin homeostasis. Exp. Dermatol. 2014, 23, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, S.; Jalkanen, M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J. Cell Biol. 1992, 116, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Kawashima, H.; Tanaka, T.; Hirose, M.; Toyama-Sorimachi, N.; Matsuzawa, Y.; Miyasaka, M. CD44 binds a chondroitin sulfate proteoglycan, aggrecan. Int. Immunol. 2001, 13, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama-Sorimachi, N.; Sorimachi, H.; Tobita, Y.; Kitamura, F.; Yagita, H.; Suzuki, K.; Miyasaka, M. A novel ligand for CD44 is serglycin, a hematopoietic cell lineage-specific proteoglycan. Possible involvement in lymphoid cell adherence and activation. J. Biol. Chem. 1995, 270, 7437–7444. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, H.; Hirose, M.; Hirose, J.; Nagakubo, D.; Plaas, A.H.; Miyasaka, M. Binding of a large chondroitin sulfate/dermatan sulfate proteoglycan, versican, to L-selectin, P-selectin, and CD44. J. Biol. Chem. 2000, 275, 35448–35456. [Google Scholar] [CrossRef] [Green Version]
- Bennett, K.L.; Jackson, D.G.; Simon, J.C.; Tanczos, E.; Peach, R.; Modrell, B.; Stamenkovic, I.; Plowman, G.; Aruffo, A. CD44 isoforms containing exon V3 are responsible for the presentation of heparin-binding growth factor. J. Cell Biol. 1995, 128, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Ishii, S.; Ford, R.; Thomas, P.; Nachman, A.; Steele, G., Jr.; Jessup, J.M. CD44 participates in the adhesion of human colorectal carcinoma cells to laminin and type IV collagen. Surg. Oncol. 1993, 2, 255–264. [Google Scholar] [CrossRef]
- Ehnis, T.; Dieterich, W.; Bauer, M.; Lampe, B.; Schuppan, D. A chondroitin/dermatan sulfate form of CD44 is a receptor for collagen XIV (undulin). Exp. Cell Res. 1996, 229, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Porsch, H.; Mehic, M.; Olofsson, B.; Heldin, P.; Heldin, C.-H. Platelet-derived growth factor beta-receptor, transforming growth factor beta type I receptor, and CD44 protein modulate each other’s signaling and stability. J. Biol. Chem. 2014, 289, 19747–19757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Heldin, C.-H.; Heldin, P. Inhibition of platelet-derived growth factor-BB-induced receptor activation and fibroblast migration by hyaluronan activation of CD44. J. Biol. Chem. 2006, 281, 26512–26519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Lee, Y.-S.; Choe, J.; Lee, H.; Kim, Y.-M.; Jeoung, D. CD44-epidermal growth factor receptor interaction mediates hyaluronic acid-promoted cell motility by activating protein kinase C signaling involving Akt, Rac1, Phox, reactive oxygen species, focal adhesion kinase, and MMP-2. J. Biol. Chem. 2008, 283, 22513–22528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghatak, S.; Misra, S.; Toole, B.P. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 2005, 280, 8875–8883. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.; Zhu, H.; Zhou, B.; Diedrich, F.; Singleton, P.A.; Hung, M.C. Hyaluronan promotes CD44v3-Vav2 interaction with Grb2-p185(HER2) and induces Rac1 and Ras signaling during ovarian tumor cell migration and growth. J. Biol. Chem. 2001, 276, 48679–48692. [Google Scholar] [CrossRef] [Green Version]
- Nagano, O.; Murakami, D.; Hartmann, D.; de Strooper, B.; Saftig, P.; Iwatsubo, T.; Nakajima, M.; Shinohara, M.; Saya, H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca(2+) influx and PKC activation. J. Cell Biol. 2004, 165, 893–902. [Google Scholar] [CrossRef]
- Suenaga, N.; Mori, H.; Itoh, Y.; Seiki, M. CD44 binding through the hemopexin-like domain is critical for its shedding by membrane-type 1 matrix metalloproteinase. Oncogene 2005, 24, 859–868. [Google Scholar] [CrossRef] [Green Version]
- Kajita, M.; Itoh, Y.; Chiba, T.; Mori, H.; Okada, A.; Kinoh, H.; Seiki, M. Membrane-type 1 matrix metalloproteinase cleaves CD44 and promotes cell migration. J. Cell Biol. 2001, 153, 893–904. [Google Scholar] [CrossRef]
- Ahrens, T.; Sleeman, J.P.; Schempp, C.M.; Howells, N.; Hofmann, M.; Ponta, H.; Herrlich, P.; Simon, J.C. Soluble CD44 inhibits melanoma tumor growth by blocking cell surface CD44 binding to hyaluronic acid. Oncogene 2001, 20, 3399–3408. [Google Scholar] [CrossRef] [Green Version]
- Murakami, D.; Okamoto, I.; Nagano, O.; Kawano, Y.; Tomita, T.; Iwatsubo, T.; de Strooper, B.; Yumoto, E.; Saya, H. Presenilin-dependent gamma-secretase activity mediates the intramembranous cleavage of CD44. Oncogene 2003, 22, 1511–1516. [Google Scholar] [CrossRef] [Green Version]
- Lammich, S.; Okochi, M.; Takeda, M.; Kaether, C.; Capell, A.; Zimmer, A.-K.; Edbauer, D.; Walter, J.; Steiner, H.; Haass, C. Presenilin-dependent intramembrane proteolysis of CD44 leads to the liberation of its intracellular domain and the secretion of an Aβ-like peptide. J. Biol. Chem. 2002, 277, 44754–44759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Mack, J.A.; Maytin, E.V. CD44 inhibits alpha-SMA gene expression via a novel G-actin/MRTF-mediated pathway that intersects with TGFbetaR/p38MAPK signaling in murine skin fibroblasts. J. Biol. Chem. 2019, 294, 12779–12794. [Google Scholar] [CrossRef]
- Hernández, D.; Miquel-Serra, L.; Docampo, M.-J.; Marco-Ramell, A.; Cabrera, J.; Fabra, A.; Bassols, A. V3 versican isoform alters the behavior of human melanoma cells by interfering with CD44/ErbB-dependent signaling. J. Biol. Chem. 2011, 286, 1475–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, R.F.; Legg, J.W.; Isacke, C.M. The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signalling events. J. Cell Sci. 2004, 117, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zöller, M. CD44, hyaluronan, the hematopoietic stem cell, and leukemia-initiating cells. Front. Immunol. 2015, 6, 235. [Google Scholar] [CrossRef] [Green Version]
- Dzwonek, J.; Wilczynski, G.M. CD44: Molecular interactions, signaling and functions in the nervous system. Front. Cell Neurosci. 2015. [Google Scholar] [CrossRef] [Green Version]
- Skelton, T.P.; Zeng, C.; Nocks, A.; Stamenkovic, I. Glycosylation provides both stimulatory and inhibitory effects on cell surface and soluble CD44 binding to hyaluronan. J. Cell Biol. 1998, 140, 431–446. [Google Scholar] [CrossRef] [Green Version]
- Hurt-Camejo, E.; Rosengren, B.; Sartipy, P.; Elfsberg, K.; Camejo, G.; Svensson, L. CD44, a cell surface chondroitin sulfate proteoglycan, mediates binding of interferon-gamma and some of its biological effects on human vascular smooth muscle cells. J. Biol. Chem. 1999, 274, 18957–18964. [Google Scholar] [CrossRef] [Green Version]
- Chetty, C.; Vanamala, S.K.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. MMP-9 induces CD44 cleavage and CD44 mediated cell migration in glioblastoma xenograft cells. Cell Signal. 2012, 24, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Stamenkovic, I. Localization of matrix metalloproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes Dev. 1999, 13, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, I.; Kawano, Y.; Murakami, D.; Sasayama, T.; Araki, N.; Miki, T.; Wong, A.J.; Saya, H. Proteolytic release of CD44 intracellular domain and its role in the CD44 signaling pathway. J. Cell Biol. 2001, 155, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Teye, K.; Numata, S.; Ishii, N.; Krol, R.P.; Tsuchisaka, A.; Hamada, T.; Koga, H.; Karashima, T.; Ohata, C.; Tsuruta, D.; et al. Isolation of all CD44 transcripts in human epidermis and regulation of their expression by various agents. PLoS ONE 2016, 11, e0160952. [Google Scholar] [CrossRef]
- Rajarajan, A.; Bloor, B.; Desai, H.; Stokes, A.; Odell, E. Variant CD44 expression by human fibroblasts. Biomarkers 2008, 13, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tammi, M.; Tammi, R. Distribution of hyaluronan and its CD44 receptor in the epithelia of human skin appendages. Histochemistry 1992, 98, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Seelentag, W.K.; Günthert, U.; Saremaslani, P.; Futo, E.; Pfaltz, M.; Heitz, P.U.; Roth, J. CD44 standard and variant isoform expression in normal human skin appendages and epidermis. Histochem. Cell Biol. 1996, 106, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Penneys, N.S. CD44 expression in normal and inflamed skin. J. Cutan. Pathol. 1993, 20, 250–253. [Google Scholar] [CrossRef]
- Man, M.; Elias, P.M.; Man, W.; Wu, Y.; Bourguignon, L.Y.W.; Feingold, K.R.; Man, M.-Q. The role of CD44 in cutaneous inflammation. Exp. Dermatol. 2009, 18, 962–968. [Google Scholar] [CrossRef]
- Lugović-Mihić, L.; Novak-Bilić, G.; Vučić, M.; Japundžić, I.; Bukvić, I. CD44 expression in human skin: High expression in irritant and allergic contact dermatitis and moderate expression in psoriasis lesions in comparison with healthy controls. Contact Dermat. 2020, 82, 297–306. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.W.; Ramez, M.; Gilad, E.; Singleton, P.A.; Man, M.-Q.; Crumrine, D.A.; Elias, P.M.; Feingold, K.R. Hyaluronan-CD44 interaction stimulates keratinocyte differentiation, lamellar body formation/secretion, and permeability barrier homeostasis. J. Investig. Dermatol. 2006, 126, 1356–1365. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, N.; Haftek, M.; Niessen, C.M.; Behne, M.J.; Furuse, M.; Moll, I.; Brandner, J.M. CD44 regulates tight-junction assembly and barrier function. J. Investig. Dermatol. 2011, 131, 932–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shatirishvili, M.; Burk, A.S.; Franz, C.M.; Pace, G.; Kastilan, T.; Breuhahn, K.; Hinterseer, E.; Dierich, A.; Bakiri, L.; Wagner, E.F.; et al. Epidermal-specific deletion of CD44 reveals a function in keratinocytes in response to mechanical stress. Cell Death Dis. 2016, 7, e2461. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.M.; Wells, A.; Thomas, D.; Stephens, P.; Steadman, R.; Phillips, A. Aging fibroblasts resist phenotypic maturation because of impaired hyaluronan-dependent CD44/epidermal growth factor receptor signaling. Am. J. Pathol. 2010, 176, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, D.; Liang, J.; Meltzer, E.B.; Gray, A.; Miura, R.; Wogensen, L.; Yamaguchi, Y.; Noble, P.W. Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 2011, 208, 1459–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansorge, H.L.; Beredjiklian, P.K.; Soslowsky, L.J. CD44 deficiency improves healing tendon mechanics and increases matrix and cytokine expression in a mouse patellar tendon injury model. J. Orthop. Res. 2009, 27, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, J.; Li, J.; DiPietro, L.; Stepp, M.A.; Sandy, J.D.; Plaas, A. Adamts5 deletion blocks murine dermal repair through CD44-mediated aggrecan accumulation and modulation of transforming growth factor β1 (TGFβ1) signaling. J. Biol. Chem. 2011, 286, 26016–26027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, D.A. Transforming growth factor-beta: A general review. Eur. Cytokine Netw. 1996, 7, 363–374. [Google Scholar]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.-H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Hinck, A.P. Structural studies of the TGF-βs and their receptors – insights into evolution of the TGF-β superfamily. FEBS Lett. 2012, 586, 1860–1870. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.; Derynck, R. Homomeric interactions between type II transforming growth factor-beta receptors. J. Biol. Chem. 1994, 269, 22868–22874. [Google Scholar] [PubMed]
- Gilboa, L.; Wells, R.G.; Lodish, H.F.; Henis, Y.I. Oligomeric structure of type I and type II transforming growth factor beta receptors: Homodimers form in the ER and persist at the plasma membrane. J. Cell Biol. 1998, 140, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Gutman, O.; Knaus, P.; Henis, Y.I. Oligomeric interactions of TGF-beta and BMP receptors. FEBS Lett. 2012, 586, 1885–1896. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Jiang, Y.; Wang, Q.; Ma, X.; Xiao, Z.; Zuo, W.; Fang, X.; Chen, Y.-G. Single-molecule imaging reveals transforming growth factor-beta-induced type II receptor dimerization. Proc. Natl. Acad. Sci. USA 2009, 106, 15679–15683. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Laiho, M.; Weis, F.M.; Boyd, F.T.; Ignotz, R.A.; Massague, J. Responsiveness to transforming growth factor-beta (TGF-beta) restored by genetic complementation between cells defective in TGF-beta receptors I and II. J. Biol. Chem. 1991, 266, 9108–9112. [Google Scholar]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [Green Version]
- Massagué, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-beta receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Huse, M.; Chen, Y.G.; Massague, J.; Kuriyan, J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 1999, 96, 425–436. [Google Scholar] [CrossRef] [Green Version]
- Huse, M.; Muir, T.W.; Xu, L.; Chen, Y.G.; Kuriyan, J.; Massague, J. The TGF beta receptor activation process: An inhibitor- to substrate-binding switch. Mol. Cell 2001, 8, 671–682. [Google Scholar] [CrossRef]
- Mulder, K.M.; Morris, S.L. Activation of p21ras by transforming growth factor beta in epithelial cells. J. Biol. Chem. 1992, 267, 5029–5031. [Google Scholar] [PubMed]
- Yan, Z.; Winawer, S.; Friedman, E. Two different signal transduction pathways can be activated by transforming growth factor beta 1 in epithelial cells. J. Biol. Chem. 1994, 269, 13231–13237. [Google Scholar] [PubMed]
- Mucsi, I.; Skorecki, K.L.; Goldberg, H.J. Extracellular signal-regulated kinase and the small GTP-binding protein, Rac, contribute to the effects of transforming growth factor-beta1 on gene expression. J. Biol. Chem. 1996, 271, 16567–16572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanafusa, H.; Ninomiya-Tsuji, J.; Masuyama, N.; Nishita, M.; Fujisawa, J.; Shibuya, H.; Matsumoto, K.; Nishida, E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J. Biol. Chem. 1999, 274, 27161–27167. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [Green Version]
- Frey, R.S.; Mulder, K.M. Involvement of extracellular signal-regulated kinase 2 and stress-activated protein kinase/Jun N-terminal kinase activation by transforming growth factor beta in the negative growth control of breast cancer cells. Cancer Res. 1997, 57, 628–633. [Google Scholar]
- Hocevar, B.A.; Brown, T.L.; Howe, P.H. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 1999, 18, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Dore, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [Green Version]
- Freudlsperger, C.; Bian, Y.; Contag Wise, S.; Burnett, J.; Coupar, J.; Yang, X.; Chen, Z.; van Waes, C. TGF-beta and NF-kappaB signal pathway cross-talk is mediated through TAK1 and SMAD7 in a subset of head and neck cancers. Oncogene 2013, 32, 1549–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingery, A.; Bradley, E.; Pederson, L.; Ruan, M.; Horwood, N.; Oursler, M. TGF-beta coordinately activates TAK1/MEK/AKT/NFkB and SMAD pathways to promote osteoclast survival. Exp. Cell Res. 2008, 314, 2725–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Edlund, S.; Landstrom, M.; Heldin, C.-H.; Aspenstrom, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkes, M.C.; Murphy, S.J.; Garamszegi, N.; Leof, E.B. Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol. Cell. Biol 2003, 23, 8878–8889. [Google Scholar] [CrossRef] [Green Version]
- Mythreye, K.; Blobe, G.C. The type III TGF-beta receptor regulates epithelial and cancer cell migration through beta-arrestin2-mediated activation of Cdc42. Proc. Natl. Acad. Sci. USA 2009, 106, 8221–8226. [Google Scholar] [CrossRef] [Green Version]
- You, H.J.; How, T.; Blobe, G.C. The type III transforming growth factor-beta receptor negatively regulates nuclear factor kappa B signaling through its interaction with beta-arrestin2. Carcinogenesis 2009, 30, 1281–1287. [Google Scholar] [CrossRef] [Green Version]
- López-Casillas, F.; Wrana, J.L.; Massagué, J. Betaglycan presents ligand to the TGF beta signaling receptor. Cell 1993, 73, 1435–1444. [Google Scholar] [CrossRef]
- Moustakas, A.; Lin, H.Y.; Henis, Y.I.; Plamondon, J.; O’Connor-McCourt, M.D.; Lodish, H.F. The transforming growth factor beta receptors types I, II, and III form hetero-oligomeric complexes in the presence of ligand. J. Biol. Chem. 1993, 268, 22215–22218. [Google Scholar]
- Varadaraj, A.; Jenkins, L.M.; Singh, P.; Chanda, A.; Snider, J.; Lee, N.Y.; Amsalem-Zafran, A.R.; Ehrlich, M.; Henis, Y.I.; Mythreye, K. TGF-β triggers rapid fibrillogenesis via a novel TβRII-dependent fibronectin-trafficking mechanism. Mol. Biol. Cell 2017, 28, 1195–1207. [Google Scholar] [CrossRef]
- Garamszegi, N.; Garamszegi, S.P.; Samavarchi-Tehrani, P.; Walford, E.; Schneiderbauer, M.M.; Wrana, J.L.; Scully, S.P. Extracellular matrix-induced transforming growth factor-beta receptor signaling dynamics. Oncogene 2010, 29, 2368–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Itin, P.; Rufli, T. In situ analysis of transforming growth factors-beta (TGF-beta 1, TGF-beta 2, TGF-beta 3) and TGF-beta type II receptor expression in basal cell carcinomas. Br. J. Dermatol. 1996, 134, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, H.; Myokai, F.; Arata, J.; Noji, S.; Taniguchi, S. Expression of type II transforming growth factor-beta receptor mRNA in human skin, as revealed by in situ hybridization. J. Dermatol. Sci. 1994, 8, 25–32. [Google Scholar] [CrossRef]
- Kudo, H.; Jinnin, M.; Asano, Y.; Trojanowska, M.; Nakayama, W.; Inoue, K.; Honda, N.; Kajihara, I.; Makino, K.; Fukushima, S.; et al. Decreased interleukin-20 expression in scleroderma skin contributes to cutaneous fibrosis. Arthritis Rheumatol. 2014, 66, 1636–1647. [Google Scholar] [CrossRef] [Green Version]
- Sowden, H.M.; Karoo, R.O.S.; Tobin, D.J. Transforming growth factor-beta receptor II is preferentially expressed in the companion layer of the human anagen hair follicle. Br. J. Dermatol. 2007, 157, 161–164. [Google Scholar] [CrossRef]
- Gold, L.I.; Sung, J.J.; Siebert, J.W.; Longaker, M.T. Type I (RI) and type II (RII) receptors for transforming growth factor-beta isoforms are expressed subsequent to transforming growth factor-beta ligands during excisional wound repair. Am. J. Pathol. 1997, 150, 209–222. [Google Scholar]
- Qin, Z.; Fisher, G.J.; Voorhees, J.J.; Quan, T. Actin cytoskeleton assembly regulates collagen production via TGF-beta type II receptor in human skin fibroblasts. J. Cell Mol. Med. 2018, 22, 4085–4096. [Google Scholar] [CrossRef] [Green Version]
- Fisher, G.J.; Shao, Y.; He, T.; Qin, Z.; Perry, D.; Voorhees, J.J.; Quan, T. Reduction of fibroblast size/mechanical force down-regulates TGF-β type II receptor: Implications for human skin aging. Aging Cell 2016, 15, 67–76. [Google Scholar] [CrossRef]
- Quan, T.; Fisher, G.J. Role of age-associated alterations of the dermal extracellular matrix microenvironment in human skin aging: A mini-review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef] [Green Version]
- March, J.T.; Golshirazi, G.; Cernisova, V.; Carr, H.; Leong, Y.; Lu-Nguyen, N.; Popplewell, L.J. Targeting TGFβ signaling to address fibrosis using antisense oligonucleotides. Biomedicines 2018, 6, 74. [Google Scholar] [CrossRef] [Green Version]
- Pohlers, D.; Brenmoehl, J.; Löffler, I.; Müller, C.K.; Leipner, C.; Schultze-Mosgau, S.; Stallmach, A.; Kinne, R.W.; Wolf, G. TGF-β and fibrosis in different organs—Molecular pathway imprints. Biochim. Biophys. Acta 2009, 1792, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penn, J.W.; Grobbelaar, A.O.; Rolfe, K.J. The role of the TGF-β family in wound healing, burns and scarring: A review. Int. J. Burns Trauma 2012, 2, 18–28. [Google Scholar] [PubMed]
- Pakyari, M.; Farrokhi, A.; Maharlooei, M.K.; Ghahary, A. Critical role of transforming growth factor beta in different phases of wound healing. Adv. Wound Care 2013, 2, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liarte, S.; Bernabé-García, Á.; Nicolás, F.J. Role of TGF—In skin chronic wounds: A keratinocyte perspective. Cells 2020, 9, 306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiritsi, D.; Nystrom, A. The role of TGFbeta in wound healing pathologies. Mech. Ageing Dev. 2018, 172, 51–58. [Google Scholar] [CrossRef]
- Lichtman, M.K.; Otero-Vinas, M.; Falanga, V. Transforming growth factor beta (TGF-beta) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef]
- Liu, J.; Johnson, K.; Li, J.; Piamonte, V.; Steffy, B.M.; Hsieh, M.H.; Ng, N.; Zhang, J.; Walker, J.R.; Ding, S.; et al. Regenerative phenotype in mice with a point mutation in transforming growth factor beta type I receptor (TGFBR1). Proc. Natl. Acad. Sci. USA 2011, 108, 14560. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.J.; Greenhalgh, D.A.; Bickenbach, J.R.; Jiang, A.; Bundman, D.S.; Krieg, T.; Derynck, R.; Roop, D.R. Expression of a dominant-negative type II transforming growth factor beta (TGF-beta) receptor in the epidermis of transgenic mice blocks TGF-beta-mediated growth inhibition. Proc. Natl. Acad. Sci. USA 1997, 94, 2386–2391. [Google Scholar] [CrossRef] [Green Version]
- Amendt, C.; Mann, A.; Schirmacher, P.; Blessing, M. Resistance of keratinocytes to TGFbeta-mediated growth restriction and apoptosis induction accelerates re-epithelialization in skin wounds. J. Cell Sci. 2002, 115, 2189–2198. [Google Scholar] [PubMed]
- Martinez-Ferrer, M.; Afshar-Sherif, A.-R.; Uwamariya, C.; de Crombrugghe, B.; Davidson, J.M.; Bhowmick, N.A. Dermal transforming growth factor-beta responsiveness mediates wound contraction and epithelial closure. Am. J. Pathol. 2010, 176, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, C.P.; Khan, K.; Hoyles, R.K.; Shiwen, X.; Leoni, P.; Chen, Y.; Eastwood, M.; Abraham, D.J. Inducible lineage-specific deletion of TbetaRII in fibroblasts defines a pivotal regulatory role during adult skin wound healing. J. Investig. Dermatol. 2009, 129, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, C.P.; Zheng, B.; Evans, L.A.; Shi-wen, X.; Ong, V.H.; Fisher, I.; Lazaridis, K.; Abraham, D.J.; Black, C.M.; de Crombrugghe, B. Fibroblast-specific expression of a kinase-deficient type II transforming growth factor beta (TGFbeta) receptor leads to paradoxical activation of TGFbeta signaling pathways with fibrosis in transgenic mice. J. Biol. Chem. 2003, 278, 25109–25119. [Google Scholar] [CrossRef] [Green Version]
- Oshima, M.; Oshima, H.; Taketo, M.M. TGF-beta receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 1996, 179, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Sonnylal, S.; Denton, C.P.; Zheng, B.; Keene, D.R.; He, R.; Adams, H.P.; Vanpelt, C.S.; Geng, Y.J.; Deng, J.M.; Behringer, R.R.; et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007, 56, 334–344. [Google Scholar] [CrossRef]
- Dudas, M.; Kim, J.; Li, W.-Y.; Nagy, A.; Larsson, J.; Karlsson, S.; Chai, Y.; Kaartinen, V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Dev. Biol. 2006, 296, 298–314. [Google Scholar] [CrossRef] [Green Version]
- Guerra, L.; Odorisio, T.; Zambruno, G.; Castiglia, D. Stromal microenvironment in type VII collagen-deficient skin: The ground for squamous cell carcinoma development. Matrix Biol. 2017, 63, 1–10. [Google Scholar] [CrossRef]
- Knaup, J.; Gruber, C.; Krammer, B.; Ziegler, V.; Bauer, J.; Verwanger, T. TGFbeta-signaling in squamous cell carcinoma occurring in recessive dystrophic epidermolysis bullosa. Anal. Cell Pathol. (Amst.) 2011, 34, 339–353. [Google Scholar] [CrossRef]
- Cammareri, P.; Rose, A.M.; Vincent, D.F.; Wang, J.; Nagano, A.; Libertini, S.; Ridgway, R.A.; Athineos, D.; Coates, P.J.; McHugh, A.; et al. Inactivation of TGFβ receptors in stem cells drives cutaneous squamous cell carcinoma. Nat. Commun. 2016, 7, 12493. [Google Scholar] [CrossRef]
- Ullrich, A.; Coussens, L.; Hayflick, J.S.; Dull, T.J.; Gray, A.; Tam, A.W.; Lee, J.; Yarden, Y.; Libermann, T.A.; Schlessinger, J. Human epidermal growth factor receptor cDNA sequence and aberrant expression of the amplified gene in A431 epidermoid carcinoma cells. Nature 1984, 309, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Piepkorn, M.; Predd, H.; Underwood, R.; Cook, P. Proliferation-differentiation relationships in the expression of heparin-binding epidermal growth factor-related factors and erbB receptors by normal and psoriatic human keratinocytes. Arch. Dermatol. Res. 2003, 295, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, E.; Mahmoudi, M.; Rahmani, F.; Yousefi, B.; Sarafnejad, A.; Kavosi, H.; Karimizadeh, E.; Jamshidi, A.; Gharibdoost, F. Attenuation of aquaporin-3 and epidermal growth factor receptor expression and activation in systemic sclerosis dermal fibroblasts. J. Cell Physiol. 2019, 234, 12876–12883. [Google Scholar] [CrossRef] [PubMed]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, K.M. A structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [Green Version]
- Normanno, N.; de Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; de Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF receptor ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Sun, L.; Carpenter, G. Epidermal growth factor activation of NF-kappaB is mediated through IkappaBalpha degradation and intracellular free calcium. Oncogene 1998, 16, 2095–2102. [Google Scholar] [CrossRef] [Green Version]
- Andl, C.D.; Mizushima, T.; Oyama, K.; Bowser, M.; Nakagawa, H.; Rustgi, A.K. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1227–G1237. [Google Scholar] [CrossRef]
- Li, Y.; Macdonald-Obermann, J.; Westfall, C.; Piwnica-Worms, D.; Pike, L.J. Quantitation of the effect of ErbB2 on epidermal growth factor receptor binding and dimerization. J. Biol. Chem. 2012, 287, 31116–31125. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.; Miura, Y.; Dimri, M.; Majumder, B.; Dodge, I.L.; Reddi, A.L.; Ghosh, A.; Fernandes, N.; Zhou, P.; Mullane-Robinson, K.; et al. Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis. J. Biol. Chem. 2003, 278, 28950–28960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umebayashi, K.; Stenmark, H.; Yoshimori, T. Ubc4/5 and c-Cbl continue to ubiquitinate EGF receptor after internalization to facilitate polyubiquitination and degradation. Mol. Biol. Cell 2008, 19, 3454–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, F.N.; Drymoussi, Z.; Delaine-Smith, R.; Kao, A.P.; Laly, A.C.; Knight, M.M.; Philpott, M.P.; Connelly, J.T. Tissue stiffening promotes keratinocyte proliferation through activation of epidermal growth factor signaling. J. Cell Sci. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Liu, S.; Yang, B.; Hajal, C.; Changede, R.; Hu, J.; Wolfenson, H.; Hone, J.; Sheetz, M.P. EGFR and HER2 activate rigidity sensing only on rigid matrices. Nat. Mater. 2017, 16, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Asthagiri, A.R. Matrix stiffening sensitizes epithelial cells to EGF and enables the loss of contact inhibition of proliferation. J. Cell Sci. 2011, 124, 1280–1287. [Google Scholar] [CrossRef] [Green Version]
- Mainiero, F.; Pepe, A.; Yeon, M.; Ren, Y.; Giancotti, F.G. The intracellular functions of alpha6beta4 integrin are regulated by EGF. J. Cell Biol. 1996, 134, 241–253. [Google Scholar] [CrossRef]
- Grassian, A.R.; Schafer, Z.T.; Brugge, J.S. ErbB2 stabilizes epidermal growth factor receptor (EGFR) expression via Erk and Sprouty2 in extracellular matrix-detached cells. J. Biol. Chem. 2011, 286, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Yarwood, S.J.; Woodgett, J.R. Extracellular matrix composition determines the transcriptional response to epidermal growth factor receptor activation. Proc. Natl. Acad. Sci. USA 2001, 98, 4472. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jeon, M.; Lee, J.; Han, J.; Oh, S.-J.; Jung, T.; Nam, S.J.; Kil, W.H.; Lee, J.E. Induction of fibronectin in response to epidermal growth factor is suppressed by silibinin through the inhibition of STAT3 in triple negative breast cancer cells. Oncol. Rep. 2014, 32, 2230–2236. [Google Scholar] [CrossRef]
- Hsu, J.-Y.; Chang, J.-Y.; Chang, K.-Y.; Chang, W.-C.; Chen, B.-K. Epidermal growth factor-induced pyruvate dehydrogenase kinase 1 expression enhances head and neck squamous cell carcinoma metastasis via up-regulation of fibronectin. FASEB J. 2017, 31, 4265–4276. [Google Scholar] [CrossRef] [Green Version]
- Santra, M.; Reed, C.C.; Iozzo, R.V. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J. Biol. Chem. 2002, 277, 35671–35681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordás, G.; Santra, M.; Reed, C.C.; Eichstetter, I.; McQuillan, D.J.; Gross, D.; Nugent, M.A.; Hajnoczky, G.; Iozzo, R.V. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J. Biol. Chem. 2000, 275, 32879–32887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbiotti, M.A.; Vallet, S.D.; Ricard-Blum, S.; Iozzo, R.V. Decorin interacting network: A comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol. 2016, 55, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.V.; Tran, K.T.; Borysenko, C.W.; Cascio, M.; Camacho, C.J.; Blair, H.C.; Bahar, I.; Wells, A. Tenascin cytotactin epidermal growth factor-like repeat binds epidermal growth factor receptor with low affinity. J. Cell Physiol. 2007, 211, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Hintermann, E.; Bilban, M.; Koshikawa, N.; Hojilla, C.; Khokha, R.; Quaranta, V. Binding to EGF receptor of a laminin-5 EGF-like fragment liberated during MMP-dependent mammary gland involution. J. Cell Biol. 2003, 161, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.; Garg, P.; Yang, S.; Gong, P.; Pallero, M.A.; Annis, D.S.; Liu, Y.; Passaniti, A.; Mann, D.; Mosher, D.F.; et al. Epidermal growth factor-like repeats of thrombospondins activate phospholipase Cgamma and increase epithelial cell migration through indirect epidermal growth factor receptor activation. J. Biol. Chem. 2009, 284, 6389–6402. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Marmorstein, L.Y. Focus on molecules: Fibulin-3 (EFEMP1). Exp. Eye Res. 2010, 90, 374–375. [Google Scholar] [CrossRef] [Green Version]
- Wolf, C.; Qian, Y.; Brooke, M.A.; Kelsell, D.P.; Franzke, C.-W. ADAM17/EGFR axis promotes transglutaminase-dependent skin barrier formation through phospholipase C γ1 and protein kinase C pathways. Sci. Rep. 2016, 6, 39780. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.R.; Werner, S.; Paus, R.; Wolf, E. Beyond wavy hairs: The epidermal growth factor receptor and its ligands in skin biology and pathology. Am. J. Pathol. 2008, 173, 14–24. [Google Scholar] [CrossRef]
- Wakita, H.; Takigawa, M. Activation of epidermal growth factor receptor promotes late terminal differentiation of cell-matrix interaction-disrupted keratinocytes. J. Biol. Chem. 1999, 274, 37285–37291. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Klarlund, J.K.; Block, E.R. Free edges in epithelia as cues for motility. Cell Adh. Migr. 2011, 5, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazie, A.R.; Spix, J.K.; Block, E.R.; Achebe, H.B.; Klarlund, J.K. Epithelial cell motility is triggered by activation of the EGF receptor through phosphatidic acid signaling. J. Cell Sci. 2006, 119, 1645. [Google Scholar] [CrossRef] [Green Version]
- Klarlund, J.K. Dual modes of motility at the leading edge of migrating epithelial cell sheets. Proc. Natl. Acad. Sci. USA 2012, 109, 15799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill, H.M.; Knudsen, B.; Moores, S.L.; Muthuswamy, S.K.; Rao, V.R.; Brugge, J.S.; Miranti, C.K. Epidermal growth factor receptor-dependent regulation of integrin-mediated signaling and cell cycle entry in epithelial cells. Mol. Cell Biol. 2004, 24, 8586–8599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.; Morton, P.E.; Takeichi, T.; Salam, A.; Roberts, N.; Proudfoot, L.E.; Mellerio, J.E.; Aminu, K.; Wellington, C.; Patil, S.N.; et al. Epithelial inflammation resulting from an inherited loss-of-function mutation in EGFR. J. Investig. Dermatol. 2014, 134, 2570–2578. [Google Scholar] [CrossRef] [Green Version]
- Gaudry, C.A.; Palka, H.L.; Dusek, R.L.; Huen, A.C.; Khandekar, M.J.; Hudson, L.G.; Green, K.J. Tyrosine-phosphorylated plakoglobin is associated with desmogleins but not desmoplakin after epidermal growth factor receptor activation. J. Biol. Chem. 2001, 276, 24871–24880. [Google Scholar] [CrossRef] [Green Version]
- Klessner, J.L.; Desai, B.V.; Amargo, E.V.; Getsios, S.; Green, K.J. EGFR and ADAMs cooperate to regulate shedding and endocytic trafficking of the desmosomal cadherin desmoglein 2. Mol. Biol. Cell 2009, 20, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Yin, T.; Getsios, S.; Caldelari, R.; Godsel, L.M.; Kowalczyk, A.P.; Muller, E.J.; Green, K.J. Mechanisms of plakoglobin-dependent adhesion: Desmosome-specific functions in assembly and regulation by epidermal growth factor receptor. J. Biol. Chem. 2005, 280, 40355–40363. [Google Scholar] [CrossRef] [Green Version]
- Lorch, J.H.; Klessner, J.; Park, J.K.; Getsios, S.; Wu, Y.L.; Stack, M.S.; Green, K.J. Epidermal growth factor receptor inhibition promotes desmosome assembly and strengthens intercellular adhesion in squamous cell carcinoma cells. J. Biol. Chem. 2004, 279, 37191–37200. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.-M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Franzke, C.-W.; Cobzaru, C.; Triantafyllopoulou, A.; Löffek, S.; Horiuchi, K.; Threadgill, D.W.; Kurz, T.; van Rooijen, N.; Bruckner-Tuderman, L.; Blobel, C.P. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J. Exp. Med. 2012, 209, 1105–1119. [Google Scholar] [CrossRef] [Green Version]
- Hitomi, K. Transglutaminases in skin epidermis. Eur. J. Dermatol. 2005, 15, 313–319. [Google Scholar]
- Blaydon, D.C.; Biancheri, P.; Di, W.-L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Brooke, M.A.; O’Toole, E.A.; Kelsell, D.P. Exoming into rare skin disease: EGFR deficiency. J. Investig. Dermatol. 2014, 134, 2486–2488. [Google Scholar] [CrossRef]
- Grasset, E.M.; Bertero, T.; Bozec, A.; Friard, J.; Bourget, I.; Pisano, S.; Lecacheur, M.; Maiel, M.; Bailleux, C.; Emelyanov, A.; et al. Matrix stiffening and EGFR cooperate to promote the collective invasion of cancer cells. Cancer Res. 2018, 78, 5229. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Commandeur, S.; van Drongelen, V.; de Gruijl, F.R.; El Ghalbzouri, A. Epidermal growth factor receptor activation and inhibition in 3D in vitro models of normal skin and human cutaneous squamous cell carcinoma. Cancer Sci. 2012, 103, 2120–2126. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Diociaiuti, A.; Steinke, H.; Nyström, A.; Schwieger-Briel, A.; Meiss, F.; Pfannenberg, C.; Bruckner-Tuderman, L.; Ruf, J.; Vito, R.D.; Hachem, M.E.; et al. EGFR inhibition for metastasized cutaneous squamous cell carcinoma in dystrophic epidermolysis bullosa. Orphanet. J. Rare Dis. 2019, 14, 1–6. [Google Scholar] [CrossRef]
- Kim, M.; Li, M.; Mather-Hillon, J.; Melbourne, W.; Tran, K.; Daniel, B.; de Souza, P.; Mallesara, G.; Murrell, D. Increased expression of EGFR in squamous cell carcinoma in recessive dystrophic epidermolysis bullosa and response to Cetuximab. Australas J. Dermatol. 2011, 52, 32. [Google Scholar]
- Filoni, A.; Cicco, G.; Lospalluti, L.; Maglietta, A.; Foti, C.; Annichiarico, G.; Resta, L.; Bonamonte, D. Morphological and morphometric analysis of cutaneous squamous cell carcinoma in patients with recessive dystrophic epidermolysis bullosa: A retrospective study. J. Eur. Acad. Dermatol. Venereol. 2019, 34, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hemidesmosomal integrin α6β4. Integrin α6β4 is a major component of hemidesmosomes. The cytoplasmic part of integrin β4 is located in the outer plaque close to the plasma membrane and is connected via plectin and bullous pemphigoid antigen 1 isoform e (BPAG1e/PB230) in the inner plaque to the intracellular keratin filaments. Extracellularly, integrin α6β4 binds laminin-332 and CD151 to stabilize hemidesmosomes. Additionally, the transmembrane collagen XVII binds integrin β4, plectin and BPAG1e as well as integrin α6 and laminin-332 in the extracellular space. Laminin-332 further links with collagen VII and together these proteins connect keratinocytes to the underlying basement membrane [51,52].
Figure 1.
Hemidesmosomal integrin α6β4. Integrin α6β4 is a major component of hemidesmosomes. The cytoplasmic part of integrin β4 is located in the outer plaque close to the plasma membrane and is connected via plectin and bullous pemphigoid antigen 1 isoform e (BPAG1e/PB230) in the inner plaque to the intracellular keratin filaments. Extracellularly, integrin α6β4 binds laminin-332 and CD151 to stabilize hemidesmosomes. Additionally, the transmembrane collagen XVII binds integrin β4, plectin and BPAG1e as well as integrin α6 and laminin-332 in the extracellular space. Laminin-332 further links with collagen VII and together these proteins connect keratinocytes to the underlying basement membrane [51,52].

Figure 2.
Epidermal integrin β1 deficiency in mice and its consequences. (a) Loss of epidermal integrin β1 decreases the laminin receptor dystroglycan and integrin α6β4, which is a major component of hemidesmosomes [102,103]; (b) Integrin β1-deficient keratinocytes have fewer hemidesmosomes and focal adhesions [102]. The skin in epidermal integrin β1-deficient in mice presents with diminished lamina densa (c) [102], deformed hair follicles resulting in hair loss (d) [104] and (e) show separation at the dermal–epidermal junction (DEJ) and dermal inflammation (f) [105]. A proposed mechanism of the latter is that loss of integrin β1 increases epidermal integrin β6, which is involved in transforming growth factor β (TGFβ) activation. Increased TGFβ signaling upregulates, among others, tenascin-C, fostering dermal cytokine/chemokine production by macrophages, which leads to immune cell (T-cells, mast cells) recruitment. Macrophages also likely cause the increase of matrix metalloproteinases (MMPs) and all together extracellular matrix (ECM) degradation, modified from and inspired by Kurbet et al. [105].
Figure 2.
Epidermal integrin β1 deficiency in mice and its consequences. (a) Loss of epidermal integrin β1 decreases the laminin receptor dystroglycan and integrin α6β4, which is a major component of hemidesmosomes [102,103]; (b) Integrin β1-deficient keratinocytes have fewer hemidesmosomes and focal adhesions [102]. The skin in epidermal integrin β1-deficient in mice presents with diminished lamina densa (c) [102], deformed hair follicles resulting in hair loss (d) [104] and (e) show separation at the dermal–epidermal junction (DEJ) and dermal inflammation (f) [105]. A proposed mechanism of the latter is that loss of integrin β1 increases epidermal integrin β6, which is involved in transforming growth factor β (TGFβ) activation. Increased TGFβ signaling upregulates, among others, tenascin-C, fostering dermal cytokine/chemokine production by macrophages, which leads to immune cell (T-cells, mast cells) recruitment. Macrophages also likely cause the increase of matrix metalloproteinases (MMPs) and all together extracellular matrix (ECM) degradation, modified from and inspired by Kurbet et al. [105].

Figure 3.
Integrin αV ligands. Integrin αV-containing integrins recognize a multitude of proteins linked to ECM assembly, inflammation or angiogenesis. The selectivity is dependent on the β-subunit [10].
Figure 3.
Integrin αV ligands. Integrin αV-containing integrins recognize a multitude of proteins linked to ECM assembly, inflammation or angiogenesis. The selectivity is dependent on the β-subunit [10].

Figure 5.
CD44 interacts with various ECM components and cell-surface receptors. CD44 acts as receptor for hyaluronan (HA) [238,239], though can also bind other ECM proteins, such as fibronectin [241], laminin [241], heparin-binding growth factors [245] or collagen I [241], IV [246] and XIV [247]. The extracellular domain of CD44 can be cleaved by MMPs or ADAMs [253,254,255]. They create a soluble form of CD44, which competes with membrane-bound CD44 for HA binding in some cells [256]. The remaining intracellular domain can be released into the cytoplasm by the presenilin-dependent-γ-secretase, where it regulates gene transcription [257,258]. CD44 also prevents the conversion of G- to F-actin. G-actin binds cytoplasmatic myocardin-related transcription factor (MRTF) and hinders it from translocating to the nucleus, where it could co-activate transcription factors (TF) [259]. CD44 interacts with the transforming growth factor β receptor (TGFβR) and the platelet-derived growth factor receptor β (PDGFRβ) and upon HA-activation negatively regulates them [248,249]. CD44 also associates with epidermal growth factor receptor (EGFR) [250,251,252]. Versican possibly competes with CD44 to bind HA and with EGF to bind EGFR and therefore blocks CD44 and EGFR signaling [260].
Figure 5.
CD44 interacts with various ECM components and cell-surface receptors. CD44 acts as receptor for hyaluronan (HA) [238,239], though can also bind other ECM proteins, such as fibronectin [241], laminin [241], heparin-binding growth factors [245] or collagen I [241], IV [246] and XIV [247]. The extracellular domain of CD44 can be cleaved by MMPs or ADAMs [253,254,255]. They create a soluble form of CD44, which competes with membrane-bound CD44 for HA binding in some cells [256]. The remaining intracellular domain can be released into the cytoplasm by the presenilin-dependent-γ-secretase, where it regulates gene transcription [257,258]. CD44 also prevents the conversion of G- to F-actin. G-actin binds cytoplasmatic myocardin-related transcription factor (MRTF) and hinders it from translocating to the nucleus, where it could co-activate transcription factors (TF) [259]. CD44 interacts with the transforming growth factor β receptor (TGFβR) and the platelet-derived growth factor receptor β (PDGFRβ) and upon HA-activation negatively regulates them [248,249]. CD44 also associates with epidermal growth factor receptor (EGFR) [250,251,252]. Versican possibly competes with CD44 to bind HA and with EGF to bind EGFR and therefore blocks CD44 and EGFR signaling [260].

Figure 6.
Epidermal growth factor (EGF) receptor (EGFR) interactions with the ECM. (a) EGFR recognizes ligands, such as EGF, transforming growth factor (TGF)-α, amphiregulin (AR), betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF), epigen (EPGN) or epiregulin (EREG) [352,353]. Upon their binding the receptor dimerizes followed by auto-phosphorylation and induction of diverse intracellular signaling events [351,352,354,355]; (b) Stiffening of the ECM, as found in tumors, causes EGFR overexpression and increased EGF bioavailability, which fuels tumorigenesis [359,360,361]. EGFR also associates with integrin α6β4 and via the Fyn kinase phosphorylates β4 integrin if integrin α6β4 is bound to laminin-332 [57,362]. This leads to hemidesmosome disruption and fosters tumor proliferation and invasion [57,362]. Furthermore, a stiff ECM increases EGFR phosphorylation, but this is also dependent on focal adhesions, since EGFR interacts with them and might be regulated by the focal adhesion kinase (FAK) [359].
Figure 6.
Epidermal growth factor (EGF) receptor (EGFR) interactions with the ECM. (a) EGFR recognizes ligands, such as EGF, transforming growth factor (TGF)-α, amphiregulin (AR), betacellulin (BTC), heparin-binding EGF-like growth factor (HB-EGF), epigen (EPGN) or epiregulin (EREG) [352,353]. Upon their binding the receptor dimerizes followed by auto-phosphorylation and induction of diverse intracellular signaling events [351,352,354,355]; (b) Stiffening of the ECM, as found in tumors, causes EGFR overexpression and increased EGF bioavailability, which fuels tumorigenesis [359,360,361]. EGFR also associates with integrin α6β4 and via the Fyn kinase phosphorylates β4 integrin if integrin α6β4 is bound to laminin-332 [57,362]. This leads to hemidesmosome disruption and fosters tumor proliferation and invasion [57,362]. Furthermore, a stiff ECM increases EGFR phosphorylation, but this is also dependent on focal adhesions, since EGFR interacts with them and might be regulated by the focal adhesion kinase (FAK) [359].

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kleiser, S.; Nyström, A. Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin. Biomolecules 2020, 10, 1170. https://doi.org/10.3390/biom10081170
AMA Style
Kleiser S, Nyström A. Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin. Biomolecules. 2020; 10(8):1170. https://doi.org/10.3390/biom10081170
Chicago/Turabian StyleKleiser, Svenja, and Alexander Nyström. 2020. "Interplay between Cell-Surface Receptors and Extracellular Matrix in Skin" Biomolecules 10, no. 8: 1170. https://doi.org/10.3390/biom10081170
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.