
Abstract
Advances in biochemical and molecular manipulation have led to increased biomass productivity and oil accumulation in the microalgae C. reinhardtii. However, scalable processes for the recovery of oil and other valuable biomolecules, such as protein, from C. reinhardtii are scarce. The use of aqueous enzymatic extraction, a non-solvent and environmentally friendly bioproduct recovery method, provides an opportunity to design an integrated process for oil and protein fractionation to reduce bioenergy and bioproducts costs. Based on the mechanistic understanding of biomolecule distribution and compartmentalization, an aqueous enzymatic treatment for the release of internally stored lipid bodies was designed. Application of a C. reinhardtii-produced protease, autolysin, for lysis of the microalgae cell wall was followed by a secondary treatment with trypsin for chloroplast disruption and lipid body release. Protein recovery after the primary treatment with autolysin indicated a 50.1 ± 4.2% release of total soluble protein and localization of lipid bodies still in the chloroplast. The development of a secondary enzyme treatment (trypsin) for chloroplast and lipid body lysis demonstrated a high percent of remaining lipids (73 ± 7%) released into the supernatant. The results indicate that the application of an enzymatic treatment scheme for protein and oil recovery is a promising alternative to traditional extraction processes.

Similar content being viewed by others

Introduction
The need to replace fossil fuels, one of the main causes of Green House Gas Emissions (GHGE) (Schenk et al. 2008) has driven new research focused on exploring renewable energy sources. Among others, ethanol production from corn and other food crops has been intensely explored, but the competition with food crops is a main concern. One alternative to food crop sources is the utilization of microalgae biomass. Microalgae cultivation has several advantages over other crops including a higher photosynthetic efficiency, higher biomass production, and faster growth rates (Mata et al. 2010). Their photosynthetic growth also contributes to CO2 sequestration (Halim et al. 2012, 2016) and reduces atmospheric air pollutants.
Lipids from algae are rich in saturated and unsaturated fatty acids such as oleic (18:1), palmitic (16:0), stearic (18:0), and linoleic (18:2) acids (Meng et al. 2009), making them ideal not only for fuel production but also as high value food products. Some microalgae species, such as Chlamydomonas reinhardtii, are known for producing lipids and proteins with potential applications in the food and pharmaceutical industries. For instance, the use of C. reinhardtii cells as host organisms for recombinant protein (RP) production is one promising application in the biopharmaceutical industry (Ahmad et al. 2020). Rapid growth rates, straightforward photo-autotrophic cultivation, unique metabolic properties, and continuous progress in RP production have increased research interest in microalgae systems (Lauersen 2019). Even though there are several advantages regarding the utilization of C. reinhardtii cells as a protein and lipid source, there are still some challenges regarding its cultivation and extraction.
Protein solubilization and extraction
Cell disruption is a critical step in a protein extraction process as proteins must first be released from internal cell compartments in a soluble form (Tan 2009). Protein extraction from algae can be challenging due to their sturdy cell wall (Safi et al. 2013) that requires disruption methods that are strong enough to lyse the cell wall while preserving protein integrity (Soto-Sierra et al. 2018, 2020). Common mechanical disruption methods, such as the French Press, pulsed electric fields, glass beads, or sonication are used to break the cell wall, followed by detergent-based total protein solubilization and extraction (Tan 2009; Soto-Sierra et al. 2018; Kulkarni and Nikolov 2018; Lam et al. 2017; Zheng et al. 2011). Conditions such as pH, temperature, and ionic strength of the aqueous solution can affect protein extractability. For example, biomass subjected to intense heat, which rapidly denatures proteins, shows poor extractability (Smith 1972). Contrarily, a slightly alkaline pH ~ 8 and temperatures between 25 and 37°C usually exhibit better protein extractability (Waniska and Kinsella 1979). Once proteins are extracted, they can be purified, used directly, or converted into valuable products such as nutraceuticals, food additives, coatings, and bioplastics.
Lipid extraction
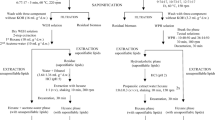
Current methods for oil extraction require long processing times, petroleum-based solvents, or energy-intensive mechanical disruption treatments (Adam et al. 2012; Deshmukh et al. 2019). In microalgae, traditional lipid extraction is performed with the aid of lipophilic extraction solvents. Currently, hexane is the most utilized solvent at an industrial scale. Biomass drying or other high-intensity cell disruption methods are required (Deshmukh et al. 2019; Ranjan et al. 2010) for lipophilic extraction solvent systems, as the immiscibility of unruptured cells do not permit solvent access to the internally stored lipid bodies (Fig. 1a). Cell disruption allows solvent to penetrate the cell and solubilize the lipids, propitiating a faster separation and recovery (Yap et al. 2014). When biphasic solvent systems are used, such Bligh & Dyer (chloroform–methanol–water), lipid recovery on unruptured cells is possible but requires long incubation times due to passive diffusion of solvents and lipids across the cell wall (Ranjan et al. 2010). Thus, cell wall disruption is a key step to increase extractable yields. When cells are ruptured, the solvent (or solvent mixture) can rapidly diffuse and solubilize lipids into the hydrophobic phase while polar cell biomolecules remain in the aqueous (hydrophilic) phase (Fig. 1a).

a Lipid solvent extraction process: (1) harvesting, (2) cell permeabilization, (3) incubation with solvent, and (4) separation and recovery. b Proposed enzymatic process; (1) harvesting, (2) autolysin treatment, (3) incubation at 35 °C for prolonged time, (4) secondary enzymatic treatment, and (5) centrifugation
It is desirable to extract proteins and lipids without significant contamination by other cellular components (Scott et al. 2010) in an energy-efficient and environmentally friendly process, avoiding drying steps and the use of large amounts of toxic substances. To address prior limitations of solvent-based extraction, a fully enzymatic process is proposed (Fig. 1b) and includes primary enzymatic treatment (using cell wall-specific enzyme) and incubation to permeabilize the cell wall, secondary enzymatic treatment to disrupt internal organelles and free lipid droplets, and recovery of respective protein and lipid streams.
Enzymatic treatment for product release
Protein recovery
Many of the naturally occurring or recombinant proteins stored in intracellular compartments cannot be secreted by the microalgae cell. Enzymatic cell lysis allows for the degradation of specific membranes and further product release, which enables for higher purification levels at early extraction stages (Andrews and Asenjo 1987; Sari et al. 2013). When sufficient, enzymatic cell wall disruption can induce osmotic lysis and further product release into the media. One of the challenges of this method is the specificity and complexity of microalgae cell walls, which implies that a treatment has to be designed for each species. While several authors have used proteases to release degraded proteins from previously lipid-extracted (permeable) cells (Sari et al. 2013, 2016; Morris et al. 2008, 2009), thus far, we have not found specific approaches for hydrolyzing microalgae cell wall while preserving the protein structure. In the case of the microalgae Chlamydomonas reinhardtii, our previous work demonstrated that autolysin treatment promotes high levels of cell permeabilization (Soto-Sierra et al. 2017) which then further facilitated protein and lipid extraction.
Lipid recovery
According to Thiam et al. (2013), lipid droplets are a dispersed phase of an oil-in-water emulsion in the cytosol-aqueous media of cells. Lipid droplets form natural emulsions inside the cells with the help of certain emulsion stabilizers such as proteins and phospholipid surfactants (Leal-Calderon et al. 2007). An emulsion can prevent lipid droplets from coalescing into larger droplets, thus preventing lipid separation from the aqueous media. For the proposed work (Fig. 1b), trypsin was selected for secondary treatment to target lipid droplets surface proteins (LDSP) entrapping the lipid bodies. This serine protease is relatively inexpensive and can cleave a wide variety of protein substrates using mild conditions (neutral pH and 37 °C), making it an attractive and economical option for cleaving C. reinhardtii chloroplasts and lipid bodies. To release the lipid bodies, several chloroplast membranes need to be digested including LDSP, chloroplast’s outer and inner membranes, and interconnected stacked thylakoid discs (Zerges and Rochaix 1998). Previous research showed that trypsin relaxed thylakoid discs and promoted membrane unstacking within spinach chloroplasts (Jennings et al. 1981). Furthermore, trypsin is usually produced in a mammal’s digestive system and has been widely used in food processing industry, thus, if still present in the protein lysate after downstream processing, this protein would not be unsafe if ingested.
The objectives of this study were to: (1) optimize the previously developed autolysin pretreatment (Soto-Sierra et al. 2017) for enhancing disruption of C. reinhardtii cell walls and enhance protein release and (2) develop a secondary enzymatic treatment for releasing intracellular lipid droplets and separating them from remaining chloroplast proteins. The proposed enzymatic treatment may increase the feasibility of algae as a feedstock by extracting more than one valuable product from C. reinhardtii biomass using mild processing conditions that should retain co-product quality. Finally, this treatment could avoid the utilization of solvents or non-GRAS chemicals.
Methods/experimental
Strain and culture medium
Algae biomass
Stock cultures of Chlamydomonas reinhardtii (CC-409 mt+) were obtained from the Chlamydomonas Resource Center, University of Minnesota. C reinhardtii cells were grown in TAP (Tris–acetate–phosphate) plates for 5 days under constant light conditions (27 µM/m2-s) and then transferred to liquid TAP media. Once the lag phase was reached (~ 1 × 107 cells per mL), biomass was centrifuged at 6000×g for 5 min, washed, and re-suspended for 48 h into the same volume of nitrogen-depleted (TAP-N) media. After the depletion period, biomass was harvested and stored at − 80 °C until use.
Mating strains
High-efficiency C. reinhardtii strains CC-620 mt+ and CC-621 mt− were kindly provided by Dr. Olson of the Division of Biology at Kansas State University. Cells were grown in solid TAP media until a high mating efficiency was achieved. After, cells were solubilized and suspended into liquid TAP media.
Characterization of the algae extract
Total protein quantification
For each total protein determination, 10 mL of biomass were centrifuged at 6000×g for 5 min. The supernatant was removed and the protein in the pellet was solubilized in a plant and algae solubilization buffer containing 0.75 mM lithium dodecyl sulfate (Amresco), 2.5 mM glycerol (Amresco), 51.4 mM TRIS base (Biosciences), and 0.02 mM EDTA (Alfa Aesar). Each sample was sonicated four times at 25% amplitude for a total of 2 min with 30 s cooling time in ice after each sonication cycle. Samples were centrifuged again under the same conditions and supernatant was recovered. Finally, lysates were diluted 10 times with carbonate–bicarbonate buffer (pH 9.6) and total soluble protein was measured using a BCA Protein Assay Kit (Pierce™) (Olson and Markwell 2007; Smith et al. 1985).
Lipid yield quantification
For total lipid quantification, a modified version of the Bligh and Dyer (Bligh and Dyer 1959) was performed. Samples (90 mL) were centrifuged at 6000×g for 5 min and the supernatant was decanted. Chloroform, methanol, and water were added to the pellet in a volume ratio of 1:2:1. Subsequently, samples were sonicated for 1 min at 25% amplitude, mixed overnight, and centrifuged at 6000×g for 5 min. The bottom lipophilic layer was extracted and filtered into pre-weighed trays. Samples were evaporated and then dried in an oven at 95°C for 1 h. Dried lipids were weighed to calculate recoverable lipid content by the following Eq. (1):
Autolysin preparation
To prepare autolysin, a modified protocol of the one proposed by Jaenicke et al. (1987) was followed. High-efficiency mating strains, CC-620 mt+ and CC-621 mt−, were cultured and placed under high-intensity LED lights (35 µM/m2 s). Three days after growth, each mating type was independently transferred into TAP-N for a final cell concentration of 1 × 107 cells/mL. After 12 h of constant mixing under high-intensity LED lights, mating tests were performed to determine mating efficiency. For the test, 200 µL of each mating strain were mixed, allowed to mate for 5 min and observed under the microscope (VWR® fluorescence inverted microscope). If approximately 95% of cells were mating, high mating efficiency was achieved and cells were ready to be mixed. Both mating strains were mixed in a clear container, placed under high light for approximately 30 min and then centrifuged at 6000×g for 5 min. Supernatants containing autolysin were filtered with a 0.45 µm PES membrane bottle-top sterile filter and stored at − 80 °C until use.
Primary enzymatic treatment using autolysin
Biomass was harvested and re-suspended in either autolysin buffer or TAP-N (nitrogen deficient) buffer as a negative control. Biomass was incubated at three different temperatures (25°C, 35°C, or 50°C) with constant mixing (250 rpm) for different pre-determined time periods. Cell counts were performed before, during, and after treatment.
TEM imaging
TEM pictures were taken at the Nanotechnology Innovation Center of Kansas State (NICKS) using Tecnai™ G2 Spirit BioTWIN (FEI Company) at 80 kV acceleration voltages. Biomass samples were fixed in Trump’s fixative overnight, post-fixed with osmium tetroxide, dehydrated in graded series of alcohol, and embedded in spur resin. Ultra-thin sections were contrasted with uranyl acetate lead citrate and observed under FEI Tecnai 12 Bio-spirit transmission electron microscope.
Quantification of percent protein released after autolysin treatment
Biomass was first incubated with autolysin at room temperature for 4 h and then for an additional 20 h at 3 different temperatures (25°C, 35°C, or 50°C). After enzymatic treatment, biomass was centrifuged at 7000×g for 5 min and supernatants collected. Total soluble protein was quantified using a BCA Protein Assay Kit (Pierce ™). The percent protein solubilized was calculated based on a total extractable protein reference. Total extractable protein was calculated following the total protein quantification procedure described in the “Total protein quantification” section and percent protein released was determined using Eq. 2:
Quantification of lipid release
Biomass/enzyme treated slurry was centrifuged at 6000×g for 5 min and supernatants and pellets were collected separately. The extracts were subjected to a modified Bligh and Dyer (Bligh and Dyer 1959) as described below, and lipid percent was calculated based on a recoverable lipid yield (Eq. 3):
Modified Bligh and Dyer extraction
Samples were centrifuged at 6000×g for 5 min. The supernatants (lysate) were collected and chloroform and methanol were layered on top in a ratio of 1:1:2 (lysate:chloroform:methanol). Samples were mixed overnight at 100 rpm in a rotary shaker, centrifuged at 6000×g for 5 min and the bottom lipophilic layer was extracted and filtered into pre-weighed trays. Finally, samples were dried at 105°C for 1 h in an air for oven, trays were reweighed, and percent lipid released was determined using Eq. 3.
Chloroplasts isolation and lipid quantification
To determine the amount of lipids remaining in the chloroplasts after autolysin treatment, biomass was treated with autolysin for 24 h. Cell lysate was centrifuged and remaining chloroplasts were isolated following the protocol published by Mason, Bricker (Mason et al. 2006). After isolation, chloroplasts were re-solubilized in storage buffer (50 mM HEPES–KOH pH 8.0 + 0.3 M sorbitol) and divided into two samples of the same volume. One sample was used to calculate dry weight of the isolated chloroplasts and the other sample was subjected to lipid extraction using a modified Bligh and Dyer (Bligh and Dyer 1959) method. Lipid content was calculated using Eq. 4:
Secondary enzymatic treatment
After cells were treated with autolysin, biomass was centrifuged and supernatants were collected for further protein recovery. The remaining pellets were subjected to a secondary enzymatic treatment with trypsin (CAS # 9002-07-7). For the treatment, cell lysate was centrifuged and re-suspended in pH 7.8 buffer containing trypsin at 2% v/w dosage. The slurry was incubated at 35 °C for 3 h, following a modified protocol of the one proposed by Hung, Vas (Hung et al. 1984). After incubation, cell lysis percent and lipid release were calculated.
Cell lysis analysis
For the quantitative analysis of cell lysis, cell suspension was loaded into a hemocytometer (10 µL) and cell counting was performed using an inverted microscope. Percent of lysed cells was calculated with Eq. 5:
Statistical analysis
One-way analysis of variance (ANOVA) was conducted for statistical analysis of the experimental data using SAS and Graph-Prism 6 software. To compare significant differences between treatments, a Tukey adjustment was made for a family-wise error rate of 0.05 (αFER = 0.05).
Results and discussion
When designing a process for protein and lipid recovery, loss or degradation of either product should be minimized. Proteins are more vulnerable to temperature and shear induced degradation. Furthermore, when associated with lipids, they promote emulsion formation, impeding lipid separation from the aqueous media. A sequential extraction where most of the protein is released first is preferable as it can minimize protein degradation as well as prevent emulsion formation that could hinder lipid recovery. Thus, the first step was to solubilize and recover proteins.
Impact of temperature and time on cell disruption and protein and lipid recovery
Prior work by Soto-Sierra, Dixon (Soto-Sierra et al. 2017) demonstrated that treating C. reinhardtii with autolysin for 4 h at 25°C was an effective method for cell lysis and resulted in ~ 20% protein release. To evaluate and optimize protein recovery after the enzymatic treatment, biomass incubation with autolysin was performed at different temperatures (25, 35, and 50°C) and extended incubation times (8, 17, and 24 h) and total protein solubilized was compared among treatments. Biomass was incubated with either autolysin or control buffer at each temperature. At each time point, biomass was centrifuged, the supernatants were collected, and total soluble protein was calculated. Results (Fig. 2a) showed that at 24 h of autolysin treatment, protein solubilization for all temperatures was significantly higher when compared to the control. A significant increase in protein solubilization of approximately 10% was observed for the control treatment at 50°C when compared to 25°C. This indicated that there was protein being solubilized by the high temperature treatment (at 50°C) rather than by autolysin, as the treatment at 50°C solubilized a significantly lower amount of protein when compared to treatments at 25°C and 35°C (data not shown). Reduced protein extractability at 50°C could be attributed to a decrease of autolysin activity at the elevated treatment temperature (Wilken and Nikolov 2016).

a Percent of total soluble protein released after 24 h of autolysin treatment at 3 different incubation temperatures and b percent of protein released at 35°C at 3 different incubation times. Percentages were calculated based on a total extractable protein reference. Error bars represent standard error for n > 3. Comparisons were made within and between groups and significant differences were corrected for multiple comparisons with Tukey adjustment and an αFER = 0.05. Different letters represent significant difference between treatments
Protein solubilization was significantly higher at 35°C when compared to autolysin treatments at 25°C and 50°C. On average, 50.1 ± 4.2% of the protein was solubilized after autolysin treatment at 35°C. Regarding incubation time, the amount of protein solubilized was approximately 15% higher for samples incubated 24 h when compared to 8 and 18 h of incubation (Fig. 2b). Based on these results, autolysin pretreatment for 24 h at 35°C was chosen as the optimum condition for cell wall disruption and protein release. In a large-scale setting, though, 24 h incubation might result in potential contamination, protein degradation by native proteases, and oxidation (Majumdar et al. 2018). Thus, further work to optimize extraction conditions regarding protein recovery, yield, and quality is necessary.
The remaining proteins, mostly photosynthetic (RuBisCO and LHC), were most likely still stored in the chloroplast along with lipids.
Effect of enhanced autolysin treatment on lipid recovery
In previous research, Soto-Sierra et al. (2017) reported that the autolysin treatment at 25°C achieved complete cell wall disruption while lipid bodies remained attached to cell remnants in the solid fraction (pellet). To determine the effect on lipid release of increasing temperature and incubation time, samples were treated with autolysin for 24 h at 35°C. TEM images of cell pellets were taken after autolysin treatment at 25°C and 35°C. Results (Fig. 3a) showed that for both temperature treatments, the majority of lipid bodies were still trapped in the solid fraction of the cell lysate. Presumably, lipid body surface proteins and phospholipids were associating with other proteins and polar biomolecules, preventing TAGs (triacylglycerols) from being released. Even though most of the lipid bodies were still contained in the solid fraction, TEM images also show an apparent reduction of lipid body size for the biomass treated at 35°C when compared to the treatment at 25°C.

a TEM images of solid fractions (pellets) after centrifugation of autolysin-treated biomass at 25°C (left) and 35°C (right). b Percent lipid released (to the supernatants) after centrifugation of autolysin-treated biomass or control (biomass incubated under same conditions but without autolysin) at 25°C or 35°C. Percentages determined by a modified Bligh and Dyer extraction method. Significant differences were found using a p value of 0.05
To further explore these results, lipid release into the supernatants after 24 h of autolysin treatment at 35°C was quantified. Results showed that the prolonged incubation time of the microalgae cells with autolysin induced significant cell disruption (Fig. 3a), which resulted in the release of 40 ± 2 % and 43 ± 1% of total lipids at 25 and 35°C (Fig. 3b), respectively. Interestingly, Fig. 3a shows that while autolysin-treated cells were significantly disrupted, most lipid bodies (red triangles) were still attached to the cell remnants (Kirchhoff et al. 2008). We suspect that the autolysin treatment promoted the release of surface and other polar lipids while the internal and non-polar lipid bodies remained attached to the chloroplast membranes (Fan et al. 2011).
After autolysin treatment at 35°C, ~ 50 ± 4% of total proteins were extracted while ~ 57–60% of the lipids remained in the solid fraction. Future studies should focus on developing a processing strategy for recovering each product into a separate stream once they have been released. Alternatively, extraction conditions could also be optimized for maximizing the solubilization of protein, which is the most soluble product, first, while keeping the lipids retained in the cells for their subsequent extraction.
Lipid content on isolated chloroplasts
To develop a solvent-free extraction system, a secondary treatment that promoted lipid body release and oil demulsification was needed. First, we aimed to understand why the majority of the lipid bodies were not being released after the cells were disrupted. Based on research regarding lipid body accumulation of C. reinhardtii cells (Fan et al. 2011) and previous TEM images (Fig. 3a), lipids can be stored in the endoplasmic reticulum and/or inside the chloroplast. If stored in the chloroplast, the previously characterized (Moellering et al. 2009) LDSP could be associating with other polar biomolecules inside this organelle, preventing lipids from being extracted. Determining where lipid bodies are attached after the enzymatic treatment would provide additional information regarding which cell structures need to be cleaved to release the lipids. Thus, the next step was to confirm if the lipid bodies were enclosed in the remaining chloroplasts and chloroplasts remnants. To do so, chloroplast remnants and thylakoids were isolated after autolysin treatment based on a modified protocol of the one proposed by Mason et al. (2006) and lipid content in the intact chloroplast plus thylakoids fractions were calculated.
The increase in lipid content (DW) indicated that chloroplasts and the disrupted thylakoids concentrated most of the lipids trapped in the solid fraction. Results (Fig. 4) showed that lipid content of the chloroplasts fraction was almost 70% (g lipids/g total dry weight) while the lipid content of intact C. reinhardtii cells was 49%. This is a 1.42-fold lipid concentration when compared to whole cells. Furthermore, the gram dry basis sum of chloroplast lipids plus lipids released after autolysin treatment was approximately ~ 0.38 g lipids/g which is about 90% of the total lipid content in a C. reinhardtii cell after 48 h of nitrogen depletion (Soto-Sierra et al. 2017). These results indicated that the majority of the lipids in the remaining biomass after autolysin treatment were stored in the chloroplasts. Possibly, stacked membranes in the chloroplasts were trapping lipid bodies. Furthermore, the amphiphilic nature of the chloroplasts could be reducing the interfacial tension between the aqueous solution and the lipid bodies, contributing to the stabilization of dispersed droplets and avoiding their association. The attachment is possibly made between LDSP and proteins or other polar molecules in the chloroplast. Consequently, the next treatment to be designed should target not only the LDSP (Moellering et al. 2009), but also proteins and other molecules present in the chloroplast. Thus, the next step was to design an aqueous enzymatic treatment to disrupt chloroplast remnants and LDSP, so attached lipids could be released.

Percent lipid content of isolated chloroplasts and whole cell biomass. Error bars represent standard error for n ≥ 3. Significant differences were found based on an α = 0.05 with different letters representing significant differences between samples. Lipid content was calculated based on modified Blight and Dyer method
Effect of a secondary enzymatic treatment on lipid release
Thus far, autolysin treatment was able to permeabilize and disrupt the cells and release 50 ± 4% of the protein by extending incubation time. To release lipid bodies from internal compartments, in this case, the chloroplasts and LDSP need to be cleaved so the lipid bodies can be released from the disrupted chloroplasts remnants. To design an efficient AEE treatment, it is crucial to make sure that proteins are being cleaved by the protease chosen. Based on preliminary data (not shown), trypsin was selected as the best fit for cleaving LDSP and other chloroplast proteins. Trypsin was selected as it can approximately cleave the ~ 260 amino acid chain of the C. reinhardtii LDSP about 20 times based on the primary structure and cleavage specificity. Trypsin treatment could also promote the release of lipid bodies attached between thylakoids by disrupting membrane stacking as it was reported in chloroplasts of plants such as spinach (Jennings et al. 1981).
Biomass was treated with autolysin and lipids were recovered as specified in “Quantification of lipid release” section. If lipids were being released from the chloroplasts, the lipid recovery in the supernatant fraction was expected to increase. Results indicated a significant increase in lipid release for samples incubated with autolysin plus trypsin treatment. Figure 5a shows that more than 30% of lipids still trapped in the solid fraction (pellet) after autolysin treatment were released by trypsin treatment. After Nile Red staining, several lipid bodies were visible in the supernatants from trypsin-treated samples (Fig. 5b) which further confirmed lipid release.

a Total lipid percent (DW) of the supernatants after incubation with TAP-N (control), autolysin, and autolysin plus trypsin. b Fluorescence microscopy imaging of lipids (yellow fluorescence) stained with Nile Red. Error bars represent standard error for n > 3. Significant differences were corrected for multiple comparisons with Tukey adjustment and an αFER = 0.05. Different letters represent significant difference between treatments
With autolysin plus trypsin treatment, ~ 73 ± 7% of total lipids stored in C. reinhardtii cells were released from the solid fraction. Several authors have also reported an increase in lipid extractability after protease treatment of diverse biological substrates, such as microalgae (Wu et al. 2017), fish (Dumay et al. 2006; Kechaou et al. 2009), maize (Tester et al. 2007), and coconut (Patil and Benjakul 2019; Senphan and Benjakul 2017). The significant increase in lipid release can be attributed, among others, to the breakdown of particular protein structures (Gbogouri et al. 2006) keeping lipids attached to the cell remnants and inside of the lipid bodies.
Protein release after secondary extraction process
Once lipid release was achieved, protein solubilization after the secondary extraction process was monitored. First, the protein release of autolysin-treated cells after incubation in buffer vs. buffer plus trypsin was compared (Fig. 6). Then, the molecular weight (MW) protein profiles (Fig. 7) were analyzed to identify proteins that remain in the cell debris/solids, solubilized, or degraded during trypsin treatment.

Additional protein release from autolysin-treated cells after secondary treatment with trypsin. The (−) control corresponded to autolysin-treated biomass resuspended in buffer without enzyme. Percentages were calculated based on a total extractable protein reference. Error bars represent standard error for n > 3. Differences were corrected for multiple comparisons with Tukey adjustment and an αFER = 0.05. Different letters represent significant difference between treatments

a Diagram showing sample collection process for gel electrophoresis analysis and b MW protein profile. MW marker (lane 1); Total protein-10X concentration- (lane 2); Supernatant after autolysin treatment-5X concentration (lane 3); Pellet-10X concentration-(lane 4) and supernatant-22.5X concentration (lane 5) after incubation in buffer at 35°C for 24 h, no trypsin; Pellet-10X concentration (lane 6) and supernatants-22.5X concentration-(lane 7) after autolysin treatment and resuspension in buffer plus trypsin; and Autolysin-17X concentration (lane 8)
Results showed that the trypsin treatment caused further solubilization of 14 ± 1% of the protein, bringing the cumulative protein release after autolysin plus trypsin treatment to ~ 64 ± 6% (Figs. 2 and 6). Even though only a small amount of the protein stored in the chloroplast was solubilized by trypsin, the specific digestion was enough to release lipids stored between the thylakoid membranes. The pellet after autolysin plus trypsin treatment (Fig. 7b, lane 6, LHC arrow) showed a decrease in band intensity of a complex of proteins of MW ~ 17 to 30 kDa, which could potentially correspond to the LHC. Solubilization of these proteins was possibly induced by the trypsin digestion. After trypsin, a slight decrease in proteins of ~ 35 kDa, ~ 45 kDa and ~ 98 kDa is also apparent (Fig. 7b, lanes 6 & 7). Moreover, the gel shows that after autolysin treatment (Fig. 7b, lane 3), high molecular weight proteins (between 98 and 198 kDa) are completely solubilized. These proteins can potentially be the glycosylated cell wall proteins, which are characterized by a high molecular weight (Mathieu-Rivet et al. 2013), and were being solubilized early on after autolysin treatment. Proteins that have not yet been solubilized can be recovered from the solid fraction using a mechanical or chemical, treatment. One advantage of preserving the proteins in the solid fraction is that it allows for the selective recovery of lipids from the liquid phase while keeping most of the proteins in the solid fraction (pellet). The separation caused by the density difference between both products, could potentially decrease steps and energy involved in the extraction process allowing for recovery of each product at higher purities.
Effect of trypsin treatment on cell structure and bioproduct release
To better understand why trypsin treatment was promoting lipid release while keeping proteins in the solid fraction, the effect of autolysin plus trypsin and autolysin treatment only on lipid release was analyzed and compared by TEM imaging. Results showed that the autolysin treatment caused the disruption of the cell wall and chloroplast envelopes (Fig. 8a, A, B). Nevertheless, numerous lipid bodies were still attached to the internal portion of the thylakoid membranes (Fig. 8a, D).


TEM images of C. reinhardtii cells incubated with (a) autolysin-control or (b) autolysin plus trypsin at 1200× (a, A, B and b, E, F), 2900× (b G) and 6400× (a, C, D and b H)) magnification. Letters indicate cellular components: S starch bodies, LB lipid bodies, T thylakoids. These images are representative of ≥ 2 replicates
For the autolysin plus trypsin-treated samples, Fig. 8b, G and H, shows an apparent decrease in membrane stacking and relaxation of thylakoid (T) membranes when compared to samples only treated with autolysin (Fig. 8a, D). This effect was previously reported (Jennings et al. 1981) when treating spinach chloroplasts with trypsin. According to Grebanier (1979), the main effect of trypsin on chloroplast membranes is to digest a small fragment from the light-harvesting protein complex. Possibly, the relaxation of the thylakoid membranes accompanied by the disruption of lipid body proteins induced the release of lipid bodies. A reduction in the amount of lipids still attached to the pellet together with the presence of empty lipid and starch bodies (Fig. 8b, G & H) after trypsin treatment confirms the abovementioned effects.
Interestingly, the autolysin plus trypsin-treated samples (Fig. 8b, E & F) showed large amounts of free starch granules in some of the TEM sections. Insoluble starch appeared to be released from the chloroplasts and sedimented at the bottom of the pellets. This is most likely due to the higher density of the starch granules (~ 1.5 g/cm3) when compared to the thylakoid fragments (~ 1 g/m3) and lipid bodies (~ 0.9 g/m3). If starch is one of the products to be recovered, the difference in density when compared to other cell components will allow this product to accumulate at the bottom of the solid phase, facilitating its recovery and further purification.
Certainly, the AEE treatment designed not only facilitates lipid and protein extraction, but also propitiates starch recovery. Further research should aim to optimize the fractionation and extraction of these three products after the enzymatic treatment.
With the primary and secondary enzymatic treatments developed, intact cells with intact cell walls were transformed into highly disrupted cells, and finally, into partially fractionated bioproducts (Fig. 9a–c).

Schematic representation of the enzymatic treatment with: a autolysin treatment for cell permeabilization, b release of cytosolic proteins, and c trypsin treatment for the release of lipid bodies and bioproducts fractionation
Conclusions
The AEE process described utilized nitrogen-deprived C. reinhardtii as a feedstock for native protein and lipids. Microalgae were initially treated with the in situ-produced enzyme, autolysin, which specifically targeted the glycoprotein-rich C. reinhardtii cell wall. Based on TEM imaging, autolysin disrupted cell walls, leaving only chloroplast membranes partially intact. Protein recovered in the supernatant following autolysin treatment for an extended incubation time at 35°C resulted in extraction of 50 ± 4% of the total extractable protein. Even though cells were highly disrupted, further degradation of cell compounds trapping the lipid bodies was necessary to release lipids from the complex chloroplast membranes.
Availability of data and materials
The datasets generated on this study are available in the figshare repository, https://doi.org/10.6084/m9.figshare.11741619.
Abbreviations
- AEE:
-
Aqueous enzymatic extraction
- AEP:
-
Aqueous enzymatic processing
- DW:
-
Lipid content
- GHGE:
-
Greenhouse gas emissions
- GRAS:
-
Generally recognized as safe
- LDSP:
-
Lipid droplet surface protein
- LED:
-
Light-emitting diode
- LHC:
-
Light-harvesting complex
- MW:
-
Molecular weight
- NICKS:
-
Nanotechnology Innovation Center of Kansas State
- RP:
-
Recombinant protein
- T:
-
Thylakoid
- TAG:
-
Triacylglycerol
- TAP:
-
Tris–acetate–phosphate
- TAP-N:
-
Nitrogen-depleted TAP media
- TEM:
-
Transmission electron microscopy
References
Adam F et al (2012) “Solvent-free” ultrasound-assisted extraction of lipids from fresh microalgae cells: a green, clean and scalable process. Biores Technol 114:457–465
Ahmad N, Mehmood MA, Malik S (2020) Recombinant protein production in microalgae: emerging Trends. Protein Pept Lett 27(2):105–110
Andrews B, Asenjo J (1987) Enzymatic lysis and disruption of microbial cells. Trends Biotechnol 5(10):273–277
Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37(8):911–917
Deshmukh S, Kumar R, Bala K (2019) Microalgae biodiesel: a review on oil extraction, fatty acid composition, properties and effect on engine performance and emissions. Fuel Process Technol 191:232–247
Dumay J et al (2006) Improvement of lipid and phospholipid recoveries from sardine (Sardina pilchardus) viscera using industrial proteases. Process Biochem 41(11):2327–2332
Fan J, Andre C, Xu C (2011) A chloroplast pathway for the de novo biosynthesis of triacylglycerol in Chlamydomonas reinhardtii. FEBS Lett 585(12):1985–1991
Gbogouri GA et al (2006) Analysis of lipids extracted from salmon (Salmo salar) heads by commercial proteolytic enzymes. Eur J Lipid Sci Technol 108(9):766–775
Grebanier AE, Steinback KE, Bogorad L (1979) Comparison of the molecular weights of proteins synthesized by isolated chloroplasts with those which appear during greening in Zea mays. Plant Physiol 63(3):436–439
Halim R, Danquah MK, Webley PA (2012) Extraction of oil from microalgae for biodiesel production: a review. Biotechnol Adv 30(3):709–732
Halim R, Webley PA, Martin GJO (2016) The CIDES process: fractionation of concentrated microalgal paste for co-production of biofuel, nutraceuticals, and high-grade protein feed. Algal Res 19:299–306
Hung N et al (1984) Relative tryptic digestion rates of food proteins. J Food Sci 49(6):1535–1542
Jaenicke L et al (1987) Cell-wall lytic enzymes (autolysins) of Chlamydomonas reinhardtii are (hydroxy)proline-specific proteases. Eur J Biochem 170(1–2):485–491
Jennings RC et al (1981) Effects of trypsin and cations on chloroplast membranes. Plant Physiol 67(2):212–215
Kechaou ES et al (2009) Enzymatic hydrolysis of cuttlefish (Sepia officinalis) and sardine (Sardina pilchardus) viscera using commercial proteases: effects on lipid distribution and amino acid composition. J Biosci Bioeng 107(2):158–164
Kirchhoff H et al (2008) Protein diffusion and macromolecular crowding in thylakoid membranes. Plant Physiol 146(4):1571–1578
Kulkarni S, Nikolov Z (2018) Process for selective extraction of pigments and functional proteins from Chlorella vulgaris. Algal Res 35:185–193
Lam GP et al (2017) Pulsed Electric Field for protein release of the microalgae Chlorella vulgaris and Neochloris oleoabundans. Algal Res 24:181–187
Lauersen KJ (2019) Eukaryotic microalgae as hosts for light-driven heterologous isoprenoid production. Planta 249(1):155–180
Leal-Calderon F, Thivilliers F, Schmitt V (2007) Structured emulsions. Curr Opin Colloid Interface Sci 12(4–5):206–212
Majumdar A et al (2018) Food degradation and foodborne diseases: a microbial approach. Microbial contamination and food degradation. Elsevier, Amsterdam, pp 109–148
Mason CB, Bricker TM, Moroney JV (2006) A rapid method for chloroplast isolation from the green alga Chlamydomonas reinhardtii. Nat Protoc 1(5):2227
Mata TM, Martins AA, Caetano NS (2010) Microalgae for biodiesel production and other applications: a review. Renew Sustain Energy Rev 14(1):217–232
Mathieu-Rivet E et al (2013) Exploring the N-glycosylation pathway in Chlamydomonas reinhardtii unravels novel complex structures. Mol Cell Proteomics 12:3160–3183
Meng X et al (2009) Biodiesel production from oleaginous microorganisms. Renew Energy 34(1):1–5
Moellering ER, Miller R, Benning C (2009) Molecular genetics of lipid metabolism in the model green alga Chlamydomonas reinhardtii. Lipids in photosynthesis. Springer, Berlin, pp 139–155
Morris HJ et al (2008) Utilisation of Chlorella vulgaris cell biomass for the production of enzymatic protein hydrolysates. Biores Technol 99(16):7723–7729
Morris HJ et al (2009) Protein hydrolysates from the alga Chlorella vulgaris 87/1 with potentialities in immunonutrition. Biotecnología Aplicada 26(2):162–165
Olson BJ, Markwell J (2007) Assays for determination of protein concentration. Curr Protoc Protein Sci 48(1):1–3
Patil U, Benjakul S (2019) Use of protease from seabass Pyloric Caeca in combination with repeated freeze-thawing cycles increases the production efficiency of virgin coconut oil. Eur J Lipid Sci Technol 121(5):1800460
Ranjan A, Patil C, Moholkar VS (2010) Mechanistic assessment of microalgal lipid extraction. Ind Eng Chem Res 49(6):2979–2985
Safi C et al (2013) Influence of microalgae cell wall characteristics on protein extractability and determination of nitrogen-to-protein conversion factors. J Appl Phycol 25(2):523–529
Sari YW, Bruins ME, Sanders JP (2013) Enzyme assisted protein extraction from rapeseed, soybean, and microalgae meals. Ind Crops Prod 43:78–83
Sari YW, Sanders JPM, Bruins ME (2016) Techno-economical evaluation of protein extraction for microalgae biorefinery. Iop C Ser Earth Env 31:012034
Schenk PM et al (2008) Ben Hankamer, second generation biofuels: high efficiency microalgae for biodiesel production. Bioenergy Res 1:20–43
Scott SA et al (2010) Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol 21(3):277–286
Senphan T, Benjakul S (2017) Comparative study on virgin coconut oil extraction using protease from hepatopancreas of pacific white shrimp and Alcalase. J Food Process Preserv 41(1):e12771
Smith AK (1972) Soybeans: chemistry and technology. Avi Pub Co., Newyork
Smith P et al (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150(1):76–85
Soto-Sierra L, Kulkarni S, Woodard SL, Nikolov ZL (2020) Processing of permeabilized Chlorella vulgaris biomass into lutein and protein rich products. J Appl Phycol 32:1697–1707
Soto-Sierra L, Dixon CK, Wilken LR (2017) Enzymatic cell disruption of the microalgae Chlamydomonas reinhardtii for lipid and protein extraction. Algal Res 25:149–159
Soto-Sierra L, Stoykova P, Nikolov ZL (2018) Extraction and fractionation of microalgae-based protein products. Algal Res 36:175–192
Tester RF et al (2007) Use of commercial protease preparations to reduce protein and lipid content of maize starch. Food Chem 105(3):926–931
Thiam AR, Farese RV Jr, Walther TC (2013) The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol 14(12):775–786
Waniska RD, Kinsella JE (1979) Foaming properties of proteins—evaluation of a column aeration apparatus using ovalbumin. J Food Sci 44(5):1398
Wilken LR, Nikolov ZL (2016) Aqueous fractionation of dry-milled corn germ for food protein production. Emerging and traditional technologies for safe, healthy and quality Food. Springer, Berlin, pp 443–461
Wu C et al (2017) Aqueous enzymatic process for cell wall degradation and lipid extraction from Nannochloropsis sp. Biores Technol 223:312–316
Yap BHJ et al (2014) A mechanistic study of algal cell disruption and its effect on lipid recovery by solvent extraction. Algal Res 5:112–120
Tan SC, Yiap BC (2009) DNA, RNA, and protein extraction: the past and the present. J Biomed Biotechnol. https://doi.org/10.1155/2009/574398
Zerges W, Rochaix J-D (1998) Low density membranes are associated with RNA-binding proteins and thylakoids in the chloroplast of Chlamydomonas reinhardtii. J Cell Biol 140(1):101–110
Zheng H et al (2011) Disruption of Chlorella vulgaris cells for the release of biodiesel-producing lipids: a comparison of grinding, ultrasonication, bead milling, enzymatic lysis, and microwaves. Appl Biochem Biotechnol 164(7):1215–1224
Acknowledgements
The authors thank Dr. Brad Olson (Department of Biology, Kansas State University) for providing the algae mating types for autolysin production and Dr. Donghai Wang (Department of Biological and Agricultural Engineering, Kansas State University) for providing use of laboratory equipment.
Funding
This material is based upon work supported by the National Science Foundation under Award No. EPS‐0903806 and matching support from the State of Kansas through the Kansas Board of Regents.
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions in conceptualizing, drafting, developing and reviewing the manuscript. The paper was reviewed and approved by all authors prior to submission for peer review.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Soto-Sierra, L., Wilken, L.R. & Dixon, C.K. Aqueous enzymatic protein and lipid release from the microalgae Chlamydomonas reinhardtii. Bioresour. Bioprocess. 7, 46 (2020). https://doi.org/10.1186/s40643-020-00328-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-020-00328-4