Development and Characterization of Orally Disintegrating Tablets Containing a Captopril-Cyclodextrin Complex

 ,
,  , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Inclusion Complex
2.3. Physical–Chemical Characterization
2.4. Development of Pharmaceutical Formulation
2.5. Study of the Release Kinetics of CAP from the Tablets
2.6. Taste Evaluation
2.7. Applied Release Theoretical Models
3. Results
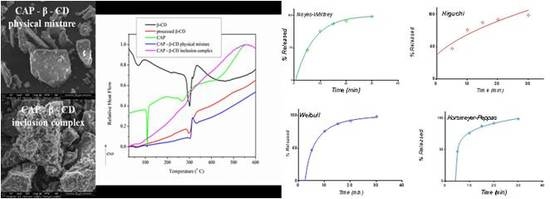
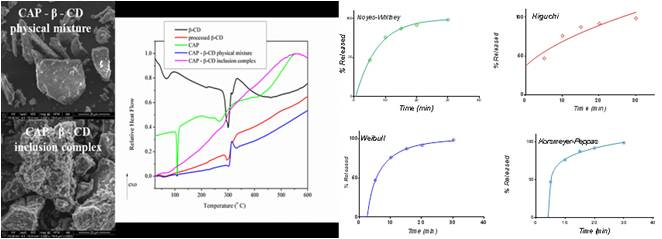
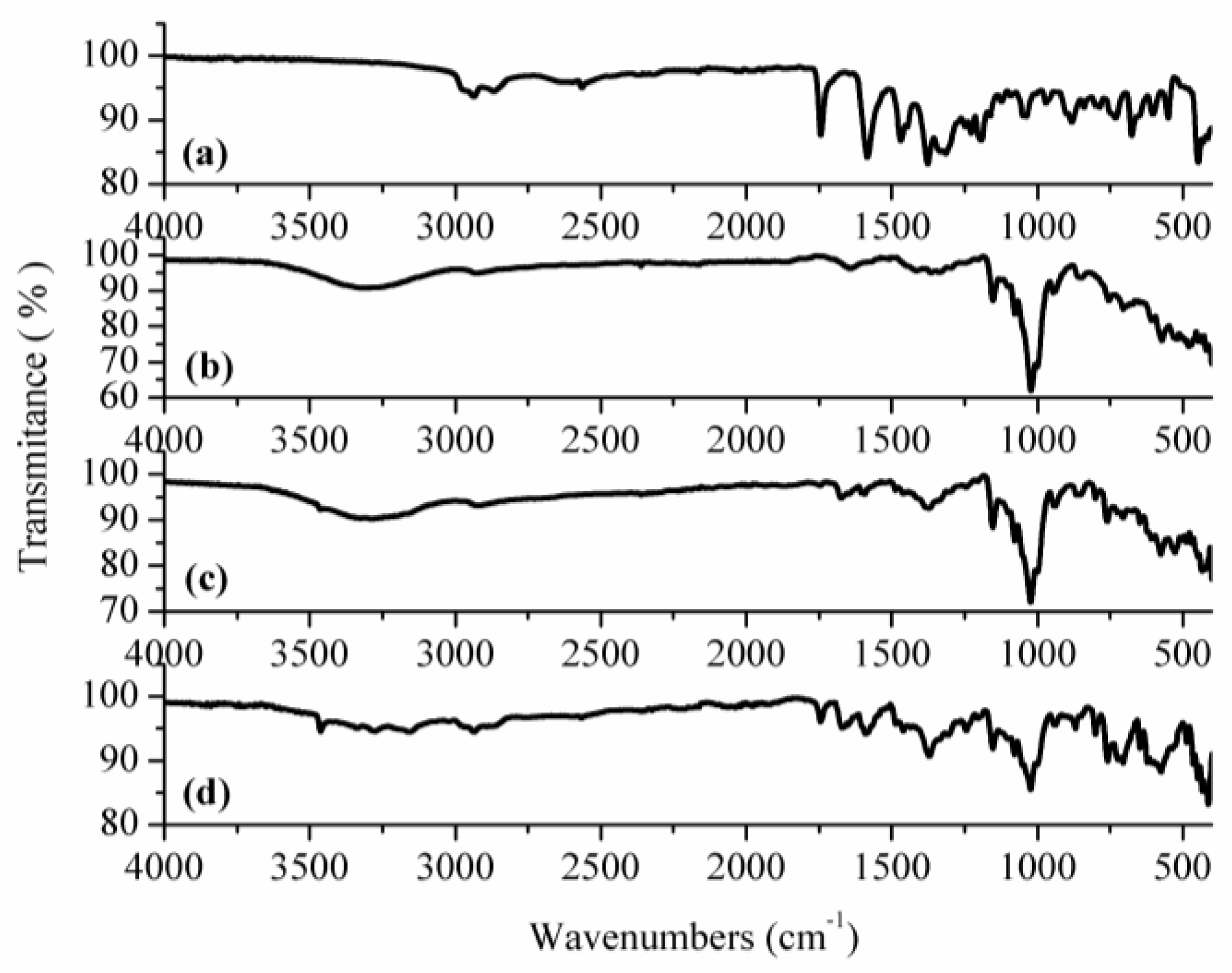
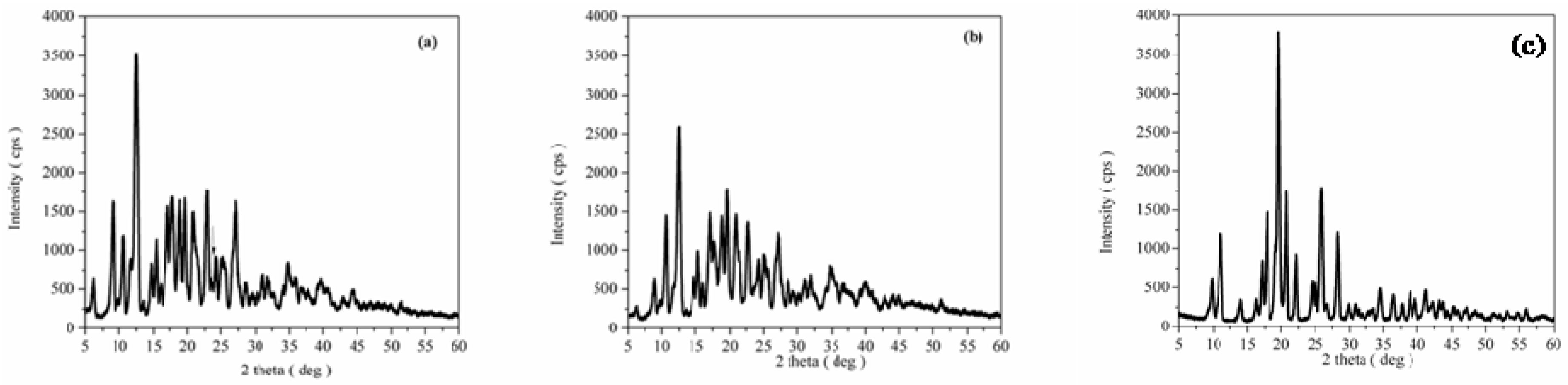
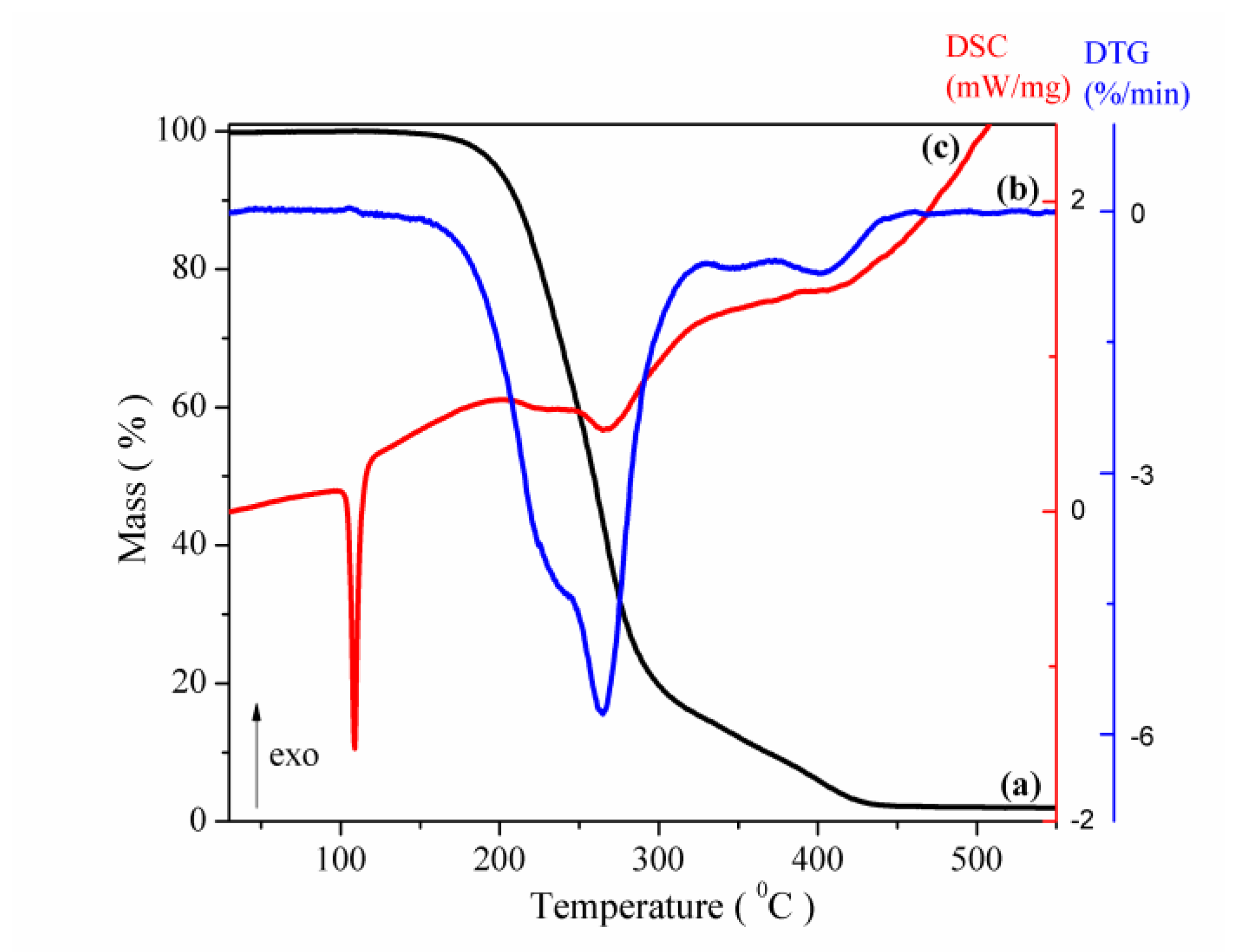
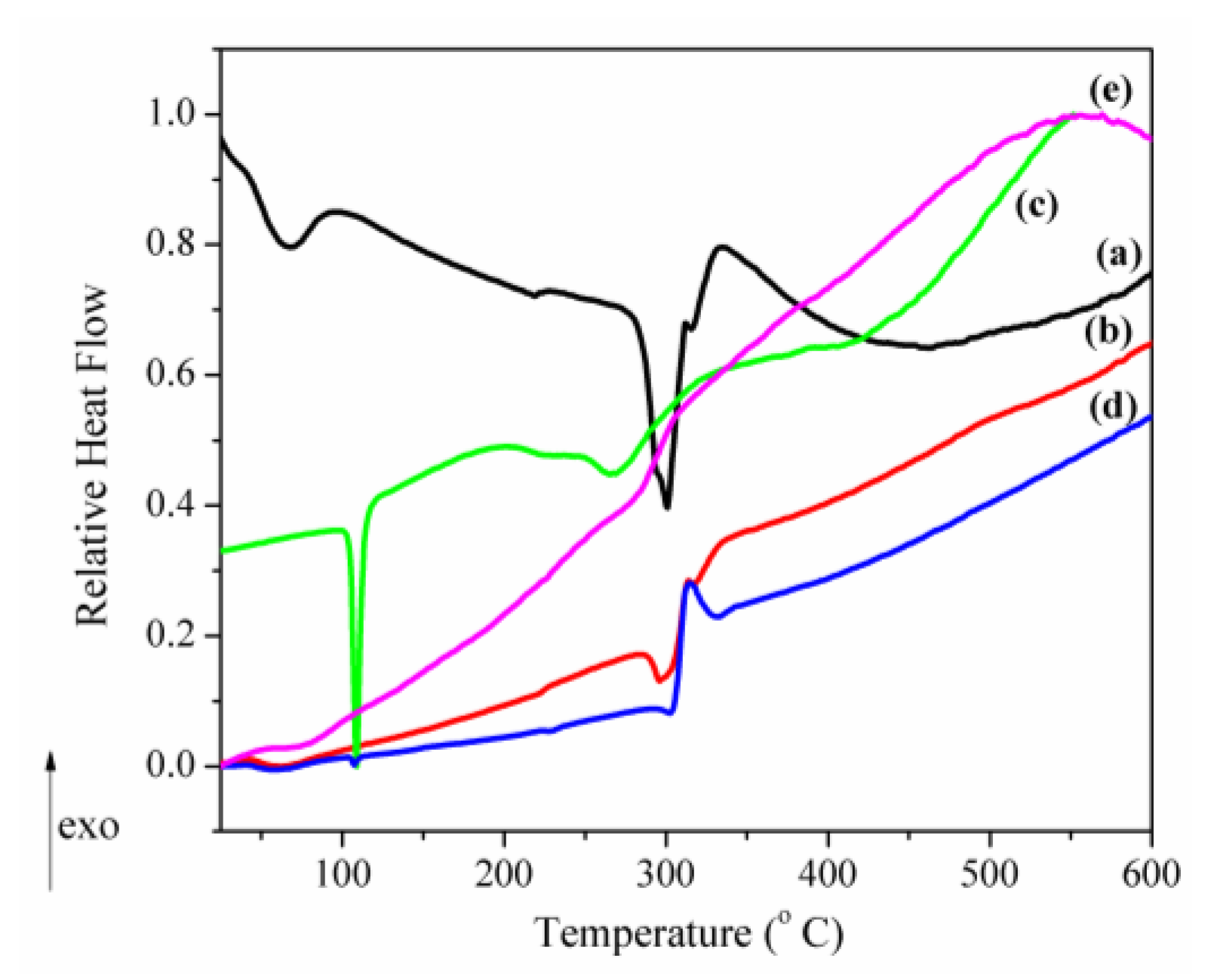
3.1. Structural, Morphological and Thermal Properties of Compounds
- (i)
- The marked attenuation (in the case of the physical mixture) or absence (in the case of the inclusion complex) of a CAP-melting endotherm. This is a sign that a consistent amount of CAP is included into the β-CD cavity, even within the 15-min mortar mixing procedure, the process being complete for the inclusion complex. The above assumption is also supported by the lower mass loss associated with “cavity-included” water in the case of the physical mixture, compared to the pure cyclodextrin.
- (ii)
- The decrease of the β-CD phase transition endotherm for both processed β-CD and its physical mixture with CAP and the absence of this endotherm for the inclusion complex.
- (iii)
- The decrease of the endo-exothermogram feature in the order β-CD > processed β-CD > physical mixture > inclusion complex. This is the thermal signature associated with the melting-decomposition of cyclodextrine; in part, the latter process takes place on behalf of the provided intramolecular oxygen. In the case of the physical mixture, this cyclodextrine oxygen may act on the CAP component. In the case of the inclusion complex, this endo-exo feature is severely attenuated.
3.2. Release Kinetics
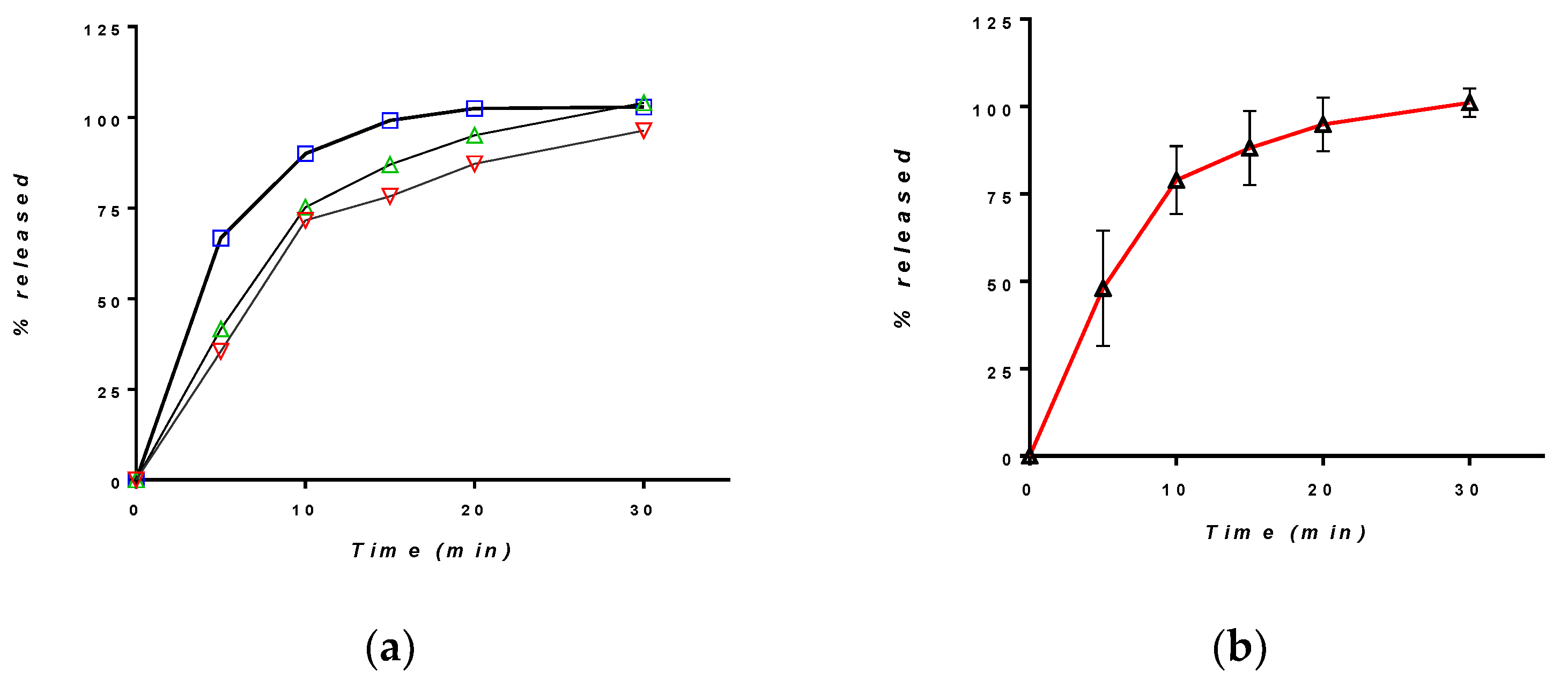
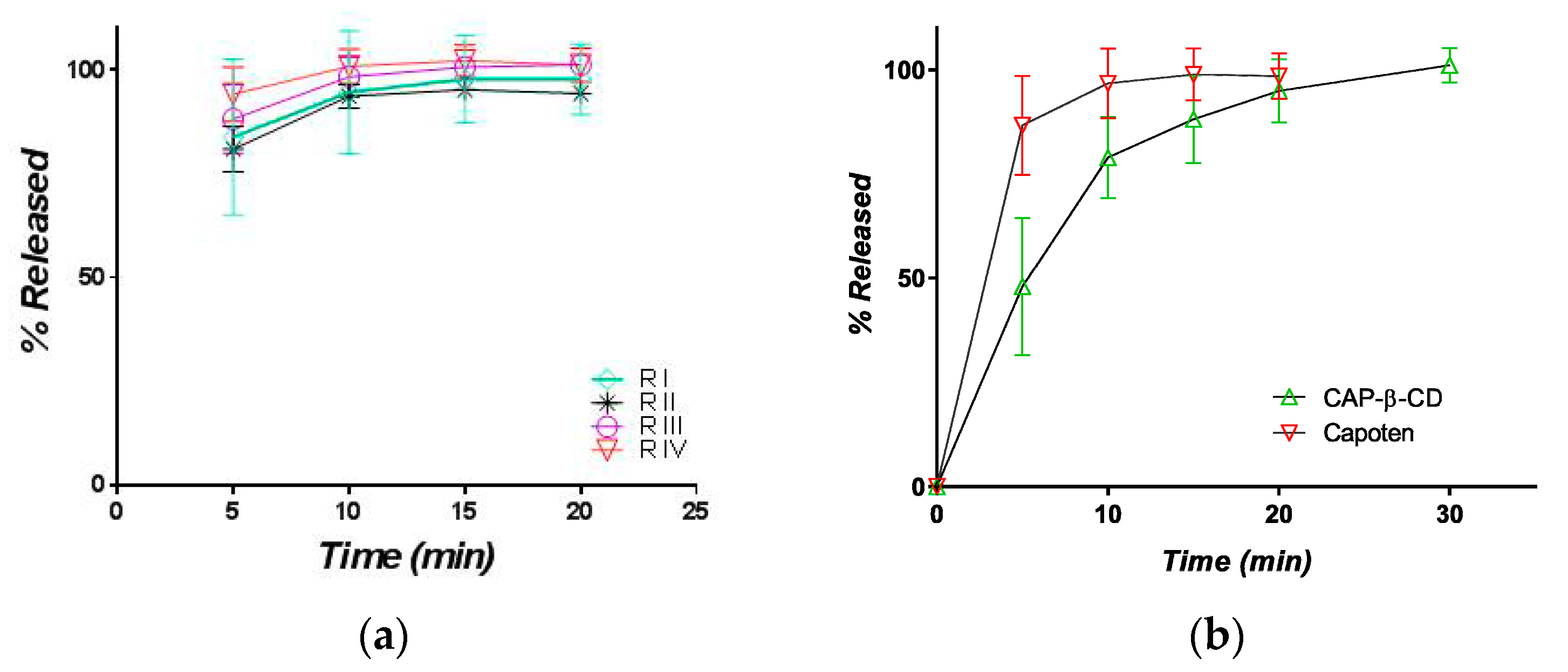
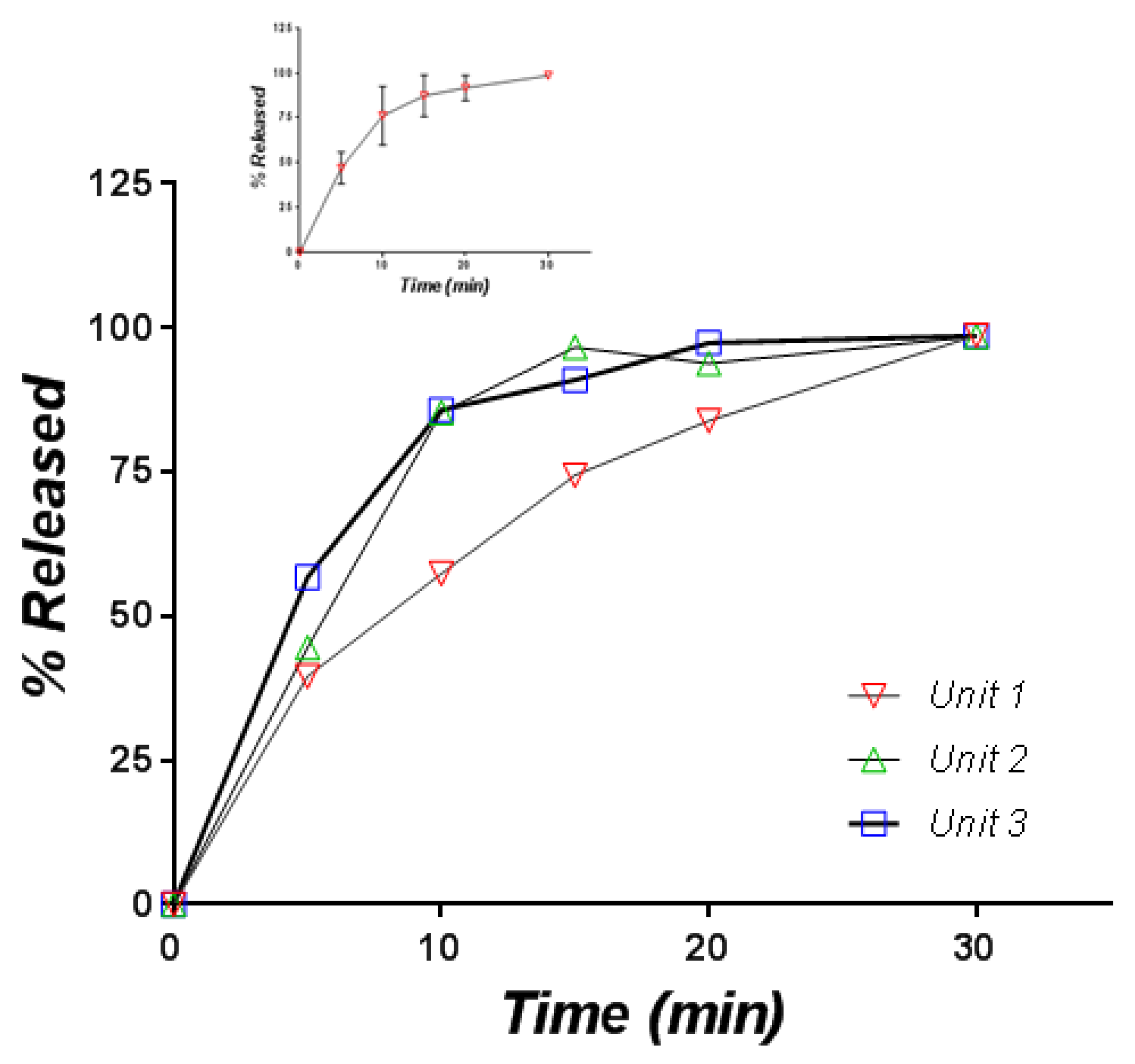
3.2.1. Release in Compendial Medium
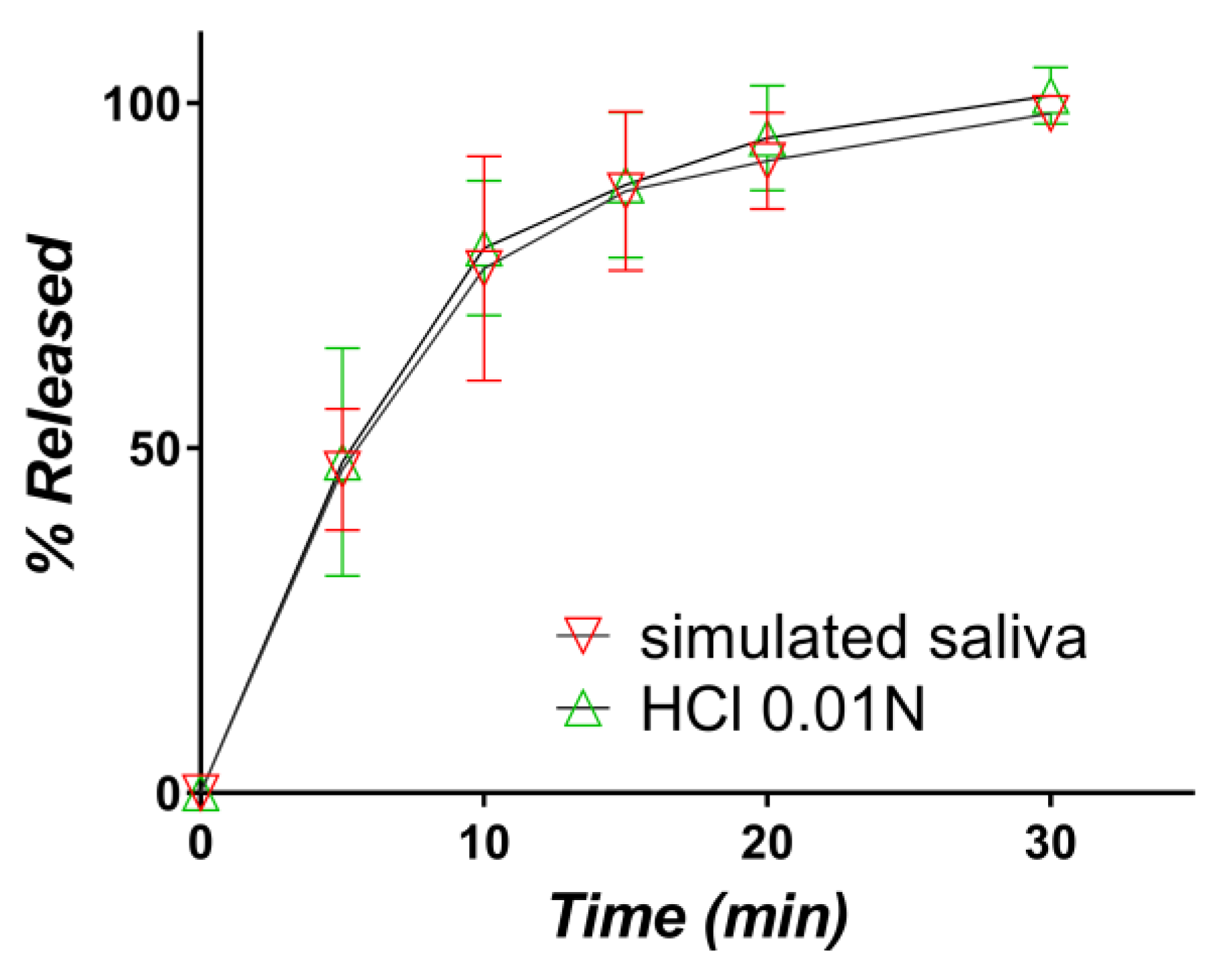
3.2.2. Release in Simulated Saliva
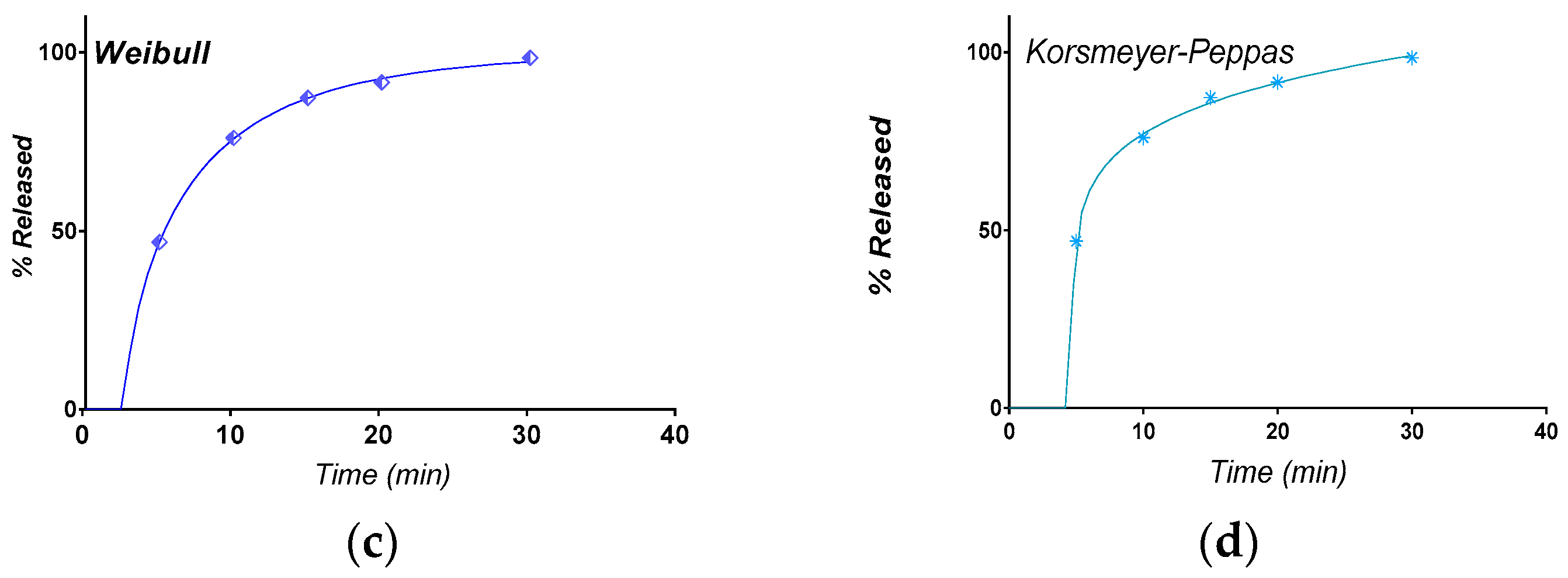
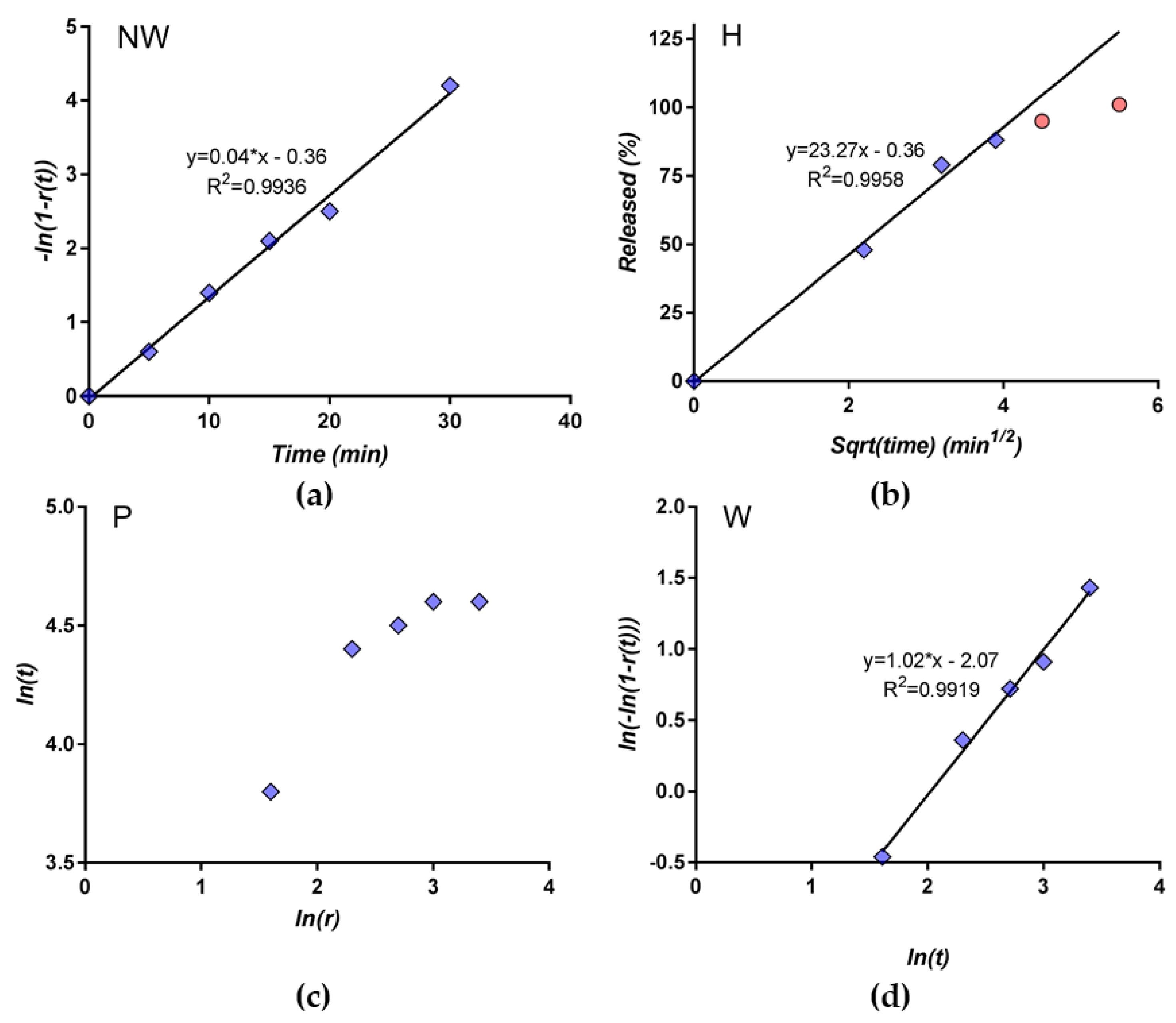
3.3. Selection of the Most Appropriate Mathematical Model
3.4. Comparison of Release Kinetics Models
3.5. Evaluation of Taste
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Duchin, K.L.; McKinstry, D.N.; Cohen, A.I.; Migdalof, B.H. Pharmacokinetics of captopril in healthy subjects and in patients with cardiovascular diseases. Clin. Pharmacokinet. 1988, 14, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.T.; Wang, S.L.; Lin, S.Y. Solid-state interaction study on the captopril/lubricants systems accelerated by grinding process. J. Phys. Chem. Sol. 2008, 69, 1007–1016. [Google Scholar] [CrossRef]
- Nur, A.O.; Zhang, J.S. Captopril floating and/or bioadhesive tablets: Design and release kinetics. Drug Dev. Ind. Pharm. 2000, 26, 965–969. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.M.; Tam, Y.K. Stability of captopril in tap water. Am. J. Heal. Syst. Pharm. 1992, 49, 612–615. [Google Scholar] [CrossRef]
- Timmins, P.; Jackson, I.M.; Wang, Y.-C.J. Factors affecting captopril stability in aqueous solution. Int. J. Pharm. 1982, 11, 329–336. [Google Scholar] [CrossRef]
- Hu, M.; Amidon, G.L. Passive and carrier-mediated intestinal absorption components of captopril. J. Pharm. Sci. 1988, 77, 1007–1011. [Google Scholar] [CrossRef]
- Hussain, N.; Nazir, T.; Ojike, F.; Lillibridge, D. Archives of Disease in Childhood: The Journal of the Royal College of Paediatrics and Child. Health; Factors affecting the oxidative degradation of captopril in oral liquid pediatric formulations: A Factorial Design Study; BMJPubl. Group: London, UK, 2013; Volume 98, p. 1136. [Google Scholar]
- Wagh, M.P.; Patel, J. Biopharmaceutical classification system: Scientific basis for biowaiver extensions. Int. J. Pharm. Pharm. Sci. 2010, 2, 12–19. [Google Scholar]
- Jardine, M.; Miles, A.; Allen, J. Dysphagia onset in older adults during unrelated hospital admission: Quantitative videofluoroscopic measures. Geriatrics 2018, 3, 66. [Google Scholar] [CrossRef] [Green Version]
- Kazerani, H.; Hajimoradi, B.; Amini, A.; Naseri, M.; Moharamzad, Y. Clinical efficacy of sublingual captopril in the treatment of hypertensive urgency. Singap. Med. J. 2009, 50, 400–402. [Google Scholar]
- Alipour, S.; Mohammadi, A.; Ahmadi, F. Captopril fast disintegrating tablets for children: Formulation and quality control by HPLC. Trends Pharm. Sci. 2017, 3, 161–168. [Google Scholar]
- Shahtalebi, M.A.; Tabbakhian, M.; Harandi, N.S. Formulation and evaluation of orally disintegrating tablets of captopril using natural super disintegrants. J. Rep. Pharm. Sci. 2014, 3, 54–64. [Google Scholar]
- Zaky, A.A.; Elewah, H. Design and optimization of captopril sublingual tablets: Enhancement of pharmacokinetic parameters in human. Al-Azhar, J. Pharm. Sci. 2016, 53, 90–107. [Google Scholar]
- Pabari, R.M.; McDermott, C.; Barlow, J.; Ramtoola, Z. Stability of an alternative extemporaneous captopril fast-dispersing tablet formulation versus an extemporaneous oral liquid formulation. Clin. Ther. 2012, 34, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Conceição, J.; Adeoye, O.; Cabral-Marques, H.M.; Lobo, J.M.S. Cyclodextrins as excipients in tablet formulations. Drug Discov. Today 2018, 23, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- de Burgos, M.; de Azevedo, M.; Tasic, L.; Fattori, J.; Rodrigues, F.H.; Cantos, F.C.; Ribeiro, L.P.; de Paula, V.; Ianzer, D.; Santos, R.A. New formulation of an old drug in hypertension treatment: The sustained release of captopril from cyclodextrin nanoparticles. Int. J. Nanomed. 2011, 6, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Hirayama, K.F.; Arima, H.; Uekama, K. Controlled release of a water soluble drug, captopril, by a combination of hydrophilic and hydrophobic derivatives. J. Control. Release 2000, 66, 271–280. [Google Scholar] [CrossRef]
- Ikeda, Y.; Motoune, S.; Matsuoka, T.; Arima, H.; Hirayama, F.; Uekama, K. Inclusion complex formation of captopril with alpha- and beta-cyclodextrins in aqueous solution: NMR spectroscopic and molecular dynamic studies. J. Pharm. Sci. 2002, 91, 2390–2398. [Google Scholar] [CrossRef]
- USP. The United States Pharmacopeia 32—The National Formulary 27; The United States Pharmacopeial Convention, Inc.: Rockville, MD, USA, 2009. [Google Scholar]
- Gohel, M.C.; Parikh, R.K.; Aghara, P.Y.; Nagori, S.A.; Delvadia, R.R.; Dabhi, M.R. Application of simplex lattice design and desirability function for the formulation development of mouth dissolving film of salbutamol sulphate. Curr. Drug Deliv. 2009, 6, 486–494. [Google Scholar] [CrossRef]
- Marques, M.R.; Loebenberg, R.; Almukainzi, M. Simulated biological fluids with possible application in dissolution testing. Dissolution Technol. 2011, 18, 15–28. [Google Scholar] [CrossRef]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J. Pharm. Sci. 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Siepmann, J.; Peppas, N. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Weibull, W. A Statistical Distribution Function of Wide Applicability. J. Appl. Mech. 1951, 18, 293–297. [Google Scholar]
- Vasilatou-Kosmidis, H. Cancer in neonates and infants. Med. Pediatr. Oncol. 2003, 41, 7–9. [Google Scholar] [CrossRef]
- Papadopoulou, V.; Kosmidis, K.; Vlachou, M.; Macheras, P. On the use of the Weibull function for the discernment of drug release mechanisms. Int. J. Pharm. 2006, 309, 44–50. [Google Scholar] [CrossRef]
- Stulzer, H.K.; Rodrigues, P.O.; Cardoso, T.M.; Matos, J.S.R.; Silva, M.A.S. Compatibility studies between captopril and pharmaceutical excipients used in tablets formulations. J. Therm. Anal. Calorim. 2008, 91, 323–328. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, Y.; Alexander, K.; Dollimore, D. The thermal analysis study of the drug captopril. Thermochim. Acta 2001, 367, 43–58. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1753. [Google Scholar] [CrossRef]
- Qiu, N.; Cheng, X.; Wang, G.; Wang, W.; Wen, J.; Zhang, Y.; Song, H.; Ma, L.; Wei, Y.; Peng, A.; et al. Inclusion complex of barbigerone with hydroxypropyl-β-cyclodextrin: Preparation and in vitro evaluation. Carbohydr. Polym. 2014, 101, 623–630. [Google Scholar] [CrossRef]
- Francesca, M.B.; Mura, M.P. Physical chemical characterization of binary systems of prilocaine hydrochloride with triacetyl-β–cyclodextrin. J. Incl. Phenom. Macrocycl. Chem. 2010, 68, 437–445. [Google Scholar] [CrossRef]
- Diogo, H.P.; Moura Ramos, J.J. The slow molecular mobility in amorphous captopril and captopril disulfide: A DSC and TSDC study. J. Non-Cryst. Solids 2015, 429, 202–207. [Google Scholar] [CrossRef]
- Sailaja, U.; Thayyil, M.S. Dielectric and Spectroscopic Investigations of Amorphous Captopril. IOSR J. Pharm. 2012, 2, 479–484. [Google Scholar] [CrossRef]
- Macedo, R.O.; Nascimento, T.G.; Aragão, C.F.S.; Gomes, A.P.B. Application of the thermal analysis in the caracterization of anti-hypertensive drugs. J. Therm. Anal. Calorim. 2000, 59, 657–661. [Google Scholar] [CrossRef]
- Bazzo, G.C.; Silva, M.A.S. Estudotermoanalítico de comprimidosrevestidoscontendo captopril através de termogravimetria (TG) e calorimetriaexploratóriadiferencial (DSC). Braz. J. Pharm. Sci. 2005, 41, 315–322. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, S. Preparation and characterization of inclusion complexes of prazosin hydrochloride with β-cyclodextrin and hydroxypropyl-β-cyclodextrin. J. Pharm. Biomed. Anal. 2006, 40, 122–127. [Google Scholar] [CrossRef]
- Yllmaz, V.T.; Karada, A.; Jgbudak, H. Thermal decomposition of β-cyclodextrin inclusion complexes of ferrocene and their derivatives. Thermochim. Acta 1995, 261, 107–118. [Google Scholar] [CrossRef]
- McClelland, C.A.; Stubbs, R.J.; Fix, J.A.; Pogany, S.A.; Zentner, G.M. Enhancement of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG CoA) reductase inhibitor efficacy through administration of a controlled-porosity osmotic pump dosage form. Pharm. Res. 1991, 8, 873–876. [Google Scholar] [CrossRef]
- Perdomo-López, I.; Rodríguez-Pérez, A.I.; Yzquierdo-Peiro, J.M.; White, A.; Estrada, E.G.; Villa, T.G.; Torres-Labandeira, J. Effect of cyclodextrins on the solubility and antimycotic activity of sertaconazole: Experimental and computacional studies. J. Pharm. Sci. 2002, 91, 2408–2415. [Google Scholar] [CrossRef]
- Esclusa-Dıaz, M.; Torres-Labandeira, J.; Kata, M.; Vila-Jato, J. Inclusion complexation of glibenclamide with 2-hydroxypropyl-b-cyclodextrin in solution and in solid state. Eur. J. Pharm. Sci. 1994, 1, 291–296. [Google Scholar] [CrossRef]
- Neama, M.H. Preparation and Evaluation of Orally Disintegrating Tablets of Captopril. Int. J. Chem. Life Sci. 2013, 2, 1213–1217. [Google Scholar]
- Imtiyaz Ali, S.T.; Jahangir, M.A.; Mazher Ahmed, M.D.; Muheem, A.; Shahab, M.A.; Bhavani, P.D.; Saleem, M.A. Development and in vitro evaluation of mouth dissolving films of captopril. Int. Res. J. Pharm. 2015, 6, 760–764. [Google Scholar] [CrossRef]
- Sunitha, H.S.; Parthiban, S.; Vikneshwiri, A.; SenthilKumar, G.P.; Tamiz Mani, T. Development and evaluation of captopril fast disintegrating or dissolving tablets by complexation techniques using guar Gum as a superdisintegrant. Int. J. Res. Pharm. Nano Sci. 2015, 4, 72–84. [Google Scholar]
- Akaike, H. Likelihood of a model and information criteria. J. Econom. 1981, 16, 3–14. [Google Scholar] [CrossRef]
- Akaike, H. Selected Papers of HirotuguAkaike; Information theory and an extension of the maximum likelihood principle; Springer: Berlin/Heidelberg, Germany, 1998; pp. 199–213. [Google Scholar]
- Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical modelling of release kinetics from supramolecular drug delivery systems. Pharmaceutics 2019, 11, 140. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Robinson, D.H. Controlled Release Kinetics of captopril from tableted microcapsules. Drug Dev. Ind. Pharm. 1988, 14, 545–560. [Google Scholar] [CrossRef]
- Preda, I.A.; Mircioiu, I.; Mircioiu, C.; Corlan, G.; Pahomi, G.; Prasacu, I.; Anuta, V. Research concerning the development of a biorelevant dissolution test for formulations containing norfloxacin. I. Modelling of in vitro release kinetics. Farmacia 2012, 60, 675–687. [Google Scholar]
- Mircioiu, I.; Anuta, V.; Purcaru, S.O.; Radulescu, F.; Miron, D.; Dumitrescu, I.B.; Ibrahim, N.; Mircioiu, C. In vitro dissolution of poorly soluble drugs in the presence of surface active agents-in vivo pharmacokinetics correlations. II. Nimesulide. Farmacia 2013, 61, 88–102. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Quantity (mg/tb) | Function |
|---|---|---|
| CAP-β-CD (1:2) complex | 286.0 | active ingredient |
| F-MELT®® | 184.0 | filler, superdisintegrant |
| CompactCel®® TC | 25.0 | dry binder |
| Magnesium stearate | 5.00 | lubricant |
| Total | 500.0 |
| Parameter | Order I | Higuchi | Korsmeyer-Peppas | Weibull |
|---|---|---|---|---|
| K | k1 = 0.143 | kH = 18.154 | kKP = 59.120 | a = 2.934 |
| Tlag | 0.487 | ‒4.285 | 4.765 | Ti = 2.614 |
| n = 0.160 | β = 0.713 | |||
| N_obs | 5 | 5 | 5 | 5 |
| Degrees of Freedom | 3 | 3 | 2 | 2 |
| Observed correlation coefficient R | 0.997 | 0.922 | 0.998 | 0.999 |
| R2 | 0.994 | 0.849 | 0.997 | 0.998 |
| R2 adjusted | 0.993 | 0.798 | 0.995 | 0.996 |
| Mean Square Error | 2.83 | 82.45 | 1.73 | 1.32 |
| Sum of squared errors | 8.49 | 247.36 | 3.46 | 2.64 |
| AIC | 14.69 | 31.55 | 12.21 | 10.86 |
| Bitter Taste | Grittiness | Residual Taste | Other Taste | Acceptance | Sulfurous Taste | |
|---|---|---|---|---|---|---|
| CAP | 4 | 3 | 4 | Burn metallic | 1 | 4 |
| CAP-β-CD | 1 | 1 | 2 | Sweet | 4 | 1 |
| CAP | 3 | 1 | 4 | Burn | 1 | 3 |
| CAP-β-CD | 1 | 3 | 2 | Sweet | 3 | 1 |
| CAP | 4 | 2 | 4 | Burn metallic | 1 | 3 |
| CAP-β-CD | 1 | 2 | 2 | Sweet | 3 | 1 |
| CAP | 3 | 3 | 4 | burn | 2 | 2 |
| CAP-β-CD | 1 | 2 | 2 | sweet | 3 | 1 |
| CAP | 2 | 3 | 3 | metallic | 3 | 1 |
| CAP-β-CD | 1 | 3 | 2 | metallic | 4 | 1 |
| CAP | 3 | 3 | 5 | metallic | 2 | 1 |
| CAP-β-CD | 1 | 2 | 2 | sweet | 5 | 1 |
| CAP | 5 | 3 | 4 | pricking | 1 | 2 |
| CAP-β -CD | 1 | 1 | 1 | - | 4 | 1 |
| CAP | 4 | 4 | 4 | astringent | 2 | 1 |
| CAP-β-CD | 1 | 2 | 2 | - | 3 | 1 |
| CAP | 4 | 3 | 4 | astringent | 2 | 2 |
| CAP-β -CD | 1 | 2 | 2 | - | 4 | 1 |
| CAP | 3 | 2 | 3 | metallic | 2 | 1 |
| CAP-β-CD | 1 | 2 | 3 | - | 4 | 1 |
| CAP | 5 | 3 | 4 | burn | 1 | 3 |
| CAP-β-CD | 1 | 2 | 2 | metallic | 4 | 1 |
| MEAN CAP | 3.64 | 2.73 | 3.91 | - | 1.64 | 2.09 |
| MEAN CAP-β-CD | 1.00 | 2.00 | 2.00 | - | 3.73 | 1.00 |
| MEDIAN CAP | 4 | 2 | 4 | - | 3 | 2 |
| MEDIAN CAP-β-CD | 1 | 4 | 2 | - | 2 | 1 |
| Paired t-test, p< | 0.000001 | 0.035 | 0.000009 | - | 0.000004 | 0.003 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musuc, A.M.; Anuta, V.; Atkinson, I.; Popa, V.T.; Sarbu, I.; Mircioiu, C.; Abdalrb, G.A.; Mitu, M.A.; Ozon, E.A. Development and Characterization of Orally Disintegrating Tablets Containing a Captopril-Cyclodextrin Complex. Pharmaceutics 2020, 12, 744. https://doi.org/10.3390/pharmaceutics12080744
Musuc AM, Anuta V, Atkinson I, Popa VT, Sarbu I, Mircioiu C, Abdalrb GA, Mitu MA, Ozon EA. Development and Characterization of Orally Disintegrating Tablets Containing a Captopril-Cyclodextrin Complex. Pharmaceutics. 2020; 12(8):744. https://doi.org/10.3390/pharmaceutics12080744
Chicago/Turabian StyleMusuc, Adina Magdalena, Valentina Anuta, Irina Atkinson, Vlad Tudor Popa, Iulian Sarbu, Constantin Mircioiu, Ghaleb Abdalameer Abdalrb, Mirela Adriana Mitu, and Emma Adriana Ozon. 2020. "Development and Characterization of Orally Disintegrating Tablets Containing a Captopril-Cyclodextrin Complex" Pharmaceutics 12, no. 8: 744. https://doi.org/10.3390/pharmaceutics12080744