A Synonymous Exonic Splice Silencer Variant in IRF6 as a Novel and Cryptic Cause of Non-Syndromic Cleft Lip and Palate

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Editorial Policies and Ethical Considerations
2.2. Patient Recruitment and Clinical Evaluation
2.3. Samples and DNA Extraction
2.4. Whole Exome Sequencing
2.5. Mutation Screening and Sanger Confirmation
2.6. In Silico Analysis
2.7. Minigene Splice Assay
- The c.921C>T variant was introduced by overlap PCR with high fidelity polymerase and the following primer sets: hIRF6-I6 forward and hIRF6-synon reverse (5′-CATGACCACTGACCTCCAGGATCAG-3′), and hIRF6-synon forward (5′-GGAGGTCAGTGGTCATGCCATTTATG-3′) and hIRF-I7 reverse. All constructs were verified by sequencing.
- Wildtype and mutant vectors were transfected into COS7 cells (CRL-1651, ATCC) by Lipofectamine 3000 (Thermo Fisher Scientific) according to the manufacturer’s protocol (n = 3 per group). Total RNA was extracted two days post transfection using QIAshredder and an RNeasy Micro Kit (Qiagen) and then cDNA was synthesized using Superscript IV (Thermo Fisher Scientific). To detect splicing patterns, PCR was carried out using pET01-ETPR04 forward (5′-GGATTCTTCTACACACCC-3′) and pET01-ETPR05 reverse (5′-GTTGACCTCGACCCACCT-3′). As a control for DNA contamination, PCR was also performed using the RNA as a template. An aliquot of each PCR was separated on a 1.2% agarose gel, stained with ethidium bromide, and the gel image capture on an C400 imager (Azure Biosystems, Dublin, CA, USA) for the quantification of individual bands. Quantification was performed in the linear range using the Azure software and manually adjusted for band size. The remainder of the amplification reaction was run on a preparative 1.2% agarose gel and the individual products were extracted using a Qiagen gel extraction kit (Qiagen) and then Sanger sequenced (Genewiz, South Plainfield, NJ, USA) using the amplifying primers.
3. Results
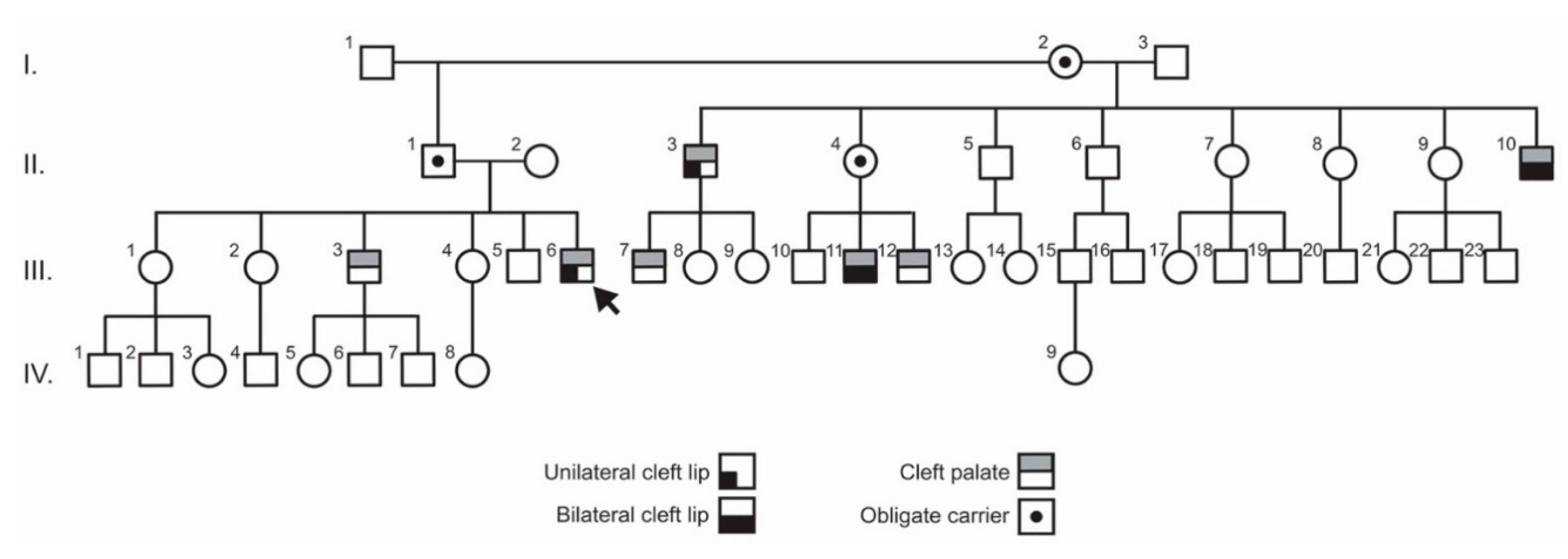
3.1. Clinical Findings
3.2. Exome Sequencing and Variant Analysis
3.3. Functional Testing of the IRF6 Variant
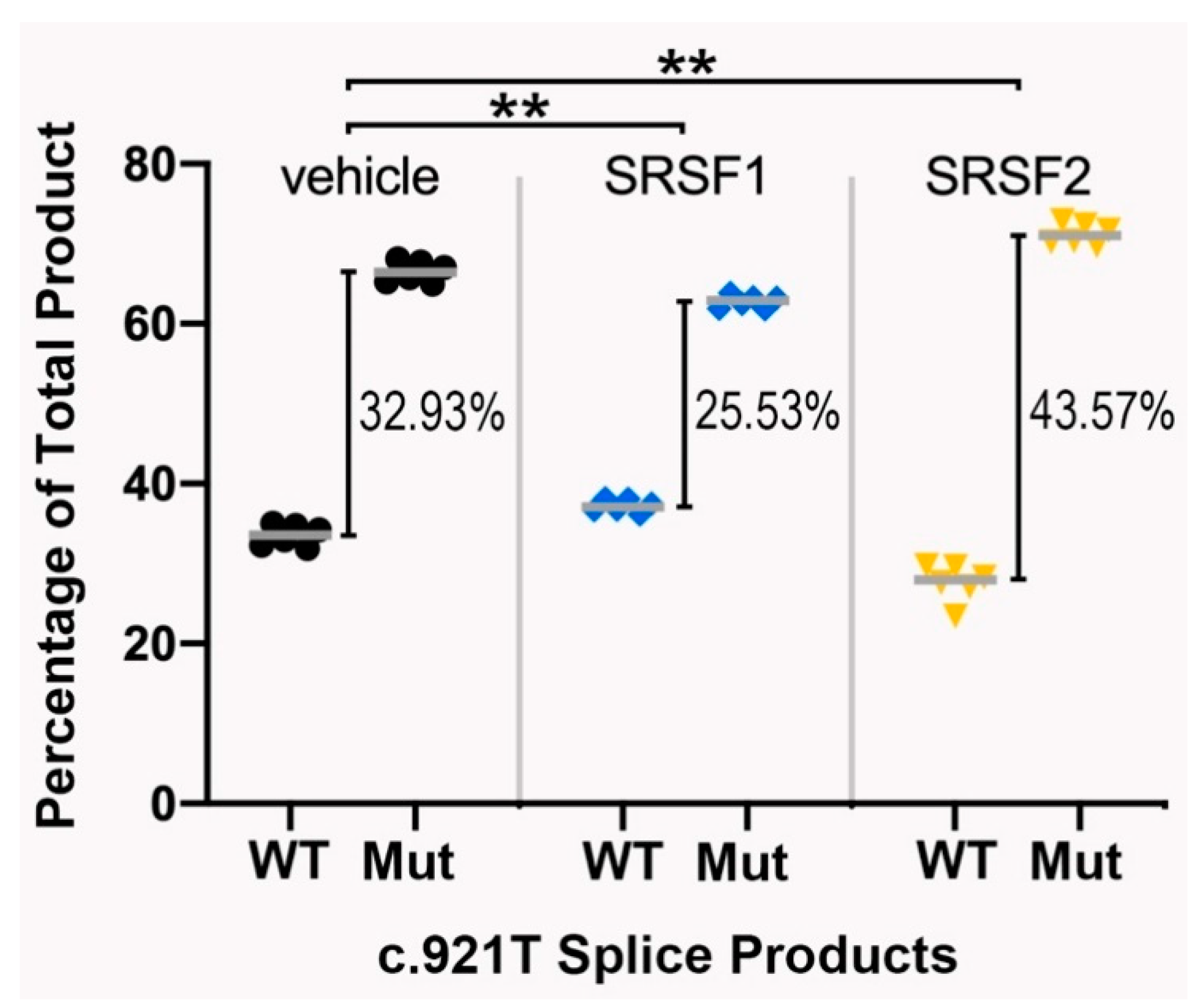
3.4. The Splicing Pattern Is Affected by Ectopic Expression of Major Splice Regulatory Factors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Fraser, F.C. Thoughts on the etiology of clefts of the palate and lip. Acta Genet. Stat. Med. 1955, 5, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Schutte, B.C.; Richardson, R.J.; Bjork, B.C.; Knight, A.S.; Watanabe, Y.; Howard, E.; de Lima, R.L.; Daack-Hirsch, S.; Sander, A.; et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat. Genet. 2002, 32, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Celli, J.; Duijf, P.; Hamel, B.C.; Bamshad, M.; Kramer, B.; Smits, A.P.; Newbury-Ecob, R.; Hennekam, R.C.; Van Buggenhout, G.; van Haeringen, A.; et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999, 99, 143–153. [Google Scholar] [CrossRef] [Green Version]
- McGrath, J.A.; Duijf, P.H.; Doetsch, V.; Irvine, A.D.; de Waal, R.; Vanmolkot, K.R.; Wessagowit, V.; Kelly, A.; Atherton, D.J.; Griffiths, W.A.; et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum. Mol. Genet. 2001, 10, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Dode, C.; Levilliers, J.; Dupont, J.M.; De Paepe, A.; Le Du, N.; Soussi-Yanicostas, N.; Coimbra, R.S.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F.; et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef] [Green Version]
- Van den Boogaard, M.J.; Dorland, M.; Beemer, F.A.; van Amstel, H.K. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat. Genet. 2000, 24, 342–343. [Google Scholar] [CrossRef]
- Neiswanger, K.; Deleyiannis, F.W.; Avila, J.R.; Cooper, M.E.; Brandon, C.A.; Vieira, A.R.; Noorchashm, N.; Weinberg, S.M.; Bardi, K.M.; Murray, J.C.; et al. Candidate genes for oral-facial clefts in Guatemalan families. Ann. Plast. Surg. 2006, 56, 518–521. [Google Scholar] [CrossRef]
- Neiswanger, K.; Walker, K.; Klotz, C.M.; Cooper, M.E.; Bardi, K.M.; Brandon, C.A.; Weinberg, S.M.; Vieira, A.R.; Martin, R.A.; Czeizel, A.E.; et al. Whorl patterns on the lower lip are associated with nonsyndromic cleft lip with or without cleft palate. Am. J. Med. Genet. Part A 2009, 149a, 2673–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, E.J.; Marazita, M.L. Genetics of cleft lip and cleft palate. Am. J. Med. Genet. C Semin Med. Genet. 2013, 163, 246–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyce, J.O.; Raj, S.; Sanchez, K.; Marazita, M.L.; Morgan, A.T.; Kilpatrick, N. Speech Phenotyping in Unaffected Family Members of Individuals With Nonsyndromic Cleft Lip With or Without Palate. Cleft Palate Craniofac J. 2019, 56, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Seto-Salvia, N.; Stanier, P. Genetics of cleft lip and/or cleft palate: Association with other common anomalies. Eur. J. med. Genet. 2014, 57, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Ghassibe, M.; Bayet, B.; Revencu, N.; Verellen-Dumoulin, C.; Gillerot, Y.; Vanwijck, R.; Vikkula, M. Interferon regulatory factor-6: A gene predisposing to isolated cleft lip with or without cleft palate in the Belgian population. Eur. J. Hum. Genet. EJHG 2005, 13, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; McIntosh, I.; Hetmanski, J.B.; Jabs, E.W.; Vander Kolk, C.A.; Wu-Chou, Y.H.; Chen, P.K.; Chong, S.S.; Yeow, V.; Jee, S.H.; et al. Association between IRF6 and nonsyndromic cleft lip with or without cleft palate in four populations. Genet. Med. Off. J. Am. Coll. Med. Genet. 2007, 9, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Rahimov, F.; Marazita, M.L.; Visel, A.; Cooper, M.E.; Hitchler, M.J.; Rubini, M.; Domann, F.E.; Govil, M.; Christensen, K.; Bille, C.; et al. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat. Genet. 2008, 40, 1341–1347. [Google Scholar] [CrossRef] [Green Version]
- Scapoli, L.; Palmieri, A.; Martinelli, M.; Pezzetti, F.; Carinci, P.; Tognon, M.; Carinci, F. Strong evidence of linkage disequilibrium between polymorphisms at the IRF6 locus and nonsyndromic cleft lip with or without cleft palate, in an Italian population. Am. J. Hum. Genet. 2005, 76, 180–183. [Google Scholar] [CrossRef] [Green Version]
- Zucchero, T.M.; Cooper, M.E.; Maher, B.S.; Daack-Hirsch, S.; Nepomuceno, B.; Ribeiro, L.; Caprau, D.; Christensen, K.; Suzuki, Y.; Machida, J.; et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N. Engl. J. Med. 2004, 351, 769–780. [Google Scholar] [CrossRef]
- Ingraham, C.R.; Kinoshita, A.; Kondo, S.; Yang, B.; Sajan, S.; Trout, K.J.; Malik, M.I.; Dunnwald, M.; Goudy, S.L.; Lovett, M.; et al. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6). Nat. Genet. 2006, 38, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Iwata, J.; Suzuki, A.; Pelikan, R.C.; Ho, T.V.; Sanchez-Lara, P.A.; Urata, M.; Dixon, M.J.; Chai, Y. Smad4-Irf6 genetic interaction and TGFbeta-mediated IRF6 signaling cascade are crucial for palatal fusion in mice. Development (Camb. Engl.) 2013, 140, 1220–1230. [Google Scholar] [CrossRef] [Green Version]
- Richardson, R.J.; Dixon, J.; Malhotra, S.; Hardman, M.J.; Knowles, L.; Boot-Handford, R.P.; Shore, P.; Whitmarsh, A.; Dixon, M.J. Irf6 is a key determinant of the keratinocyte proliferation-differentiation switch. Nat. Genet. 2006, 38, 1329–1334. [Google Scholar] [CrossRef]
- Richardson, R.J.; Dixon, J.; Jiang, R.; Dixon, M.J. Integration of IRF6 and Jagged2 signalling is essential for controlling palatal adhesion and fusion competence. Hum. Mol. Genet. 2009, 18, 2632–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parada-Sanchez, M.T.; Chu, E.Y.; Cox, L.L.; Undurty, S.S.; Standley, J.M.; Murray, J.C.; Cox, T.C. Disrupted IRF6-NME1/2 Complexes as a Cause of Cleft Lip/Palate. J. Dent. Res. 2017, 96, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinodoz, M.; Royer-Bertrand, B.; Cisarova, K.; Di Gioia, S.A.; Superti-Furga, A.; Rivolta, C. DOMINO: Using Machine Learning to Predict Genes Associated with Dominant Disorders. Am. J. Hum. Genet. 2017, 101, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Bretschneider, H.; Gandhi, S.; Deshwar, A.G.; Zuberi, K.; Frey, B.J. COSSMO: Predicting competitive alternative splice site selection using deep learning. Bioinformatics 2018, 34, i429–i437. [Google Scholar] [CrossRef]
- Anczukow, O.; Buisson, M.; Salles, M.J.; Triboulet, S.; Longy, M.; Lidereau, R.; Sinilnikova, O.M.; Mazoyer, S. Unclassified variants identified in BRCA1 exon 11: Consequences on splicing. Genes Chromosomes Cancer 2008, 47, 418–426. [Google Scholar] [CrossRef]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Frose, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef]
- Gartner, J.J.; Parker, S.C.; Prickett, T.D.; Dutton-Regester, K.; Stitzel, M.L.; Lin, J.C.; Davis, S.; Simhadri, V.L.; Jha, S.; Katagiri, N.; et al. Whole-genome sequencing identifies a recurrent functional synonymous mutation in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 13481–13486. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.V.; Steffensen, A.Y.; Jonson, L.; Andersen, M.K.; Ejlertsen, B.; Nielsen, F.C. The silent mutation nucleotide 744 G --> A, Lys172Lys, in exon 6 of BRCA2 results in exon skipping. Breast Cancer Res. Treat. 2010, 119, 547–550. [Google Scholar] [CrossRef] [Green Version]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science (New York N.Y.) 2007, 315, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montera, M.; Piaggio, F.; Marchese, C.; Gismondi, V.; Stella, A.; Resta, N.; Varesco, L.; Guanti, G.; Mareni, C. A silent mutation in exon 14 of the APC gene is associated with exon skipping in a FAP family. J. Med. Genet. 2001, 38, 863–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecina-Slaus, N.; Majic, Z.; Musani, V.; Zeljko, M.; Cupic, H. Report on mutation in exon 15 of the APC gene in a case of brain metastasis. J. Neuro-Oncol. 2010, 97, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raponi, M.; Kralovicova, J.; Copson, E.; Divina, P.; Eccles, D.; Johnson, P.; Baralle, D.; Vorechovsky, I. Prediction of single-nucleotide substitutions that result in exon skipping: Identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat. 2011, 32, 436–444. [Google Scholar] [CrossRef] [Green Version]
- Supek, F.; Minana, B.; Valcarcel, J.; Gabaldon, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [Green Version]
- Toscano, C.; Raimundo, S.; Klein, K.; Eichelbaum, M.; Schwab, M.; Zanger, U.M. A silent mutation (2939G>A, exon 6; CYP2D6*59) leading to impaired expression and function of CYP2D6. Pharmacogn. Genom. 2006, 16, 767–770. [Google Scholar] [CrossRef]
- Bartoszewski, R.A.; Jablonsky, M.; Bartoszewska, S.; Stevenson, L.; Dai, Q.; Kappes, J.; Collawn, J.F.; Bebok, Z. A synonymous single nucleotide polymorphism in DeltaF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J. Biolog. Chem. 2010, 285, 28741–28748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazrak, A.; Fu, L.; Bali, V.; Bartoszewski, R.; Rab, A.; Havasi, V.; Keiles, S.; Kappes, J.; Kumar, R.; Lefkowitz, E.; et al. The silent codon change I507-ATC->ATT contributes to the severity of the DeltaF508 CFTR channel dysfunction. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 4630–4645. [Google Scholar] [CrossRef] [Green Version]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hebuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Baek, J.; Liany, H.; Foo, J.N.; Kim, K.M.; Yang, S.C.; Liu, J.; Song, K. A Synonymous Variant in IL10RA Affects RNA Splicing in Paediatric Patients with Refractory Inflammatory Bowel Disease. J. Crohns Colitis 2016, 10, 1366–1371. [Google Scholar] [CrossRef] [Green Version]
- Nackley, A.G.; Shabalina, S.A.; Tchivileva, I.E.; Satterfield, K.; Korchynskyi, O.; Makarov, S.S.; Maixner, W.; Diatchenko, L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science (New York, N.Y.) 2006, 314, 1930–1933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterne-Weiler, T.; Sanford, J.R. Exon identity crisis: Disease-causing mutations that disrupt the splicing code. Genome Biol. 2014, 15, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, P.; Gaudon, K.; Fournier, E.; Jackson, C.; Bauche, S.; Haddad, H.; Koenig, J.; Echenne, B.; Hantai, D.; Eymard, B. A synonymous CHRNE mutation responsible for an aberrant splicing leading to congenital myasthenic syndrome. Neuromuscul. Disord. NMD 2007, 17, 409–414. [Google Scholar] [CrossRef]
- Vidal, C.; Cachia, A.; Xuereb-Anastasi, A. Effects of a synonymous variant in exon 9 of the CD44 gene on pre-mRNA splicing in a family with osteoporosis. Bone 2009, 45, 736–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmley, J.L.; Chamary, J.V.; Hurst, L.D. Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol. Biol. Evolut. 2006, 23, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Chamary, J.V.; Parmley, J.L.; Hurst, L.D. Hearing silence: Non-neutral evolution at synonymous sites in mammals. Nat. Rev. Genet. 2006, 7, 98–108. [Google Scholar] [CrossRef]
- Savisaar, R.; Hurst, L.D. Exonic splice regulation imposes strong selection at synonymous sites. Genome Res. 2018, 28, 1442–1454. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Nielsen, R. Mutation-selection models of codon substitution and their use to estimate selective strengths on codon usage. Mol. Biol. Evolut. 2008, 25, 568–579. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.A.; Wilke, C.O. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 2008, 134, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Hua, Y.; Sahashi, K.; Rigo, F.; Hung, G.; Horev, G.; Bennett, C.F.; Krainer, A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 2011, 478, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Passini, M.A.; Bu, J.; Richards, A.M.; Kinnecom, C.; Sardi, S.P.; Stanek, L.M.; Hua, Y.; Rigo, F.; Matson, J.; Hung, G.; et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011, 3, 72ra18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramowicz, A.; Gos, M. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeboom, R.G.; Supek, F.; Lehner, B. The rules and impact of nonsense-mediated mRNA decay in human cancers. Nat. Genet. 2016, 48, 1112–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Zahler, A.M.; Damgaard, C.K.; Kjems, J.; Caputi, M. SC35 and heterogeneous nuclear ribonucleoprotein A/B proteins bind to a juxtaposed exonic splicing enhancer/exonic splicing silencer element to regulate HIV-1 tat exon 2 splicing. J. Biolog. Chem. 2004, 279, 10077–10084. [Google Scholar] [CrossRef] [Green Version]
- Letra, A.; Menezes, R.; Cooper, M.E.; Fonseca, R.F.; Tropp, S.; Govil, M.; Granjeiro, J.M.; Imoehl, S.R.; Mansilla, M.A.; Murray, J.C.; et al. CRISPLD2 variants including a C471T silent mutation may contribute to nonsyndromic cleft lip with or without cleft palate. Cleft Palate Craniofac J. 2011, 48, 363–370. [Google Scholar] [CrossRef] [Green Version]
- Kumari, P.; Singh, S.K.; Raman, R. TGFbeta3, MSX1, and MMP3 as Candidates for NSCL+/-P in an Indian Population. Cleft Palate Craniofac J. 2019, 56, 363–372. [Google Scholar] [CrossRef]
- Gaczkowska, A.; Biedziak, B.; Budner, M.; Zadurska, M.; Lasota, A.; Hozyasz, K.K.; Dabrowska, J.; Wojcicki, P.; Szponar-Zurowska, A.; Zukowski, K.; et al. PAX7 nucleotide variants and the risk of non-syndromic orofacial clefts in the Polish population. Oral. Dis. 2019, 25, 1608–1618. [Google Scholar] [CrossRef]
- Ke, S.; Shang, S.; Kalachikov, S.M.; Morozova, I.; Yu, L.; Russo, J.J.; Ju, J.; Chasin, L.A. Quantitative evaluation of all hexamers as exonic splicing elements. Genome Res. 2011, 21, 1360–1374. [Google Scholar] [CrossRef] [Green Version]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Groß, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Yang, X.; Hu, X.; Li, S. Fifty-four novel mutations in the NF1 gene and integrated analyses of the mutations that modulate splicing. Int. J. Mol. Med. 2014, 34, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Wakatsuki, R.; Kato, T.; Okano, T.; Yamanishi, S.; Mayumi, N.; Tanaka, M.; Ogura, Y.; Kanegane, H.; Nonoyama, S.; et al. A synonymous splice site mutation in IL2RG gene causes late-onset combined immunodeficiency. Int. J. Hematol. 2019, 109, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Platt, C.D.; Massaad, M.J.; Cangemi, B.; Schmidt, B.; Aldhekri, H.; Geha, R.S. Janus kinase 3 deficiency caused by a homozygous synonymous exonic mutation that creates a dominant splice site. J. Allergy Clin. Immunol. 2017, 140, 268–271.e266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Patel, P.N.; Gorham, J.M.; McDonough, B.; DePalma, S.R.; Adler, E.E.; Lam, L.; MacRae, C.A.; Mohiuddin, S.M.; Fatkin, D.; et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, 7689–7694. [Google Scholar] [CrossRef] [Green Version]
- Odaira, K.; Tamura, S.; Suzuki, N.; Kakihara, M.; Hattori, Y.; Tokoro, M.; Suzuki, S.; Takagi, A.; Katsumi, A.; Hayakawa, F.; et al. Apparent synonymous mutation F9 c.87A>G causes secretion failure by in-frame mutation with aberrant splicing. Thromb. Res. 2019, 179, 95–103. [Google Scholar] [CrossRef]
- Thomassen, M.; Blanco, A.; Montagna, M.; Hansen, T.V.; Pedersen, I.S.; Gutiérrez-Enríquez, S.; Menéndez, M.; Fachal, L.; Santamariña, M.; Steffensen, A.Y.; et al. Characterization of BRCA1 and BRCA2 splicing variants: A collaborative report by ENIGMA consortium members. Breast Cancer Res. Treat. 2012, 132, 1009–1023. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Genomic Coordinate | Gene Name | Location | Type | Protein Change | CADD Scaled | DOMINO Class (Probability) | gnomAD Allele Count (MAF) | Associated Disease |
|---|---|---|---|---|---|---|---|---|
| chr8:g.13251073G>C | DLC1 | Coding | Missense | p.Pro435Ala | 23.4 | LD (0.706) | 0 | Nephrosis |
| chr1:g.155838558G>A | SYT11 | Coding | Synonymous | p.Arg279Arg | 11.89 | EDR (0.478) | 0 | |
| chr1:g.114940211G>T | TRIM33 | 3’UTR | SNP | 0.742 | VLD (0.997) | 0 | ||
| chr3:g.149684103G>A | PFN2 | 3’UTR | SNP | 16.21 | VLD (0.948) | 0 | ||
| chr1:g.16262494_16262499dupTGTCCC | SPEN | Coding | Duplication | p.Val3254_Pro3255dup | VLD (1) | 0 | ||
| chr5:g.149922536G>A | NDST1 | Intronic | Splice | 14.37 | VLD (0.964) | 0 | Mental retardation | |
| chr11:g.156907027_156907033delCCTGCTT | ARHGEF11 | Intronic | Deletion | LD (0.794) | 0 | |||
| chr7:g.137597556G>C | CREB3L2 | Intronic | Splice | 5.642 | LD (0.616) | 0 | Fibromyxoid sarcoma | |
| chr8:g.38947723delT | ADAM9 | Intronic | Deletion | EDR (0.584) | 0 | Cone-rod dystrophy | ||
| chr18:g.59936640dupC | KIAA1468 | Intronic | Splice | VLD (0.959) | 0 | |||
| chr12:g.64882264delT | TBK1 | Intronic | Splice | VLD (0.882) | 0 | Frontotemporal dementia/ALS | ||
| chr9:g.73458046dupA | TRPM3 | Intronic | Splice | EDR (0.585) | 0 | Intellectual disability/epilepsy | ||
| chr19:g.10798342G>A | ILF3 | Coding | Missense | p.Gly798Arg | 24.2 | LD (0.677) | 2 / 247274 (8.09 × 10−6) | |
| chr1:g.209963979G>A | IRF6 | Coding | Synonymous | p.Ser307Ser | 14.48 | LD (0.733) | 3 / 251352 (1.19 × 10−5) | Orofacial clefting |
| chr8:g.66647047A>G | PDE7A | Coding | Synonymous | p.Cys226Cys | 12.85 | EDR (0.407) | 3 / 250766 (1.20 × 10−5) | |
| chr12:g.6094200G>A | VWF | Intronic | SNP | 5.659 | LD (0.617) | 6 / 251458 (2.39 × 10−5) | von Willebrand disease | |
| chr2:g.240085511C>T | HDAC4 | Coding | Missense | p.Arg200His | 25 | VLD (0.999) | 7 / 251428 (2.78 × 10−5) | Brachydactyly, mental retardation |
| chr8:g.26484831G>A | DPYSL2 | Intronic | SNP | 0.659 | VLD (0.995) | 9 / 282698 (3.18 × 10−5) | ||
| chr1:g.16268440_16268443delTAAA | SPEN/ZBTB17 | 3’UTR | Deletion | VLD (1) | 7 / 218068 (3.21 × 10−5) | |||
| chr7:g.98555591G>T | TRRAP | Intronic | SNP | 0.096 | VLD (1) | 10 / 248972 (4.02 × 10−5) | Developmental delay/autism, dysmorphic facies | |
| chr1:g.150551341G>A | MCL1 | Coding | Synonymous | p.Arg222Arg | 12.07 | VLD (0.997) | 13 / 281774 (4.61 × 10−5) | |
| chr17:g.78113791C>A | EIF4A3 | Intronic | SNP | 0.049 | VLD (0.999) | 12 / 244608 (4.91 × 10−5) | Robin sequence, cleft mandible, limb anomalies | |
| chr1:g.154527884G>A | UBE2Q1 | Intronic | SNP | 4.602 | LD (0.727) | 13 / 251300 (5.17 × 10−5) | ||
| chr1:g.155948197T>G | ARHGEF2 | Coding | Missense | p.Thr8Pro | 22.4 | VLD (0.916) | 14 / 167764 (8.35 × 10−5) | Neurodevelopmental disorder, brain malformations |
| chr10:g.33196031T>C | ITGB1 | coding | Missense | p.Lys791Arg | 22.6 | VLD (0.996) | 26 / 279598 (9.30 × 10−5) | |
| chr12:g.52911558C>T | KRT5 | intronic | SNP | 5.636 | EDR (0.589) | 28 / 282824 (9.90 × 10−5) | epidermolysis bullosa | |
| chr17:g.72759561G>A | SLC9A3R1 | coding | Missense | p.Arg220Lys | 14.76 | VLD (0.865) | 25 / 250294 (9.99 × 10−5) | Nephrolithiasis/osteoporosis, hypophosphatemia |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sylvester, B.; Brindopke, F.; Suzuki, A.; Giron, M.; Auslander, A.; Maas, R.L.; Tsai, B.; Gao, H.; Magee, W., III; Cox, T.C.; et al. A Synonymous Exonic Splice Silencer Variant in IRF6 as a Novel and Cryptic Cause of Non-Syndromic Cleft Lip and Palate. Genes 2020, 11, 903. https://doi.org/10.3390/genes11080903
Sylvester B, Brindopke F, Suzuki A, Giron M, Auslander A, Maas RL, Tsai B, Gao H, Magee W III, Cox TC, et al. A Synonymous Exonic Splice Silencer Variant in IRF6 as a Novel and Cryptic Cause of Non-Syndromic Cleft Lip and Palate. Genes. 2020; 11(8):903. https://doi.org/10.3390/genes11080903
Chicago/Turabian StyleSylvester, Beau, Frederick Brindopke, Akiko Suzuki, Melissa Giron, Allyn Auslander, Richard L. Maas, Becky Tsai, Hanlin Gao, William Magee, III, Timothy C. Cox, and et al. 2020. "A Synonymous Exonic Splice Silencer Variant in IRF6 as a Novel and Cryptic Cause of Non-Syndromic Cleft Lip and Palate" Genes 11, no. 8: 903. https://doi.org/10.3390/genes11080903