
Abstract
Background
Decline in mitochondrial function occurs with aging and may increase mortality. We discuss mitochondrial contribution to Covid-19 sepsis, specifically the complex interaction of innate immune function, viral replication, hyper-inflammatory state, and HIF-α/Sirtuin pathways.
Methods
Articles from PubMed/Medline searches were reviewed using the combination of terms “SARS-CoV-2, Covid-19, sepsis, mitochondria, aging, and immunometabolism”.
Results
Evidence indicates that mitochondria in senescent cells may be dysfunctional and unable to keep up with hypermetabolic demands associated with Covid-19 sepsis. Mitochondrial proteins may serve as damage-associated molecular pattern (DAMP) activating innate immunity. Disruption in normal oxidative phosphorylation pathways contributes to elevated ROS which activates sepsis cascade through HIF-α/Sirtuin pathway. Viral–mitochondrial interaction may be necessary for replication and increased viral load. Hypoxia and hyper-inflammatory state contribute to increased mortality associated with Covid-19 sepsis.
Conclusions
Aging is associated with worse outcomes in sepsis. Modulating Sirtuin activity is emerging as therapeutic agent in sepsis. HIF-α, levels of mitochondrial DNA, and other mitochondrial DAMP molecules may also serve as useful biomarker and need to be investigated. These mechanisms should be explored specifically for Covid-19-related sepsis. Understanding newly discovered regulatory mechanisms may lead to the development of novel diagnostic and therapeutic targets.
Similar content being viewed by others


Introduction
Sepsis is defined as a life-threatening organ dysfunction caused by dysregulated host response to an infection [1]. Despite advances in antibiotic therapy and critical care management of sepsis, multi-organ failure with morbidity and mortality continues to remain high. This is clearly demonstrated with the current Covid-19 pandemic where infection with SARS-CoV-2, corona virus-associated sepsis has led to a catastrophic increase in morbidity and mortality affecting people of all nations, ethnicities, sex and age groups [2]. Particularly effected are frail elderly populations, in densely populated community and nursing homes facilities. The most common risk factors are elderly patients with multiple comorbidities such as cardiopulmonary diseases, diabetes, obesity, and hypertension. This current Covid 19 pandemic crisis has established that our understanding of the pathophysiology of sepsis remains incomplete and further research is needed to fill these gaps.
Biochemical features unique to sepsis associated with the Covid-19 has established a state of acute inflammation with cytokine storm associated with destructive downstream effects manifested as profound persistent hypoxia, acidosis, hypercoagulability, and altered aerobic glycolytic metabolism (elevated LDH and lactic acidosis). The organs typically affected are lungs, kidneys, liver, heart, brain, and gastrointestinal tract. This is manifested as hypoxia with respiratory failure, renal failure requiring hemodialysis, liver failure with elevated transaminases, heart failure with associated cardiomyopathy and arrhythmias, encephalopathy, and enteric symptoms with diarrhea and mesenteric ischemia [3].
This brief review discusses the current developments in the Covid-19 pathogenesis and the possible role of mitochondria in the dynamics of sepsis. Excessive Covid-19 virus-related sepsis and mortality may be due to decline in mitochondrial efficiency and respiratory chain functions with aging [4, 5]. Furthermore, recent evidence also indicates that the virus may manipulate the mitochondria to maintain its life cycle. The resulting tissue hypoxia along with increased inflammation, viral load, and other baseline risk factors contributes to an increased risk for severe sepsis. This association may explain for high mortality associated in elderly patients with rapid onset, profound, and resistant vital organ failure. Autopsies have confirmed hyper-inflammatory state with organ fibrosis particularly in cells with high metabolism and mitochondrial volume such as pneumocytes, cardiomyocytes, endothelial cells, hepatocytes, renal cells, and neurons.
Mitochondrial function in health
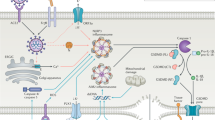
Mitochondria is a vital organelle present in all cells except mature erythrocytes, with key cellular functions involving oxidative phosphorylation, ATP synthesis, intracellular calcium regulation, thermoregulation, regulation of ROS, lipid metabolism, iron metabolism, autophagy, apoptosis, and regulation of certain hormones such as cortisol, estrogen, and triiodothyronine [5,6,7]. Elderly patients with high proportion of senescent tissue and immune cells with suboptimal mitochondria may be especially susceptible and unable to keep up with the sudden hypermetabolic demands associated with sepsis [8]. Further mitochondria also have a role and are associated with cellular senescence, chronic inflammation (inflammaging), decline in innate immunity, and the age-dependent decline in stem cell activity [9] (Fig. 1).

Covid 19 virus and possible mitochondrial dynamics, complex interplay in immunometabolism. Dysfunctional mitochondrial oxidative phosphorylation leads to accumulation of reactive oxygen species (ROS). There is activation of aerobic glycolysis, pentose phosphate pathway, and lactic acid formation, further increasing the ROS. This leads to mitochondrial membrane damage and release of DAMP factors. Mitochondria may also contribute to the formation of double-membrane vesicle which is critical in corona virus life cycle. Innate immunity is activated with downstream hyper-inflammatory cascade. HIF-α/Sirtuin pathways are also activated by cellular hypoxia and ROS and activate aerobic glycolysis and innate immune system. All these factors lead to multiple organ dysfunction and increase sepsis-related mortality. TLR Toll-like receptor is a PRR, RIG 1 retinoic acid inducible is a cytosolic PRR, IL interleukins, TNF tumor necrosis factor, IFN interferons, NLRP3 NOD like receptor is a PRR
As people age, their cells accumulate replication errors and mutations in the mitochondrial DNA (mtDNA) [8]. Oxidative damage to the cells by the reactive oxygen species (ROS) such as hydrogen peroxide (H2O2), superoxides, and hydroxyl radicals also accumulate with aging. The mitochondrial respiratory chain is the source of these ROS and they also have the mechanism for elimination with enzymes that have scavenger functions, for example: superoxide dismutase. Mitochondrial dysfunction is also associated with toxic exposures and chronic anoxic conditions [7]. With a higher proportion of senescent cells in the elderly, deficiencies in the respiratory chain functions may develop eventually and may be associated with a decline in mitochondrial respiratory efficiency [9].
Mitochondrial dysfunction in sepsis (Fig. 1)
Sepsis is an abnormal pathological state and consists of an initial acute hyper-inflammatory phase, to fight the infection followed by an immune-tolerant, hibernating phase to aid with recovery. Different metabolic processes and reprogramming unique to each phase have been described and play a role in the different, varied clinical manifestations in the two phases. Hyper-inflammatory phase is characterized by increased aerobic glycolysis (Warburg effect). This phase occurs as the host tissues reacts, with the aim of ramping up energy production, activating the innate immune cells to increase its pathogen killing capacity and control the spread of the infection and defend against the invading microbes [5, 10,11,12]. The late hypo-inflammatory, immune-tolerant phase is characterized by increased fatty acid oxidation in immune cells [10, 11]. A recent analysis of differential gene expression in the SARS-CoV-2 infected lung cell lines demonstrates upregulation of genes involved in mitochondrial cytokine/inflammatory signaling and downregulation in the mitochondrial organization, respiration, and autophagy genes [13]. These findings provide evidence that mitochondrial disruption hinders an effective immune response, increases inflammation, and severity in the Covid-19-related sepsis.
Metabolic and immune consequences, and release of DAMP (Fig. 1)
A key feature of early sepsis is hypoxia. At the cellular level, hypoxia correlates with a switch to aerobic (glycolytic) respiration with alteration of normal oxidative phosphorylation and mitochondrial function due to reduced activity of complex I–IV and complex V of electron transport chain. There is increased oxygen consumption, elevated ATP production, and hyperglycemia. In addition, due to increased cell turnover, there is increased requirement for nucleotides and increase in pentose phosphate pathway [4, 5, 10, 12]. These altered metabolic pathways result in production of excessive cytoplasmic and mitochondrial ROS. Failure to clear the ROS by senescent mitochondria may impair and damage mitochondrial structure, lipid membranes, biogenesis units, and alter the intact mitochondrial DNA [5, 14]. Other mitochondrial factors exacerbating and causing elevated ROS include altered lipid peroxidation, glutathione (GSH) depletion, and altered iron metabolism [15]. Loss of mitochondrial membrane integrity may release mtDNA in the systemic circulation. These circulating mtDNA fragments are also known as danger-associated molecular patterns (DAMP) and are similar to the pathogen associated molecular patterns (PAMP) which are released by microbes and identified by pathogen recognition receptors (PRR) on the immune cells. Nakahira et al. in a prospective study of 200 patients were first to demonstrate the association of elevated cell-free, plasma mtDNA levels with increased ICU-related mortality. They suggested mtDNA as a DAMP capable of propagating the inflammatory response seen in sepsis and other inflammatory conditions and a predictive biomarker for ICU-related mortality [16]. Other DAMP proteins associated with mitochondrial derangement include mtROS, mt formyl peptides, Cytochrome C, cardiolipin, carbamoyl phosphate synthetase, heat shock proteins (HSP), and high-mobility group box proteins-1 (HMBG-1) [5, 6, 17, 18] (Fig. 1).
These endogenously derived DAMP molecules from senescent aged, disrupted mitochondria can activate the innate immune system similar to the PAMP molecules obtained from virus particles. The activation of the PRR (family of Toll-like receptors) results in the activation of immune cells consisting of neutrophils, macrophages, dendritic cells, and natural killer cells. Activated innate immunity initiates the transcription of genes which lead to the production of signal molecules such as prostaglandins, pro-inflammatory cytokines, chemokines, and interleukins, and also activates the complement system, thus evoking the inflammatory cascade [16, 19].
DAMP have also shown to activate the (Nuclear factor kappa-light chain enhancer of B cell) NF-Kβ pathway, activation of inflammasommes, and (stimulator of interferon genes) STING pathways. These mechanisms exacerbate and compound tissue dysfunction involving cardiovascular, immune, metabolic, endocrine, coagulation, and thermoregulatory pathways, and are responsible for the systemic effects and multi-organ failure [5, 16, 19,20,21]. Previous reports from septic patients and septic animal models indicate decrease in intact mtDNA and an increase in circulating free mtDNA fragments [16, 18].
Role of HIF-α/Sirtuins in sepsis (Fig. 1)
Recent investigations have highlighted mechanisms in acute inflammation and sepsis cascade by HIF-α (hypoxia-inducible factor-Iα)–Sirtuins (silent mating type information regulator) signaling pathway [10, 11, 22]. Tissue hypoxia is a central feature of sepsis. There is upregulation of HIF-α signaling in tissue hypoxia. These signaling molecules and transcription factors are involved in both the hyper-inflammatory phase (elevated HIF-α/low Sirtuins) and the second immune-tolerant, recovery phase of sepsis (elevated Sirtuins/low HIF-α).
Sirtuins (types 1–7) are a highly conserved family of proteins known for their anti-inflammatory and anti-oxidant properties. These proteins belong to the (nicotinamide adenine dinucleotide) NAD+ dependent class III histone deacetylase (HDAC) family of enzymes and different types are present in different compartments of cells such as cytoplasm, mitochondria, and nucleus. Sirtuins are (NAD+) energy sensors of a cell and are considered as guardians of homeostasis. Sirtuins type 3, 4, 5 are localized to the mitochondria [10, 11, 22] (Fig. 1). NAD+ levels decline with aging and are further reduced in chronically stressed cells such as patients with diabetes, hypertension, smokers, and obesity thereby reducing the activity of Sirtuin. These groups of patients have the highest risk for severe sepsis and mortality as currently observed in the Covid-19 pandemic [23]. Recent experiments on SARS-CoV-2 infected murine, human lung cell lines, and autopsy specimens suggest upregulation of poly (ADP) ribose polymerase (PARP) family of genes which are involved in the consumption of NAD+. Overexpression of these PARP genes consume and depress the levels of NAD+ and are overexpressed in these cells [24]. Reduced activity and depletion of NAD+ further attenuate protective function of Sirtuin [23, 24].
Hypoxic cells upregulate the transcription factor, HIFα [12, 20]. In general, stabilization of HIFα and an increase in its hyper-inflammatory activity occur with loss or dysfunctional protective action of the cellular Sirtuins. This has been demonstrated in the hyper-inflammatory stage of animal models of sepsis [10,11,12, 22].
During this initial hyper-inflammatory phase with associated tissue hypoxia, the PRR (Toll-like receptors) upregulate a number of proliferative pathways such as NF-ĸB, hyper activation of NLRP3 inflammasommes, mitogen-activated protein kinase (MAPK), and mammalian target of rapamycin (mTOR) that trigger the cytokine storm [23,24,25].
Along with increased proliferative activity and increased cell turn over, the accompanying aerobic glycolytic pathway and dysfunctional oxidative phosphorylation release ROS which results in downregulation of Sirtuins. Low Sirtuin activity itself promotes elevated ROS due to inefficient clearance. Elevated ROS also downregulates prolyl hydroxylase (PHD). These enzymes inactivate HIF-α. Taken together, these factors stabilize HIF-α and prevent it from undergoing ubiquitination by E3 ligases (von Hippel Lindau factor, VHL) for proteasomal degradation. Stabilized HIF-α translocates to the nucleus and activates key genes upregulating glycolysis and production of pro-inflammatory cytokines and maintains the vicious cycle of a hyper-inflammatory state [10,11,12, 22]. Key HIF-α targets include glucose transporter 1 (Glut1), hexokinase (HK), lactic dehydrogenase (LDH), pyruvate dehydrogenase kinase (PDHK), and cyclo-oxygenase (COX-2). These enzymes shift metabolism away from oxidative phosphorylation and promoting glycolytic signature and further fueling anabolic pathways [10,11,12, 22] (Fig. 1).
During the second phase of sepsis, a hypo-metabolic state will ensue with decreased oxygen consumption, resumption of mitochondrial respiration, and ATP production. Majority of the mortality associated with sepsis occurs during this phase. This hypo-metabolic state allows cells to enter an anti-inflammatory hibernating state allowing for slow recovery of cellular function. However, this cyto-protective, immunosuppressive state may also be detrimental to clear infections [10]. Moreover, secondary nosocomial infections may occur during this phase. For example: clostridium difficile infections, and secondary bacterial and candida infections. Often these infections are multidrug resistant and increase sepsis-related morality in the setting of the hypo-inflammatory phase [10]. Biochemically, this phase is associated with reduced activity of HIF-α and upregulation of Sirtuin activity. Sirtuins with their deacetylase function inactivate NF-ĸB, HIF-α, high-mobility group box proteins-1 (HMBG-1), and other DAMP molecules, thus decreasing the overall cytokine load [23,24,25]. Sirtuins also coordinate switch from glucose to fatty acid oxidation during this phase. The molecular mechanisms for this remain unclear [10, 11, 14, 22, 25]. With cell recovery, mitochondrial biogenesis is initiated and there are upregulation of markers of mitochondrial biogenesis such as PAR gamma coactivator-1α (PGC-1α), Transcription factor A for mitochondria (TFAM), and nuclear respiratory factors (NRF-1) [9, 15, 23].
Mitochondria localization may be necessary for SARS-CoV-2 replication
SARS-COV-2 virus uses ACE2 receptor for cell entry and TMPRSS2 for spike protein (S protein) priming [26]. Variations of ACE 2 receptors in different ethnic populations may play a role in the virulence and transmissibility of this virus [27].
SARS-CoV-2 protein and human–protein interactions that connect multiple complex biological processes such as replication, protein trafficking, and ubiquitination have been observed. As a group SARS-CoV viruses produce accessory proteins called open reading frames (ORF) which interact with mitochondrial outer membrane receptors. One particular interaction involves ORF-9 interaction with mitochondrial antiviral signaling systems (MAVS, TOMM.70) [28]. MAVS is a mitochondrial import receptor and also functions as cytoplasmic viral recognition receptor. ORF has shown to suppress MAVS activity, thus limiting the initial host cell, innate immune, interferon, and antiviral response [28,29,30]. Similar other SARS-CoV-2 virus non-structural protein (NSP4, NSP5, NSP7, and NSP8) interaction with human mitochondrial membranes and matrix functions has been demonstrated. These viral proteins have been demonstrated to associate with RNA processing, electron transport and mitochondrial signaling, and trafficking proteins. Thus, viral–mitochondrial interactions may disrupt both the membrane integrity and functional aspects of the mitochondria [28].
Once inside the cytoplasm, the SARS-CoV-2 virus has to replicate from a single-stranded RNA (ss RNA) through an intermediate double-stranded RNA (ds RNA). This predisposes the virus to the antagonism through their recognition by Toll-like receptors, RIG-1 receptors, and mitochondrial antiviral signaling systems (MAVS) and, thus, activation of innate immunity as described earlier. SARS-CoV-2 virus evades this detection by forming double-membrane vesicles (DMV) around its dsRNA, thus shielding it from detection. Viruses need intracellular organelles such as mitochondria and endoplasmic reticulum for their replication and dissemination [31, 32].
Based on computational machine learning models, it has recently been demonstrated by Wu et al. that the SARS-CoV-2 RNA genome and all sub-genomic RNAs are enriched in the host mitochondrial matrix and nucleolus [33]. Open reading frame (ORF) from corona viral genome has been identified in the mitochondria. Based on these models, they predicted that mitochondrial residency may be required to the formation of double-membrane vesicles which is a critical in corona virus being able to replicate un-abated by evading cellular defenses. In other words, the virus simply may hijack the mitochondria and use its machinery for its own replication and sustenance [33]. In doing so, it may damage the mt DNA and this may cause leakage of the mt DNA into the cytoplasm with then acts as a trigger for activation of innate immunity [34]. Previously, an association between high mitochondrial viral RNA and decrease in mitochondrial functional integrity has been demonstrated with HIV RNA virus [35]. This hypothesis needs further validation with SARS-CoV-2. These findings suggest that mitochondrial involvement by the SARS-CoV-2 RNA virus may actually assist with virulence and transmissibility. Mechanisms by which the virus gains access into the mitochondria and cause dysfunction remain to be investigated.
Covid-19 and biomarkers associated with high mortality in the elderly, Fig. 2
Several viral and host-related biomarkers are emerging to be critical in determining the severity of infection in Covid-19 sepsis. The major question remains whether these biomarkers perform consistently, can be tested and replicated uniformly, and can be validated by pooling of the currently available data.

Venn diagram: depicting the risk for severe sepsis and increased mortality in Covid-19 pandemic. A combination of: (a) elderly patients with comorbidities living in close knit communities and densely populated areas, (b) associated mitochondrial dysfunction, and risk factors such as diabetes, hypertension, lung diseases, and activation of innate immunity, and (c) an increased viral load; all together set a perfect storm for increased viral shedding and transmissibility with risk for severe sepsis and high mortality
As the current SARS-CoV-2 virus continues to spread in the USA and worldwide, high quantitative viral load emerges as a critical factor in epidemiology, and also to predict the clinical course in Covid-19 infection [36,37,38,39,40]. Key observations with current Covid-19 pandemic demonstrate a high mortality in elderly patients with sepsis. Increased quantitative viral load represents a high level of viral replication and is a surrogate marker for infectivity. This association may be related to its interaction and mitochondria dysfunction in the elderly. Previously, studies have attempted to ascertain an association between viral loads and increased severity with prolonged hospitalization on patients infected with influenza, rhino, and other respiratory virus [36, 37, 40]. Real-time PCR assays (rRT-PCR) to detect respiratory viruses such as group 2 coronaviruses (HCoV-OC43 and HCoV-HKU1), and SARS associated coronavirus have demonstrated that a high viral load measured on nasopharyngeal samples at the point of hospitalization was associated with longer length of stay in adult patients [36, 40]. Similar to recent literature on SARS-COV-2 virus, Covid-19 pandemic has suggested that higher viral load values from nasopharyngeal swabs remain a significant factor and are associated with prolonged virus shedding period and severe clinical outcomes [38, 39]. Elevated viral loads may be noted in both asymptomatic and symptomatic patients. This remains a cause of concern, as asymptomatic carriers may be silent spreaders in the population. Viral load may be a useful marker for both detecting super spreaders and also disease severity and prognosis in the symptomatic patients [38, 39].
Early determination of quantitative viral load also has other therapeutic benefits, such as early implementation of convalescent plasma, antiviral, and corticosteroids. Convalescent plasma from donors who recovered from Covid-19 infection could potentially improve the clinical outcomes by decreasing the severity in Covid-19 cases [41, 42].
A persistently elevated viral load may thus be associated with worse metabolic and mitochondrial dysfunction with a prolonged hyper-inflammatory phase as described earlier. This hypothesis may explain the clinical variation observed with Covid-19 patients worldwide, i.e., higher mortality in elderly, especially in a close knit population such as nursing homes suggestive of mitochondrial dysfunction associated with high viral load (Fig. 2). Further epidemiological studies and assays on Covid-19 viral loads are needed to confirm this hypothesis.
Other host-related biomarkers which are consistently observed and correlated in various populations across the world from the current pandemic are elevated levels of d-dimer, IL-6 concentration, and lymphopenia. These portend severity of the inflammation and correlate with poor prognosis and increased mortality [41, 43, 44].
Future directions: potential mitochondrial-related diagnostic biomarkers and therapeutic targets
The role HIF-α, as a biomarker, for severity in sepsis is unclear and continues to be investigated [10,11,12, 25]. Sepsis is dynamic process and the levels of HIF-α may vary depending on the phase of the disease and also with the different pathogens [12]. A previous study demonstrated that HIF-1 RNA expression was noted to be elevated in septic patients, but no difference was noted in survivors and non-survivors [45]. A clinical trial (NCT02163473) investigating HIF-α as a potential biomarker has been completed and results are awaited.
Levels of mt DNA and other mt DAMP molecules may also serve as useful biomarker and need to be investigated in the context of SARS-CoV-2 infection [6, 9, 16, 17, 19]. As described earlier, mtDNA may serve as a DAMP capable of propagating the inflammatory response seen in sepsis and a predictive biomarker for ICU-related mortality [16]. Recent evidence in rodent models has demonstrated that neutralizing mtDNA with the nucleic acid scavengers such as hexadimethrine bromide (HDMBr) could reduce the severity of multi-organ dysfunction and may improve survival [46]. These mechanisms should be explored specifically for Covid-19-related sepsis.
Modulating Sirtuin activity levels and thus mitochondrial bioenergetics with activating and inhibiting agents are emerging as therapeutic agents in experimental models of sepsis [10, 11]. However, it has to be timed correctly in specific phases of sepsis to have beneficial effects. Drugs that can increase the NAD+ levels and thus elevate the levels of the Sirtuin in the hyper-inflammatory stage of sepsis may be used to attenuate the cytokine storm and may improve survival. Previously, experiments on rodents have demonstrated that NAD+ boosters attenuated the NF-ĸB activity and improved endothelial function and decreased microvascular inflammation [47]. Sirtuin activating agents such as resveratrol (a naturally occurring stilbenoid), NAD+ precursors such as nicotinamide riboside (NR) and nicotinamide mononucleotide (NMN), and enzymes that regulate NAD+ levels such as CD38, CD157, and NAMPT have all been studied in experiments and demonstrated to increase Sirtuin activity [11, 47, 48].
Similarly, reducing the Sirtuin activity with inhibitors (EX-527) during the second hypo-inflammatory stages may reverse the hibernating endothelium and vasculature, improve neutrophilic function, clear the debris, aid with effective oxidative phosphorylation and mitochondrial biogenesis, and healing, and may improve survival [11, 49].
There have been recent developments and interest in molecules which can target chronic inflammation associated with aging, obesity, and metabolic syndrome known as inflammaging. These molecules are also labelled as geroprotectors [23]. Some of these molecules may have favorable effects in prophylaxis and adjuvant therapies in sepsis, and are worth mentioning briefly. It should be emphasized that these are empirical therapies which have not been confirmed in human trials on Covid-19 sepsis. Mammalian target of rapamycin (mTOR) signaling pathways have demonstrated to control immune functions such as antigen presentation, activation, and cytokine production. Modulating mTOR activity with its inhibitor rapamycin has shown some improvement in the immune functions in the elderly [23]. Similarly, metformin in addition to its antiglycemic and favorable effects in metabolic syndrome also lowers AMPK activity and inhibits mTOR pathway. Metformin may improve metabolic derangements, improve mitochondrial function, and decrease cytokine production [23, 48]. NAD+ boosters such as resveratrol, a naturally occurring polyphenol, found in red grapes and blueberries, activate Sirtuin activity and attenuate the NF-ĸB activity, improve endothelial function, and decrease microvascular inflammation [47].
Disrupted circadian rhythms, and increased gut permeability with associated intestinal dysbiosis occur in sepsis. Melatonin and probiotics, molecules with anti-inflammatory and anti-oxidant activity, may improve mitochondrial bioenergetics and survival in sepsis [50].
Conclusion
Covid-19 has shed light on the vulnerability of the aging population to pandemics and global challenges of new diseases. Developing therapies and vaccines requires a comprehensive and deep understanding of pathogenesis and molecular mechanisms of these emerging viruses. Mitochondrial dysfunction continues to be explored as the cause for the excess mortality associated with this virus. We have highlighted a few mechanisms by which the virus–mitochondrial interactions may play a role in viral replication, modulating innate immunity, hyper-inflammatory state, and end organ damage.
Based on the emerging evidence, it is suggestive that the mitochondrial dysfunction may be the primary event. Interactions of the SARS-CoV-2 virus proteins such as ORF and NSP proteins with host cell mitochondrial proteins lead to loss of membrane integrity and also cause dysfunction in the bioenergetics of the mitochondria. What follows is a secondary damaging event with the release of mt DNA, elevated ROS; loss of MAVS associated protective, antiviral interferon production, activation of the NF-Kβ pathway, inflammasommes, and innate immunity with hyper-inflammatory state and cytokine storm.
Mitochondrial mechanisms may explain and is reflected in the particular susceptibility of the elderly population with comorbidities. These patients have dysfunctional, senescent cells, and mitochondria. Involvement of the virus may create an imbalance that is difficult to overcome with resulting excess mortality. Infected younger patients in physiologically good health may escape this devastation due to healthy tissues and adequate mitochondrial dynamics. Furthermore, it becomes apparent that mitochondria may be necessary for the virus to replicate (formation of double-membrane vesicles) and propagate its life cycle.
In conclusion, the relevance of mitochondrial dysfunction in pathogenesis of multi-organ failure associated with Covid-19 sepsis continues to be under active investigation. Many questions remain unanswered. The current Covid-19 pandemic exposed our gaps in the knowledge of sepsis and hopefully reinvigorated the scientific community. Further research is urgently required to shed light and elucidate mechanisms in Covid-19 sepsis. Understanding these regulatory mechanisms may lead to development of novel diagnostic and therapeutic targets to combat any future pandemic crisis.
References
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315:801–10. https://doi.org/10.1001/jama.2016.0287.
Fauci AS, Lane HC, Redfield RR. Covid-19—navigating the uncharted. N Engl J Med. 2020;382:1268–9. https://doi.org/10.1056/NEJMe2002387(Epub 2020 Feb 28).
Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. 2020. https://doi.org/10.1001/jama.2020.1585(Epub ahead of print).
Brealey D, Brand M, Hargreaves I, et al. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360:219–23.
Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence. 2014;5:66–72. https://doi.org/10.4161/viru.26907(Epub 2013 Nov 1. Review).
Yao X, Carlson D, Sun Y, et al. Mitochondrial ROS induces cardiac inflammation via a pathway through mtDNA damage in a pneumonia-related sepsis model. PLoS ONE. 2015;10:e0139416. https://doi.org/10.1371/journal.pone.0139416(eCollection 2015).
van der Bliek AM, Sedensky MM, Morgan PG. Cell biology of the mitochondrion. Genetics. 2017;207:843–71. https://doi.org/10.1534/genetics.117.300262.
Park CB, Larsson NG. Mitochondrial DNA mutations in disease and aging. J Cell Biol. 2011;193:809–18. https://doi.org/10.1083/jcb.201010024(Epub 2011 May 23. Review).
Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61:654–66. https://doi.org/10.1016/j.molcel.2016.01.028(Review).
Wang X, Buechler NL, Woodruff AG, et al. Sirtuins and immuno-metabolism of sepsis. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19092738(Review).
Vachharajani V, McCall CE. Sirtuins: potential therapeutic targets for regulating acute inflammatory response? Expert Opin Ther Targets. 2020;24:489–97. https://doi.org/10.1080/14728222.2020.1743268(Epub 2020 Mar 26).
Fitzpatrick SF. Immunometabolism and sepsis: a role for HIF? Front Mol Biosci. 2019;6:85. https://doi.org/10.3389/fmolb.2019.00085(eCollection 2019. Review).
Singh K, Chen YC, Judy JT, et al. network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS-CoV-2 infection. Preprint. bioRxiv. 2020;2020.05.13.092536. https://doi.org/10.1101/2020.05.13.092536. Published 14 May 2020.
Shokolenko I, Venediktova N, Bochkareva A, et al. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009;37:2539–48. https://doi.org/10.1093/nar/gkp100(Epub 2009 Mar 5).
Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79. https://doi.org/10.1038/cdd.2015.158(Epub 2016 Jan 22. Review).
Nakahira K, Kyung SY, Rogers AJ, et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. 2013;10:e1001577. https://doi.org/10.1371/journal.pmed.1001577.
Gentile LF, Moldawer LL. DAMPs, PAMPs, and the origins of SIRS in bacterial sepsis. Shock. 2013;39:113–4. https://doi.org/10.1097/SHK.0b013e318277109c(No abstract available).
Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. https://doi.org/10.1038/nature08780.
Harrington JS, Choi AMK, Nakahira K. Mitochondrial DNA in sepsis. Curr Opin Crit Care. 2017;23:284–90. https://doi.org/10.1097/MCC.0000000000000427.
West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–7. https://doi.org/10.1038/nature14156(Epub 2015 Feb 2).
Samy RP, Lim LH. DAMPs and influenza virus infection in ageing. Ageing Res Rev. 2015;24:83–97. https://doi.org/10.1016/j.arr.2015.07.005(Epub 2015 Jul 19. Review).
Li L, Chen Z, Fu W, et al. Emerging evidence concerning the role of sirtuins in sepsis. Crit Care Res Pract. 2018;2018:5489571. https://doi.org/10.1155/2018/5489571(eCollection 2018. Review).
Mueller AL, McNamara MS, Sinclair DA. Why does COVID-19 disproportionately affect older people? Aging. 2020;12:9959–81. https://doi.org/10.18632/aging.103344.
Heer CD, Sanderson DJ, Alhammad YMO, et al. Coronavirus and PARP expression dysregulate the NAD Metabolome: a potentially actionable component of innate immunity. Preprint. bioRxiv. 2020;2020.04.17.047480. https://doi.org/10.1101/2020.04.17.047480. Published 30 Apr 2020.
Vanderhaeghen T, Vandewalle J, Libert C. Hypoxia-inducible factors in metabolic reprogramming during sepsis. FEBS J. 2020;287:1478–95. https://doi.org/10.1111/febs.15222(Epub 2020 Feb 3. Review).
Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052.
Cao Y, Li L, Feng Z, et al. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020;6:11. https://doi.org/10.1038/s41421-020-0147-1.
Gordon DE, Jang GM, Bouhaddou M, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–68. https://doi.org/10.1038/s41586-020-2286-9.
Kopecky-Bromberg SA, Martínez-Sobrido L, Frieman M, et al. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–57. https://doi.org/10.1128/JVI.01782-06.
Shi CS, Qi HY, Boularan C, et al. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J Immunol. 2014;193:3080–9. https://doi.org/10.4049/jimmunol.1303196.
Knoops K, Kikkert M, Worm SH, et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008;6:e226. https://doi.org/10.1371/journal.pbio.0060226.
Hagemeijer MC, Vonk AM, Monastyrska I, Rottier PJ, de Haan CA. Visualizing coronavirus RNA synthesis in time by using click chemistry. J Virol. 2012;86:5808–16. https://doi.org/10.1128/JVI.07207-11.
Wu KE, Fazal FM, Parker KR, et al. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020;11(102–108):e3. https://doi.org/10.1016/j.cels.2020.06.008(published online ahead of print, 2020 Jun 20).
Singh KK, Chaubey G, Chen JY, Suravajhala P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am J Physiol Cell Physiol. 2020;319:C258–67. https://doi.org/10.1152/ajpcell.00224.2020.
Somasundaran M, Zapp ML, Beattie LK, et al. Localization of HIV RNA in mitochondria of infected cells: potential role in cytopathogenicity. J Cell Biol. 1994;126:1353–60. https://doi.org/10.1083/jcb.126.6.1353.
Clark TW, Ewings S, Medina MJ, et al. Viral load is strongly associated with length of stay in adults hospitalised with viral acute respiratory illness. J Infect. 2016;73:598–606. https://doi.org/10.1016/j.jinf.2016.09.001(Epub 2016 Sep 9).
Granados A, Peci A, McGeer A, Gubbay JB. Influenza and rhinovirus viral load and disease severity in upper respiratory tract infections. J Clin Virol. 2017;86:14–9. https://doi.org/10.1016/j.jcv.2016.11.008(Epub 2016 Nov 22).
Zou L, Ruan F, Huang M, et al. SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N Engl J Med. 2020;382(12):1177–9. https://doi.org/10.1056/NEJMc2001737.
Liu Y, Yan LM, Wan L, et al. Viral dynamics in mild and severe cases of COVID-19. Lancet Infect Dis. 2020;20(6):656–7. https://doi.org/10.1016/S1473-3099(20)30232-2.
Chu CM, Poon LL, Cheng VC, et al. Initial viral load and the outcomes of SARS. CMAJ. 2004;171:1349–52. https://doi.org/10.1503/cmaj.1040398.
Vabret N, Britton GJ, Gruber C, et al. Immunology of COVID-19: current state of the science. Immunity. 2020;52:910–41. https://doi.org/10.1016/j.immuni.2020.05.002.
Duan K, Liu B, Li C, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc Natl Acad Sci USA. 2020;117:9490–6. https://doi.org/10.1073/pnas.2004168117.
Liu J, Li S, Liu J, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. 2020;55:102763. https://doi.org/10.1016/j.ebiom.2020.102763.
Tan L, Wang Q, Zhang D, et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther. 2020;5:33. https://doi.org/10.1038/s41392-020-0148-4.
Textoris J, Beaufils N, Quintana G, et al. Hypoxia-inducible factor (HIF1α) gene expression in human shock states. Crit Care. 2012;16:R120. https://doi.org/10.1186/cc11414.
Aswani A, Manson J, Itagaki K, et al. Scavenging circulating mitochondrial DNA as a potential therapeutic option for multiple organ dysfunction in trauma hemorrhage. Front Immunol. 2018;9:891. https://doi.org/10.3389/fimmu.2018.00891.
Wang X, Buechler NL, Yoza BK, et al. Resveratrol attenuates microvascular inflammation in sepsis via SIRT-1-Induced modulation of adhesion molecules in ob/ob mice. Obesity. 2015;23:1209–17. https://doi.org/10.1002/oby.21086.
Bonkowski MS, Sinclair DA. Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol. 2016;17:679–90. https://doi.org/10.1038/nrm.2016.93.
Vachharajani VT, Liu T, Brown CM, et al. SIRT1 inhibition during the hypoinflammatory phenotype of sepsis enhances immunity and improves outcome. J Leukoc Biol. 2014;96:785–96. https://doi.org/10.1189/jlb.3MA0114-034RR.
Anderson G, Reiter RJ. Melatonin: roles in influenza, Covid-19, and other viral infections. Rev Med Virol. 2020;30:e2109. https://doi.org/10.1002/rmv.2109.
Acknowledgements
The author has no conflict of interest to disclose or any funding for this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Shenoy, S. Coronavirus (Covid-19) sepsis: revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 69, 1077–1085 (2020). https://doi.org/10.1007/s00011-020-01389-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-020-01389-z