A Comparison of “Bottom-Up” and “Top-Down” Approaches to the Synthesis of Pt/C Electrocatalysts

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of Pt/C via the Polyol Process
2.3. Synthesis of Pt/C via EDPAC
2.4. Physical Characterization
2.5. Electrochemical Measurements
3. Results and Discussion
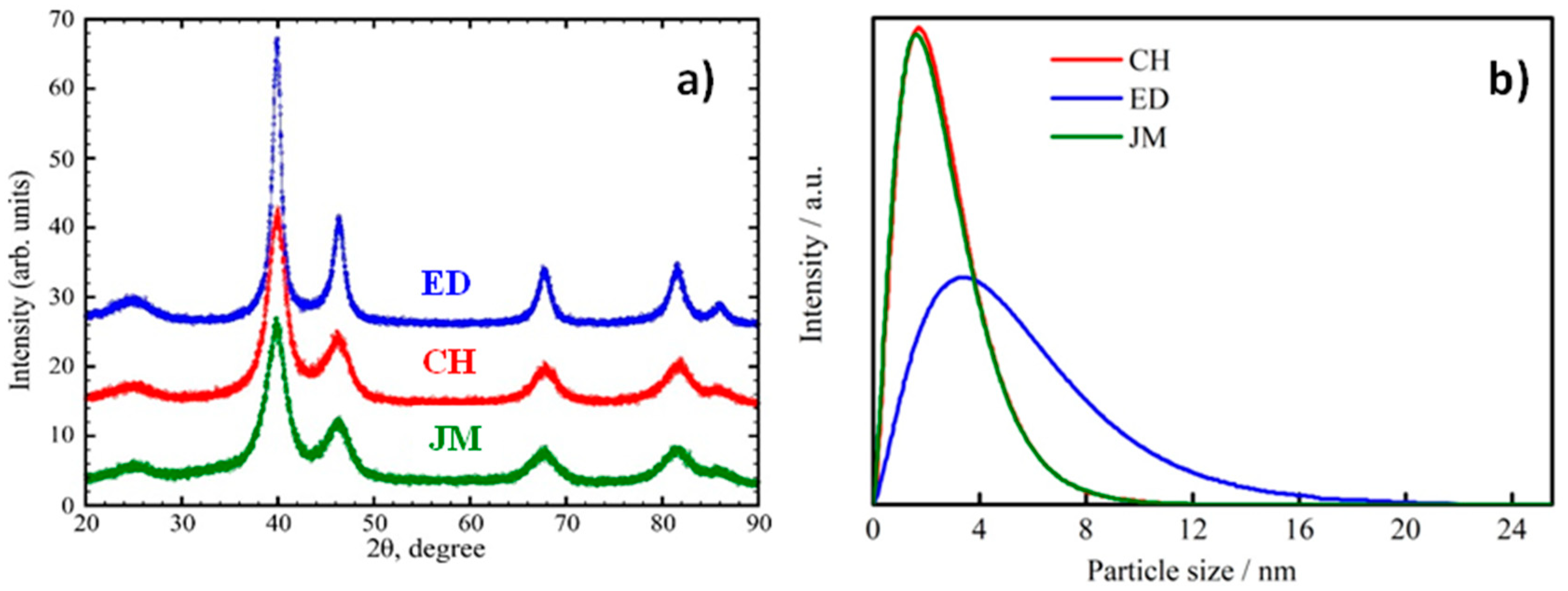
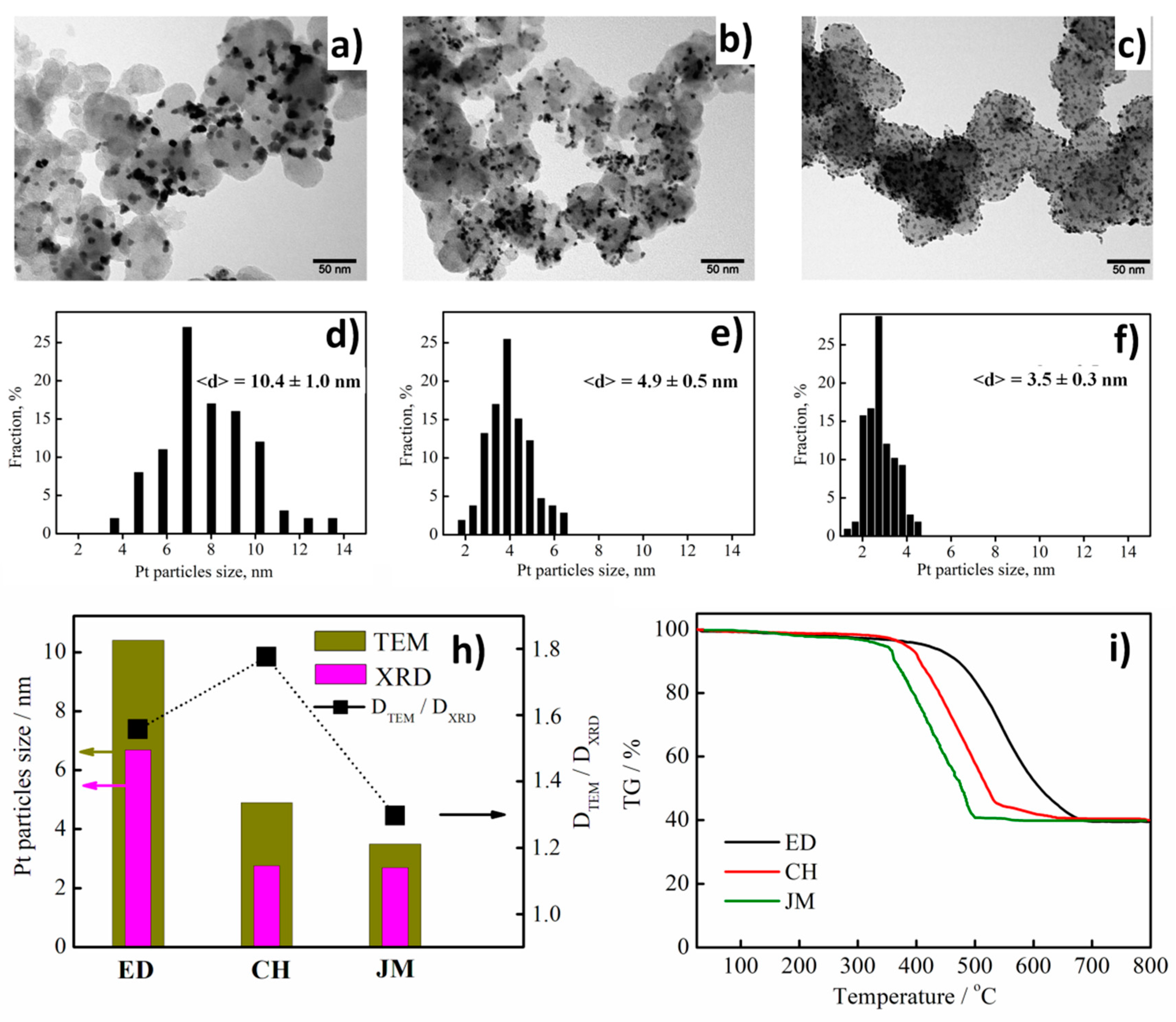
3.1. Physical Characterization of Pt/C Electrocatalysts
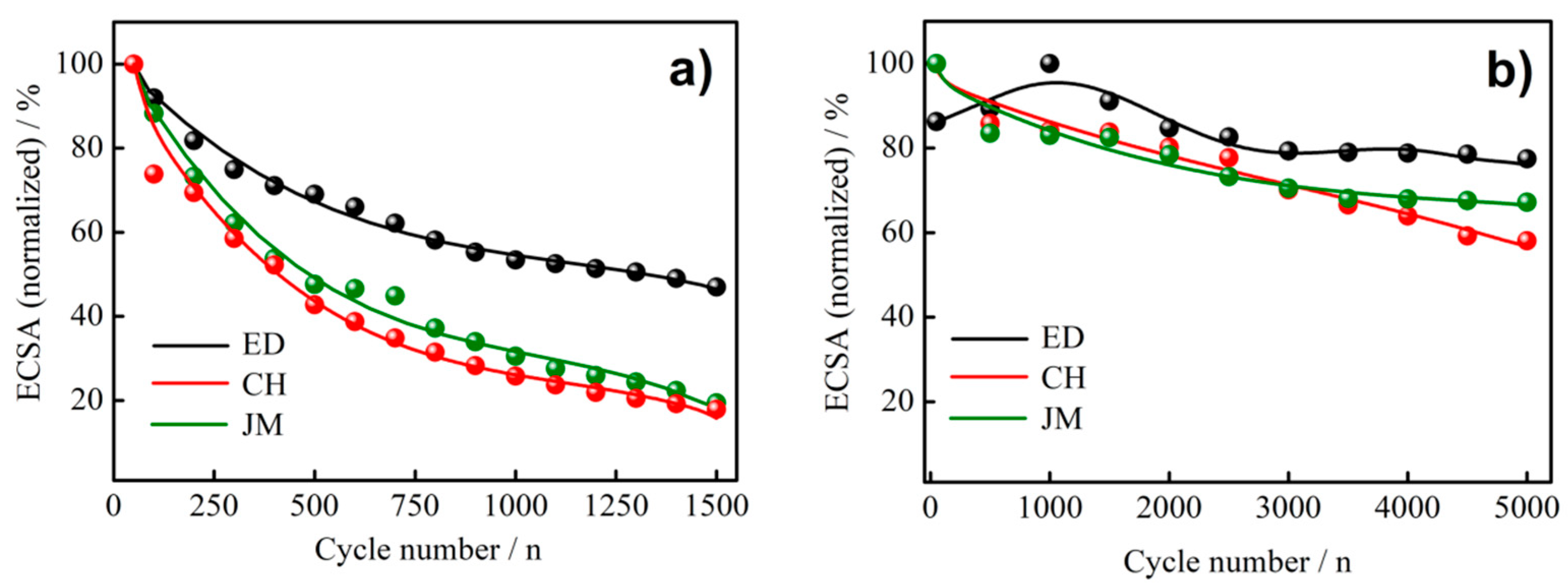
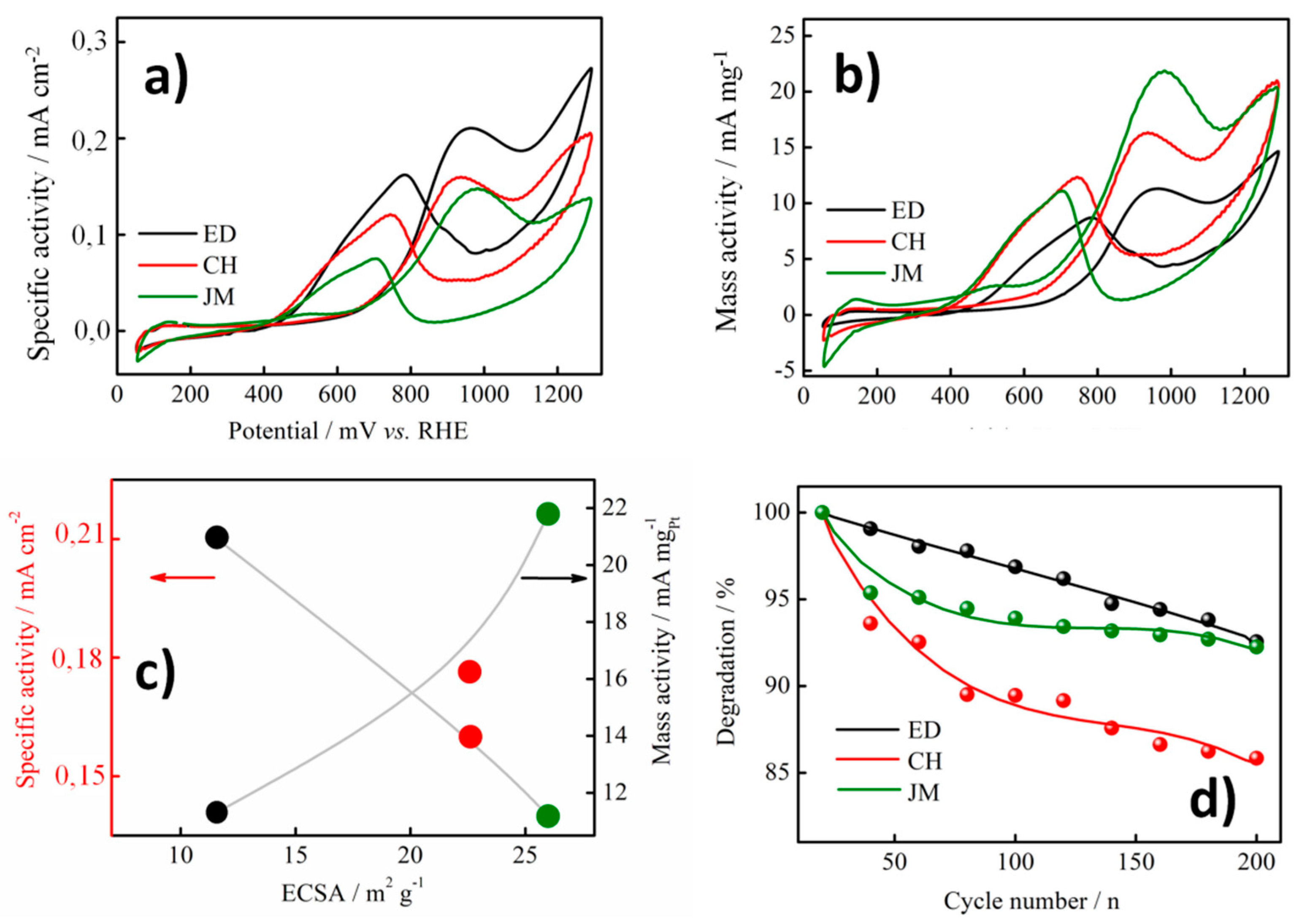
3.2. Electrochemical Measurements
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, C.-Y.; Zhang, D.; Zhao, Z.; Xia, Z. Covalent Organic Framework Electrocatalysts for Clean Energy Conversion. Adv. Mater. 2017, 30, 1703646. [Google Scholar] [CrossRef]
- Jung, N.; Chung, D.Y.; Ryu, J.; Yoo, S.J.; Sung, Y.-E. Pt-Based nanoarchitecture and catalyst design for fuel cell applications. Nano Today 2014, 9, 433–456. [Google Scholar] [CrossRef]
- Antolini, E. Structural parameters of supported fuel cell catalysts: The effect of particle size, inter-particle distance and metal loading on catalytic activity and fuel cell performance. Appl. Catal. B Environ. 2016, 181, 298–313. [Google Scholar] [CrossRef]
- Park, Y.-C.; Tokiwa, H.; Kakinuma, K.; Watanabe, M.; Uchida, M. Effects of carbon supports on Pt distribution, ionomer coverage and cathode performance for polymer electrolyte fuel cells. J. Power Sour. 2016, 315, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sour. 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Han, B.C.; Miranda, C.R.; Ceder, G. Effect of particle size and surface structure on adsorption of O and OH on platinum nanoparticles: A first-principles study. Phys. Rev. B 2008, 77, 075410. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.-S.; Vidal-Iglesias, F.J.; Solla-Gullón, J.; Sun, S.-G.; Feliu, J.M. Role of surface defect sites: From Pt model surfaces to shape-controlled nanoparticles. Chem. Sci. 2012, 3, 136–147. [Google Scholar] [CrossRef]
- Chen, Z.; Higgins, D.; Yu, A.; Zhang, L.; Zhang, J. A review on non-precious metal electrocatalysts for PEM fuel cells. Energy Environ. Sci. 2011, 4, 3167–3192. [Google Scholar] [CrossRef]
- Brouzgou, A.; Song, S.; Tsiakaras, P. Low and non-platinum electrocatalysts for PEMFCs: Current status, challenges and prospects. Appl. Catal. B Environ. 2012, 127, 371–388. [Google Scholar] [CrossRef]
- Pryadchenko, V.V.; Srabionyan, V.V.; Kurzin, A.A.; Bulat, N.V.; Shemet, D.B.; Avakyan, L.A.; Belenov, S.V.; Volochaev, V.A.; Zizak, I.; Guterman, V.E.; et al. Bimetallic PtCu core-shell nanoparticles in PtCu/C electrocatalysts: Structural and electrochemical characterization. Appl. Catal. A Gen. 2016, 525, 226–236. [Google Scholar] [CrossRef]
- Yang, H. Platinum-Based Electrocatalysts with Core-Shell Nanostructures. Angew. Chem. Int. Ed. 2011, 50, 2674–2676. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Wu, M.; Ning, S.; Huang, L.; Kanga, X.; Chen, S. Ru@Pt Core-Shell Nanoparticles: Impact of the Atomic Ordering of the Ru Metal Core on the Electrocatalytic Activity of the Pt Shell. ACS Sustain. Chem. Eng. 2019, 7, 9007–9016. [Google Scholar] [CrossRef]
- Sharma, G.; Kumar, D.; Kumar, A.; Al-Muhtaseb, A.H.; Pathania, D.; Naushad, M.; Mola, G.T. Revolution from monometallic to trimetallic nanoparticle composites, various synthesis methods and their applications: A review. Mater. Sci. Eng. C 2017, 71, 1216–1230. [Google Scholar] [CrossRef] [PubMed]
- Jamkhande, P.G.; Ghule, N.W.; Bamer, A.H.; Kalaskar, M.G. Metal nanoparticles synthesis: An overview on methods of preparation, advantages and disadvantages, and applications. J. Drug Deliv. Sci. Technol. 2019, 53, 101174. [Google Scholar] [CrossRef]
- Alegre, C.; Gálvez, M.E.; Moliner, R.; Baglio, V.; Aricò, A.; Lázaro, M.J.; Gresa, C.A. Towards an optimal synthesis route for the preparation of highly mesoporous carbon xerogel-supported Pt catalysts for the oxygen reduction reaction. Appl. Catal. B Environ. 2014, 147, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Fernández, P.; Rojas, S.; Ocón, P.; Gómez de la Fuente, J.L.; San Fabian, J.; Sanza, J.; Peña, M.A.; Garcia-Garcia, F.J.; Terreros, P.; Fierro, J.L.G. Influence of the Preparation Route of Bimetallic Pt-Au Nanoparticle Electrocatalysts for the Oxygen Reduction Reaction. J. Phys. Chem. C 2007, 111, 2913–2923. [Google Scholar] [CrossRef]
- Leontyev, I.; Kuriganova, A.; Kudryavtsev, Y.; Dkhil, B.; Smirnova, N. New life of a forgotten method: Electrochemical route toward highly efficient Pt/C catalysts for low-temperature fuel cells. Appl. Catal. A Gen. 2012, 431, 120–125. [Google Scholar] [CrossRef]
- Shinohara, K.; Ohma, A.; Iiyama, A.; Yoshida, T.; Daimaru, A. Membrane and Catalyst Performance Targets for Automotive Fuel Cells by FCCJ Membrane, Catalyst, MEA WG. ECS Meet. Abstr. 2011, 41, 775–784. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, X.-Z.; Hin, J.N.C.; Wang, H.; Friedrich, K.A.; Schulze, M. A review of platinum-based catalyst layer degradation in proton exchange membrane fuel cells. J. Power Sour. 2009, 194, 588–600. [Google Scholar] [CrossRef]
- Leontyev, I.N.; Kuriganova, A.B.; Leontyev, N.G.; Hennet, L.; Rakhmatullin, A.; Smirnova, N.V.; Dmitriev, V. Size dependence of the lattice parameters of carbon supported platinum nanoparticles: X-Ray diffraction analysis and theoretical considerations. RSC Adv. 2014, 4, 35959–35965. [Google Scholar] [CrossRef]
- Leontyev, I.N.; Belenov, S.V.; Guterman, V.E.; Haghi-Ashtiani, P.; Shaganov, A.P.; Dkhil, B. Catalytic Activity of Carbon-Supported Pt Nanoelectrocatalysts. Why Reducing the Size of Pt Nanoparticles is Not Always Beneficial. J. Phys. Chem. C 2011, 115, 5429–5434. [Google Scholar] [CrossRef]
- Leontyev, I.N.; Kuriganova, A.B.; Allix, M.; Rakhmatullin, A.; Timoshenko, P.E.; Maslova, O.A.; Mikheykin, A.S.; Smirnova, N.V. On the Evaluation of the Average Crystalline Size and Surface Area of Platinum Catalyst Nanoparticles. Phys. Status Solidi B 2018, 255. [Google Scholar] [CrossRef]
- Yanson, A.I.; Rodriguez, P.; Garcia-Araez, N.; Mom, R.V.; Tichelaar, F.D.; Koper, M.T.M. Cathodic Corrosion: A Quick, Clean, and Versatile Method for the Synthesis of Metallic Nanoparticles. Angew. Chem. Int. Ed. 2011, 50, 6346–6350. [Google Scholar] [CrossRef] [PubMed]
- Doronkin, D.E.; Kuriganova, A.B.; Leontyev, I.N.; Baier, S.; Lichtenberg, H.; Smirnova, N.V.; Grunwaldt, J.-D. Electrochemically Synthesized Pt/Al2O3 Oxidation Catalysts. Catal. Lett. 2015, 146, 452–463. [Google Scholar] [CrossRef]
- Novikova, K.; Kuriganova, A.; Leontyev, I.; Gerasimova, E.; Maslova, O.; Rakhmatullin, A.; Smirnova, N.V.; Dobrovolsky, Y. Influence of Carbon Support on Catalytic Layer Performance of Proton Exchange Membrane Fuel Cells. Electrocatalysis 2017, 9, 22–30. [Google Scholar] [CrossRef]
- Cherevko, S.; Topalov, A.A.; Zeradjanin, A.R.; Katsounaros, I.; Mayrhofer, K.J.J. Gold dissolution: Towards understanding of noble metal corrosion. RSC Adv. 2013, 3, 16516. [Google Scholar] [CrossRef]
- Cherevko, S.; Zeradjanin, A.R.; Topalov, A.A.; Kulyk, N.; Katsounaros, I.; Mayrhofer, K.J.J. Dissolution of Noble Metals during Oxygen Evolution in Acidic Media. ChemCatChem 2014, 6, 2219–2223. [Google Scholar] [CrossRef]
- Katsounaros, I.; Meier, J.C.; Klemm, S.O.; Topalov, A.A.; Biedermann, P.U.; Auinger, M.; Mayrhofer, K.J.J. The effective surface pH during reactions at the solid–liquid interface. Electrochem. Commun. 2011, 13, 634–637. [Google Scholar] [CrossRef]
- Klemm, S.O.; Karschin, A.; Schuppert, A.K.; Topalov, A.A.; Mingers, A.M.; Katsounaros, I.; Mayrhofer, K.J.J. Time and potential resolved dissolution analysis of rhodium using a microelectrochemical flow cell coupled to an ICP-MS. J. Electroanal. Chem. 2012, 677, 50–55. [Google Scholar] [CrossRef]
- Topalov, A.A.; Cherevko, S.; Zeradjanin, A.R.; Meier, J.C.; Katsounaros, I.; Mayrhofer, K.J.J. Towards a comprehensive understanding of platinum dissolution in acidic media. Chem. Sci. 2014, 5, 631–638. [Google Scholar] [CrossRef] [Green Version]
- Topalov, A.A.; Katsounaros, I.; Auinger, M.; Cherevko, S.; Meier, J.C.; Klemm, S.O.; Mayrhofer, K. Dissolution of Platinum: Limits for the Deployment of Electrochemical Energy Conversion? Angew. Chem. Int. Ed. 2012, 51, 12613–12615. [Google Scholar] [CrossRef] [Green Version]
- Kasatkin, V.E.; Tytik, D.L.; Revina, A.A.; Busev, S.A.; Abaturov, M.A.; Vysotskii, V.V.; Roldugin, V.I.; Kazanskii, L.P.; Kuz’Min, V.I.; Gadzaov, A.; et al. Electrochemical synthesis of iron and platinum nanoparticles in deionized water. Prot. Met. Phys. Chem. Surf. 2015, 51, 973–979. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, C.; Yang, X.; Feng, L.; Gallogly, E.B.; Wang, R. Studies on how to obtain the best catalytic activity of Pt/C catalyst by three reduction routes for methanol electro-oxidation. Electrochem. Commun. 2011, 13, 314–316. [Google Scholar] [CrossRef]
- Qi, J.; Jiang, L.; Jing, M.; Tang, Q.; Sun, G. Preparation of Pt/C via a polyol process—Investigation on carbon support adding sequence. Int. J. Hydrogen Energy 2011, 36, 10490–10501. [Google Scholar] [CrossRef]
- Comignani, V.; Sieben, J.M.; Sanchez, M.D.; Duarte, M.M.E. Influence of carbon support properties on the electrocatalytic activity of PtRuCu nanoparticles for methanol and ethanol oxidation. Int. J. Hydrogen Energy 2017, 42, 24785–24796. [Google Scholar] [CrossRef]
- Fraga, M.A.; Jordão, E.; Mendes, M.J.; Freitas, M.M.A.; Faria, J.L.; Figueiredo, J.L. Properties of Carbon-Supported Platinum Catalysts: Role of Carbon Surface Sites. J. Catal. 2002, 209, 355–364. [Google Scholar] [CrossRef]
- Gurrath, M.; Kuretzky, T.; Boehm, H.P.; Okhlopkova, L.B.; Lisitsyn, A.S.; Likholobov, V.A. Palladium catalysts on activated carbon supports. Carbon 2000, 38, 1241–1255. [Google Scholar] [CrossRef]
- López-Cudero, A.; Solla-Gullón, J.; Herrero, E.; Aldaz, A.; Feliu, J.M. CO electrooxidation on carbon supported platinum nanoparticles: Effect of aggregation. J. Electroanal. Chem. 2010, 644, 117–126. [Google Scholar] [CrossRef]
- Maillard, F.; Schreier, S.; Hanzlik, M.; Savinova, E.R.; Weinkauf, S.; Stimming, U. Influence of particle agglomeration on the catalytic activity of carbon-supported Pt nanoparticles in CO monolayer oxidation. Phys. Chem. Chem. Phys. 2005, 7, 385–393. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Watt-Smith, M.J.; Friedrich, J.M.; Rigby, S.P.; Ralph, T.R.; Walsh, F.C. Determination of the electrochemically active surface area of Pt/C PEM fuel cell electrodes using different adsorbates. J. Phys. D Appl. Phys. 2008, 41, 174004. [Google Scholar] [CrossRef]
- Li, W.; Lane, A.M. Resolving the HUPD and HOPD by DEMS to determine the ECSA of Pt electrodes in PEM fuel cells. Electrochem. Commun. 2011, 13, 913–916. [Google Scholar] [CrossRef]
- Thomas, J.M. Handbook of Heterogeneous Catalysis, 2nd Completely Revised and Enlarged Edition; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley: Hoboken, NJ, USA, 2008; Volume 8. [Google Scholar]
- Abe, K.; Uchida, H.; Inukai, J. Electro-Oxidation of CO Saturated in 0.1 M HClO4 on Basal and Stepped Pt Single-Crystal Electrodes at Room Temperature Accompanied by Surface Reconstruction. Surfaces 2019, 2, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Arenz, M.; Mayrhofer, K.J.J.; Stamenković, V.; Blizanac, B.B.; Tomoyuki, T.; Ross, P.N.; Marković, N.M. The Effect of the Particle Size on the Kinetics of CO Electrooxidation on High Surface Area Pt Catalysts. J. Am. Chem. Soc. 2005, 127, 6819–6829. [Google Scholar] [CrossRef]
- Farias, M.J.S.; Busó-Rogero, C.; Vidal-Iglesias, F.J.; Solla-Gullón, J.; Camara, G.A.; Feliu, J.M. Mobility and Oxidation of Adsorbed CO on Shape-Controlled Pt Nanoparticles in Acidic Medium. Langmuir 2017, 33, 865–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Duan, Z.; Henkelman, G. Computational design of CO-tolerant Pt3M anode electrocatalysts for proton-exchange membrane fuel cells. Phys. Chem. Chem. Phys. 2019, 21, 4046–4052. [Google Scholar] [CrossRef] [PubMed]
- Maillard, F.; Savinova, E.R.; Stimming, U. CO monolayer oxidation on Pt nanoparticles: Further insights into the particle size effects. J. Electroanal. Chem. 2007, 599, 221–232. [Google Scholar] [CrossRef]
- Lafforgue, C.; Zadick, A.; Dubau, L.; Maillard, F.; Chatenet, M. Selected Review of the Degradation of Pt and Pd-based Carbon-supported Electrocatalysts for Alkaline Fuel Cells: Towards Mechanisms of Degradation. Fuel Cells 2018, 18, 229–238. [Google Scholar] [CrossRef]
- Weber, P.; Werheid, M.; Janssen, M.; Oezaslan, M. Fundamental Insights in Degradation Mechanisms of Pt/C Nanoparticles for the ORR. ECS. Transactions 2018, 86, 433–445. [Google Scholar]
- Kuriganova, A.; Leontyeva, D.V.; Smirnova, N. On the mechanism of electrochemical dispersion of platinum under the action of alternating current. Russ. Chem. Bull. 2015, 64, 2769–2775. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, J.; Wang, X.; Zhang, X.; Toyoda, H.; Nagatsu, M.; Meng, Y. High performance of carbon nanowall supported Pt catalyst for methanol electro-oxidation. Carbon 2012, 50, 3731–3738. [Google Scholar] [CrossRef]
- Wang, X.X.; Tan, Z.H.; Zeng, M.; Wang, J.N. Carbon nanocages: A new support material for Pt catalyst with remarkably high durability. Sci. Rep. 2014, 4, 4437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, Y.; Wang, X. Nanoporous bimetallic Pt-Au alloy nanocomposites with superior catalytic activity towards electro-oxidation of methanol and formic acid. Nanoscale 2011, 3, 1663–1674. [Google Scholar] [CrossRef]
- Garsany, Y.; Singer, I.L.; Swider-Lyons, K.E. Impact of film drying procedures on RDE characterization of Pt/VC electrocatalysts. J. Electroanal. Chem. 2011, 662, 396–406. [Google Scholar] [CrossRef]
- Basu, D.; Basu, S. Synthesis and characterization of Pt-Au/C catalyst for glucose electro-oxidation for the application in direct glucose fuel cell. Int. J. Hydrogen Energy 2011, 36, 14923–14929. [Google Scholar] [CrossRef]
- Fu, T.; Fang, J.; Wang, C.; Zhao, J. Hollow porous nanoparticles with Pt skin on a Ag-Pt alloy structure as a highly active electrocatalyst for the oxygen reduction reaction. J. Mater. Chem. A 2016, 4, 8803–8811. [Google Scholar] [CrossRef]
- Saleh, F.S.; Easton, E.B. Assessment of the ethanol oxidation activity and durability of Pt catalysts with or without a carbon support using Electrochemical Impedance Spectroscopy. J. Power Sour. 2014, 246, 392–401. [Google Scholar] [CrossRef]
- Sohn, H.; Xiao, Q.; Seubsai, A.; Ye, Y.; Lee, J.; Han, H.; Park, S.; Chen, G.; Lu, Y. Thermally Robust Porous Bimetallic (NixPt1–x) Alloy Mesocrystals within Carbon Framework: High-Performance Catalysts for Oxygen Reduction and Hydrogenation Reactions. ACS Appl. Mater. Interfaces 2019, 11, 21435–21444. [Google Scholar] [CrossRef]
- Jiang, Z.-J.; Jiang, Z.-J.; Meng, Y. High catalytic performance of Pt nanoparticles on plasma treated carbon nanotubes for electrooxidation of ethanol in a basic solution. Appl. Surf. Sci. 2011, 257, 2923–2928. [Google Scholar] [CrossRef]
- Jung, J.H.; Park, H.J.; Kim, J.; Hur, S.H. Highly durable Pt/graphene oxide and Pt/C hybrid catalyst for polymer electrolyte membrane fuel cell. J. Power Sour. 2014, 248, 1156–1162. [Google Scholar] [CrossRef]
- Zeng, M.; Wang, X.X.; Tan, Z.H.; Huang, X.X.; Wang, J.N. Remarkable durability of Pt-Ir alloy catalysts supported on graphitic carbon nanocages. J. Power Sour. 2014, 264, 272–281. [Google Scholar] [CrossRef]
- Sakthivel, M.; Drillet, J.-F. An extensive study about influence of the carbon support morphology on Pt activity and stability for oxygen reduction reaction. Appl. Catal. B Environ. 2018, 231, 62–72. [Google Scholar] [CrossRef]
- Song, J.; Li, G.; Qiao, J. Ultrafine porous carbon fiber and its supported platinum catalyst for enhancing performance of proton exchange membrane fuel cells. Electrochim. Acta 2015, 177, 174–180. [Google Scholar] [CrossRef]
- Naidoo, Q.-L.; Naidoo, S.; Petrik, L.; Nechaev, A.; Ndungu, P. The influence of carbon based supports and the role of synthesis procedures on the formation of platinum and platinum-ruthenium clusters and nanoparticles for the development of highly active fuel cell catalysts. Int. J. Hydrogen Energy 2012, 37, 9459–9469. [Google Scholar] [CrossRef]
- Liu, Z.; Hong, L.; Tham, M.P.; Lim, T.H.; Jiang, H. Nanostructured Pt/C and Pd/C catalysts for direct formic acid fuel cells. J. Power Sour. 2006, 161, 831–835. [Google Scholar] [CrossRef]
- Alekseenko, A.A.; Guterman, V.E.; Volochaev, V.A.; Belenov, S.V.; Vladimir, G. Effect of wet synthesis conditions on the microstructure and active surface area of Pt/C catalysts. Inorg. Mater. 2015, 51, 1258–1263. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, S.; Zhou, W.; Wang, G.; Jiang, L.; Li, W.; Song, S.; Liu, J.; Sun, G.; Xin, Q. Novel synthesis of highly active Pt/C cathode electrocatalyst for direct methanol fuel cell. Chem. Commun. 2003, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Lebègue, E.; Baranton, S.; Coutanceau, C. Polyol synthesis of nanosized Pt/C electrocatalysts assisted by pulse microwave activation. J. Power Sour. 2011, 196, 920–927. [Google Scholar] [CrossRef]
- Gu, Z.; Balbuena, P.B. Absorption of Atomic Oxygen into Subsurfaces of Pt(100) and Pt(111): Density Functional Theory Study. J. Phys. Chem. C 2007, 111, 9877–9883. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Accelerated Ageing, Mode 1 | Soft Ageing, Mode 2 |
|---|---|---|
| Number of cycles | 1500 | 5000 |
| Electrolyte | 0.5 М H2SO4 | 0.5 М H2SO4 |
| Temperature (°C) | 22–23 | 22–23 |
| Potential range (V) | 0.05–1.35 | 0.6–1.0 |
| Potential sweep rate (mV s−1) | 50 | 100 |
| ECSA measurement | Every 100 cycles | Every 500 cycles |
| Sample | ED | CH | JM |
|---|---|---|---|
| Dav (nm) | 4.74 | 2.55 | 2.48 |
| ΔDav | 0.86 | 0.5 | 0.2 |
| D111 (nm) | 6.68 | 2.76 | 2.70 |
| D200 (nm) | 4.71 | 2.08 | 2.01 |
| D200/D111 | 0.78 | 0.77 | 0.83 |
| <D> (nm) | 5.56 | 2.69 | 2.68 |
| σ (nm) | 3.49 | 1.63 | 1.69 |
| Sgeom (m2 g−1) | 28.13 | 59.96 | 58.14 |
| a (Å) | 3.9153 | 3.9133 | 3.9151 |
| Sample Preparation Method | Particle Size, nm | ECSA (m2 g−1)/Technique | Ref. |
|---|---|---|---|
| JM (40 wt % Pt/C) | 3.0 | 50.9/HUPD | [52] |
| 3.5 | 46.4/HUPD | [53] | |
| - | 39.4/HUPD | [54] | |
| 3.1 | 49 ± 1/HUPD | [55] | |
| 4.6 | 60.8/HUPD | [56] | |
| 3.0 | 74.5/CO stripping | [57] | |
| 3.5 | 29.0/HUPD | [58] | |
| - | 63.9/HUPD | [59] | |
| 3.09 | 43.9/HUPD | [60] | |
| 3.0 | 54.21/HUPD | [61] | |
| 3.0 | 46.4/HUPD | [62] | |
| 5.1 | 18.17/HUPD | [63] | |
| 3.4 | 36.3/HUPD | [64] | |
| 3.76 | 190.0/HUPD | [65] | |
| 3.5 | 18.0 ± 0.1/HUPD 1 | This work | |
| 22.0 ± 0.2/HUPD 2 | This work | ||
| 26.0 ± 0.5/CO stripping 3 | This work | ||
| 30 wt % Pt/C/polyol process | 3.6 | - | [66] |
| 19 wt % Pt/C/polyol process | 2.0 | 99.0 ± 10/HUPD | [67] |
| 40 wt % Pt/C/polyol process | 2.9 | - | [68] |
| 40 wt % Pt/C/microwave-assisted polyol synthesis | 2.6 ± 0.7 | - | [69] |
| 40 wt % Pt/C/polyol synthesis | 3.1 | 58.6/HUPD | [49] |
| 40 wt % Pt/C/polyol synthesis | 2.9 | 53.0/HUPD | [34] |
| CH (40 wt % Pt/C)/polyol synthesis | 2.55 (XRD) | 13.0 ± 0.1/HUPD 1 | This work |
| 15.0 ± 0.2/HUPD 2 | This work | ||
| 22.0 ± 0.2/CO stripping 3 | This work |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuriganova, A.; Faddeev, N.; Gorshenkov, M.; Kuznetsov, D.; Leontyev, I.; Smirnova, N. A Comparison of “Bottom-Up” and “Top-Down” Approaches to the Synthesis of Pt/C Electrocatalysts. Processes 2020, 8, 947. https://doi.org/10.3390/pr8080947
Kuriganova A, Faddeev N, Gorshenkov M, Kuznetsov D, Leontyev I, Smirnova N. A Comparison of “Bottom-Up” and “Top-Down” Approaches to the Synthesis of Pt/C Electrocatalysts. Processes. 2020; 8(8):947. https://doi.org/10.3390/pr8080947
Chicago/Turabian StyleKuriganova, Alexandra, Nikita Faddeev, Mikhail Gorshenkov, Dmitri Kuznetsov, Igor Leontyev, and Nina Smirnova. 2020. "A Comparison of “Bottom-Up” and “Top-Down” Approaches to the Synthesis of Pt/C Electrocatalysts" Processes 8, no. 8: 947. https://doi.org/10.3390/pr8080947