
Abstract
Multiple system atrophy (MSA) is a progressive neurodegenerative disease variably associated with motor, nonmotor, and autonomic symptoms, resulting from putaminal and cerebellar degeneration and associated with glial cytoplasmic inclusions enriched with α-synuclein in oligodendrocytes and neurons. Although symptomatic treatment of MSA can provide significant improvements in quality of life, the benefit is often partial, limited by adverse effects, and fails to treat the underlying cause. Consistent with the multisystem nature of the disease and evidence that motor symptoms, autonomic failure, and depression drive patient assessments of quality of life, treatment is best achieved through a coordinated multidisciplinary approach driven by the patient’s priorities and goals of care. Research into disease-modifying therapies is ongoing with a particular focus on synuclein-targeted therapies among others. This review focuses on both current management and emerging therapies for this devastating disease.
Similar content being viewed by others

Introduction
Multiple system atrophy (MSA) is a rare, progressive neurodegenerative disease with estimated incidence of 0.6 to 0.7 cases per 100,000 person-years and prevalence of 3.4 to 4.9 cases per 100,000 [1,2,3,4]. The mean age of symptom onset is reported between 54 and 63 years, depending on the study, and the estimated survival from symptom onset is 6 to 11 years (median survival, ~ 9.5 years) [5,6,7,8]. MSA comprises a variable combination of autonomic dysfunction, parkinsonism, and ataxia, with the predominance of one or other motor syndrome defining parkinsonian (MSA-P) or cerebellar subtypes (MSA-C) [9]. Currently available lab tests or imaging do not provide additional diagnostic or prognostic sensitivity or specificity beyond that of a thorough clinical assessment. Some fluid biomarkers and imaging findings, however, are thought to be supportive [10, 11]. Commensurate with the multisystem nature of the disease, effective management requires both evidence-based and off-label multidisciplinary treatment with the patient at the center of an interdisciplinary team of well-coordinated providers including the neurologist, consulting physicians, rehabilitation therapists (physical, occupational, and speech–language), dietician, social worker, and potential research staff. Such treatment is driven largely by personalized, symptomatic management of the patient’s specific constellation of symptoms. There are currently no approved disease-modifying therapies, although research in a variety of mechanistically driven areas is ongoing and the patient should be given the opportunity to participate in clinical trials. This article provides an overview of the pathology and pathogenesis, clinical presentation, evaluation, treatment, and management, including both evidence-based and off-label therapies. We also discuss ongoing research efforts to develop potential symptomatic and disease-modifying therapies for this complex and devastating disease.
Pathology and Pathogenesis
Neuropathologically, MSA exhibits putaminal, pontine, and cerebellar atrophy [12] that is associated with shared and differing relative patterns of degeneration consistent with the specific clinical subtype. MSA-P is typically associated with prominent caudal putamen and caudate involvement with preferential degeneration of GABAergic medium spiny neurons, as well as substantia nigra involvement with degeneration of dopaminergic striatonigral neurons [13, 14]. In contrast, MSA-C tends to show more prominent degeneration of the dentate and inferior olivary nuclei, as well as the basis pontis, cerebellar vermis and hemispheres, and cerebellopontine fibers [13]. Both subtypes share involvement of the motor and supplementary motor cortex. The pathology of the locus coeruleus and vagus and Onuf’s nuclei, and degeneration of the intermediolateral spinal columns, contribute to autonomic dysfunction [15, 16].
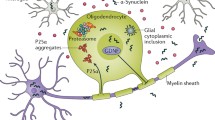
MSA histopathology is characterized by oligodendroglial cytoplasmic inclusions (GCIs), or Papp–Lantos bodies, that contain abundant pathologic α-synuclein aggregates and other proteins (Fig. 1) [13, 17,18,19,20]. Similar neuronal inclusions have also been described in the cytoplasm (NCIs) and nuclei (NNIs), but are much less common [17, 21]. GCI pathology is a defining feature of “definite” MSA and correlates with neuronal loss and disease duration [22]. Neuronal loss, axonal degeneration, astrogliosis, and microglial activation are widespread and variably involve the putamen, substantia nigra, pons, inferior olivary nucleus, cerebellum, and brainstem depending on the MSA subtype [23].

(A) Glial cytoplasmic inclusions (arrows) in oligodendrocytes (hematoxylin and eosin stain). (B) α-Synuclein staining (brown) in GCIs
The presence of α-synuclein aggregates in oligodendrocyte is a notable distinguishing feature of MSA pathology as compared to other synucleinopathies, such as Parkinson disease and dementia with Lewy bodies, in which α-synuclein deposits are predominantly neuronal. The physiological role of α-synuclein remains an active area of research, and evidence suggests roles in synaptic vesicular function and neurotransmitter release [24]. The role of α-synuclein in glial cells has yet to be elucidated. Pathologically, α-synuclein is phosphorylated and self-aggregates to form fibrils, aggregates, and larger intracellular inclusions recognized as Lewy bodies [25]. A wealth of evidence indicates that pathological forms of α-synuclein (i.e., fibrils or aggregates) are secreted or released from neurons, extracellularly, and may propagate α-synuclein pathology from cell to cell in a prion-like fashion [26,27,28]. Further evidence suggests the presence of different conformational “strains” of α-synuclein fibrils that result in preferential degeneration of certain cell types and provide an explanation for the clinical diversity among synucleinopathies [29, 30]. Human brain homogenates from MSA patients have been shown to “seed” pathological α-synuclein fibrils in cultured cells and neuronal deposits in transgenic mice expressing human α-synuclein [31, 32]. Although different α-synuclein “strains” have been shown to target distinct cellular populations and cell types within the brain, and recapitulate some of the selective targeting observed in human synucleinopathies, a major limitation of these models is that they exhibit only neuronal α-synuclein pathology. Understanding how or why α-synuclein pathology targets mainly oligodendrocytes has yet to be elucidated. Nevertheless, these findings have led to significant efforts toward disease-modifying therapies focused on reduction or clearance of pathological α-synuclein [33].
Clinical Symptoms
Motor Symptoms
Historically, a variety of eponymous or general anatomic terms have been used to refer to MSA, often denoting the predominant constellation of symptoms. These terms included Shy–Drager syndrome, striatonigral degeneration, and olivopontocerebellar atrophy, which tended to refer to patients with predominant autonomic dysfunction, parkinsonism features, or cerebellar dysfunction, respectively. The discovery that these seemingly different syndromes shared a common pathology of oligodendroglial cytoplasmic inclusions (GCI) motivated efforts to modernize terminology. The first consensus definition was established in 1998 and revised in 2007 [34, 35].
The core motor symptoms of MSA are parkinsonism and ataxia. Modern categorization includes 2 subtypes: the MSA-parkinsonism subtype (MSA-P) with predominant symptoms of tremor, rigidity, and bradykinesia that are poorly responsive to levodopa and the MSA-cerebellar subtype (MSA-C) with predominant symptoms of gait and limb ataxia, scanning dysarthria, and cerebellar oculomotor dysfunction, though parkinsonian features are sometimes also present. Generally, symptom presentation is more symmetric and tremor less prominent compared to that in Parkinson disease. MSA tremor has been described as having a higher frequency and lower amplitude as compared to Parkinson disease, with a “jerky,” stimulus-sensitive, myoclonic component in the setting of sustained posture holding, referred to as polyminimyoclonus [36, 37]. Initial presenting symptoms of MSA vary widely and overlap with those of other parkinsonian disorders and sporadic adult-onset ataxias. As such, delay in diagnosis and misdiagnosis are common [38,39,40].
Beyond the core motor symptoms of MSA, a variety of additional motor symptoms are common. Dystonia affects over 40% of patients [41] and includes limb, orofacial, cervical, and truncal dystonias. Limb dystonias can lead to significant disability, painful contractures, and early striatal hands or toe deformities. Anterocollis, Pisa syndrome, and camptocormia, characterized by abnormal forward or lateral flexion of the trunk during standing or walking that is relieved by lying down, are particularly associated with MSA, although these can be seen in other forms of parkinsonism [42, 43]. In addition, restless leg syndrome (RLS) has been described as affecting between 5 and 30% of patients [44, 45]. Other suggestive features include hyperreflexia, Babinski sign, early dysarthria, dysphagia, inspiratory sigh, and stridor [9]. The speech in MSA in particular is characterized by a mixed, spastic–hypokinetic (or ataxia, if MSA-C) dysarthria, sometimes with a vocal “quiver” or “flutter” [46, 47]. Dysphagia occurs earlier and is more severe than that seen in Parkinson disease.
Nonmotor, Cognitive, and Affective Symptoms
MSA has significant cognitive and affective symptoms, often referred to as “nonmotor” symptoms, many of which play a prominent role in patients’ quality of life and should be a major focus of clinical assessment and therapy [48]. Although dementia is an exclusion criterion for a clinical diagnosis of MSA, single or multidomain cognitive impairment is not uncommon and frank dementia is reported in up to 10 to 15% of patients [49]. Executive dysfunction is most common and consistent with frontal–subcortical pathology seen in MSA and other parkinsonian disorders.
Depressive symptoms and anxiety are common in MSA and more prevalent and severe than in Parkinson disease, and correlate with poor quality of life [50,51,52,53,54]. Emotional lability or pseudobulbar affect, characterized by uncontrolled, sudden, inappropriate laughter or crying, may also occur [55].
Autonomic Symptoms
Prominent autonomic symptoms are a key defining feature of MSA, but have variable presentations. The clinical criteria for MSA require the presence of either orthostatic hypotension or significant urinary dysfunction such as urgency, increased frequency, incontinence, or retention (needing catheterization) with erectile dysfunction in males that is not otherwise explained by diseases such as diabetes or autonomic neuropathy [35]. It is important to note that significant orthostatic hypotension may be lacking in a given patient despite the presence of other prominent autonomic symptoms. Additional autonomic features may include gastrointestinal symptoms such as gastroparesis, constipation, or diarrhea; heat and cold intolerance; flushing, diaphoresis, hypohidrosis, or global anhidrosis; and acrocyanosis characterized by bluish discoloration of the fingers or toes [4, 56, 57]. Urinary dysfunction, in particular, is a prominent and common early autonomic symptom in MSA with incomplete bladder emptying, urinary retention, and other voiding difficulties [58, 59]. In males, erectile dysfunction can be a telltale sign of early MSA even in the absence (i.e., preceding) of orthostatic symptoms [60, 61].
Autonomic symptoms are the earliest symptom in up to 50% of patients with early or severe autonomic dysfunction carrying a more guarded prognosis [62, 63]. Clinicians should rule out secondary causes of dysautonomia such as diabetes or other common maladies such as benign prostatic hypertrophy (BPH) in males or bladder prolapse in females that may contribute to urinary dysfunction. In addition, primary autonomic failure (PAF) should only be entertained as an alternative diagnosis after 5 years of isolated autonomic disturbances as some patients with presumptive PAF diagnosis will phenoconvert to MSA in time [64]. As olfactory function is relatively preserved in PAF compared to PD and MSA, this may be a clinically useful distinguishing feature [65].
Sleep Issues and Disordered Breathing
A variety of sleep-related problems are common in MSA. REM sleep behavior disorder (RBD), characterized by recurrent dream enactment with excessive motor behavior (i.e., thrashing, punching, kicking) [66], is frequent not only in MSA but other α-synucleinopathies, Parkinson disease, and dementia with Lewy bodies, with over 90% of patients affected [4]. RBD contributes to poor sleep quality and, if severe, may result in injury to self or bedpartners. RBD symptoms may precede the onset of MSA by several years and “burn-out” later in disease [67]. Central and obstructive sleep apnea are also common in MSA [68, 69]. Laryngeal stridor [70, 71] is a particularly concerning symptom that may play a central role in survival through oxygen desaturations, respiratory failure, and sudden death during sleep [69]. Stridor may be a presenting feature of MSA and is defined as a strained, high-pitched, harsh respiratory sound, mainly inspiratory, occurring only during sleep or during both sleep and wakefulness, that is caused by laryngeal dysfunction leading to narrowing of the rima glottides [70]. Diagnosis can be made clinically if present or with the aid of a recording or bed partner, and confirmed through laryngoscopy, sleep endoscopy, or video polysomnography. Given the prevalence of these symptoms, polysomnography should be considered in the initial workup for patients with MSA even in the absence of reported symptoms.
Diagnosis
The diagnosis of MSA is primarily clinical and based on the most recent consensus criteria [35]. Three levels of diagnostic certainty were established: definite, probable, and possible (Table 1). Whereas a diagnosis of definite MSA requires postmortem histopathologic confirmation, probable MSA requires autonomic failure (orthostatic hypotension defined by a drop of 30 mmHg systolic and 15 mmHg diastolic from supine to within 3 min of standing, or urinary incontinence/retention and erectile dysfunction in males) and either parkinsonism that is poorly levodopa-responsive or a predominant cerebellar syndrome. Possible MSA includes less stringent criteria: parkinsonism or cerebellar syndrome plus at least one feature suggesting autonomic dysfunction (i.e., urinary urgency, frequency, or incomplete emptying; erectile dysfunction in men, or orthostatic hypotension, though only requiring a 20-point drop in systolic and a 10-point drop in diastolic blood pressure). At least one additional feature is required such as hyperreflexia/Babinski sign or stridor, or imaging findings consistent with MSA such as putaminal atrophy, FDG–PET hypometabolism, or evidence of striatal presynaptic dopaminergic denervation on SPECT or PET. With the 2008 revised criteria, improved sensitivity was achieved in early disease for the “probable MSA” category [72]. It should be noted, however, that despite the criteria’s > 90% sensitivity in late disease and the relative improvement in early probable disease, sensitivity remains low at 41%, warranting continued efforts to improve diagnosis for early disease [72].
Despite an aggressive effort to identify reliable diagnostic biomarkers for MSA, to date there are no approved serum or cerebral spinal fluid-based tests to aid clinical diagnosis or to assess disease progression. Neuroimaging remains a primary diagnostic modality and is often helpful. Supportive findings on brain imaging include MRI- or CT-based evidence of pontine and/or cerebellar atrophy (Fig. 2). In MSA-C, there is predominant olivopontocerebellar atrophy, with loss of pontocerebellar fibers and gliosis best seen on T2-weighted MRI imaging, sometimes producing the characteristic “hot cross bun” sign [11]. In MSA-parkinsonism, MRI may show evidence of putaminal atrophy with decreased T2 signal in the posterior putamen and a hyperintense T2 rim, also known as a putaminal rim sign [73], which is typically observed at 1.5-T MRI strength but is unreliable with the more commonly used 3-T scanners today. These findings correlate with increased free-water [74, 75] and iron deposition within the putamen (as seen on MRI susceptibility-weighted imaging) [76, 77]. In addition, FDG–PET has been shown to help with MSA diagnosis and shows hypometabolism of the putamen, pons, and cerebellum [78]. Dopamine transporter (DAT) PET and SPECT imaging, although not specific for MSA, shows reduced striatal binding consistent with presynaptic dopaminergic denervation [79].

MRI brain imaging in MSA-P (A–C) demonstrates putaminal atrophy (small arrow) with hypointense T1/T2 signal and hyperintense T2 rim sign (large arrows). (C) SWI shows hemosiderin dense posterior putamen. (D) Olivopontocerebelllar atrophy in MSA-C. (E) T2 image with pontine atrophy, gliosis, and “hot cross bun” sign
Cardiac sympathetic neuroimaging may also help distinguish MSA from Parkinson disease. In a meta-analysis, cardiac iodine-123 meta-iodobenzylguanidine (MIBG) scintigraphy showed high sensitivity and specificity for distinguishing PD from other forms of parkinsonism [80]. MSA patients are usually found to have normal cardiac postganglionic sympathetic innervation, although a third of patients have some degree of cardiac sympathetic denervation [81, 82]. By contrast, Parkinson disease patients have altered sympathetic denervation in virtually all cases [83,84,85,86]. As a result, a normal MIBG scan can reliably exclude PD but not MSA [84, 85]. Additional diagnostic studies may include olfactory testing, autonomic test such as tilt table and sweat tests (e.g., QSART), urodynamic testing, and anal sphincter EMG and manometry, as well as gastric emptying and polysomnography.
Despite a lack of clinically reliable diagnostic biopsy-based or body fluid biomarkers, several investigational biomarkers have shown potential value and a combination of biomarkers may provide increased sensitivity and specificity [10]. A recent meta-analysis suggests that a combination of α-synuclein, neurofilament light chain (NFL), and total tau may have diagnostic value. A reduction of α-synuclein and an increase in NFL in CSF has the potential to distinguish MSA patients from PD and healthy controls [10]. In addition, a reduction of total tau (t-tau) in CSF may distinguish MSA from healthy controls, and an elevation of t-tau could distinguish MSA from PD. Additional CSF biomarkers under study include uric acid, homocysteine, phosphorylated tau, and coenzyme Q10. Investigational CSF biomarkers also include microRNAs (miRNAs), with recent data suggesting that a combination of increased expression of miR-19a, miR-19b, miR-24, and miR-34c was able to distinguish MSA patients from healthy controls [87]. In addition, serum and plasma have been investigated for potential biomarkers. Another meta-analysis found increased plasma α-synuclein among PD patients which may have value in MSA patients as well [88].
Finally, skin biopsy has shown promise as a potential investigational biomarker. Evidence suggests that cutaneous α-synuclein deposition in unmyelinated somatosensory fibers of the subepidermal plexus, but not in dermal autonomic fibers, can distinguish MSA from other subtypes of synucleinopathy [89]. In addition, several recent studies utilizing the ultrasensitive real-time quaking induced conversion (RT-QuIC) assay claim the ability to detect α-synuclein aggregation in tissues such as olfactory mucosa [90] and the submandibular gland [91] with high sensitivity and specificity for α-synucleinopathies including Parkinson disease and MSA.
Symptomatic Management
To date there are no approved disease-modifying therapies for MSA; thus, treatment is focused mainly on symptom management tailored to the specific patient (Table 2) and importantly involves a multidisciplinary approach with rehab and other allied healthcare partners. As a multisystem disorder, MSA often requires care from several disciplines including neurology, cardiology, gastroenterology, urology, sleep medicine, pulmonary, psychiatry, neuropsychology, dietary, palliative care, and hospice. Social services and support are critical and should include engaging social work, care partners, and discussion of life goals, end-of-life planning, and advanced directives.
While ideal, multidisciplinary care for MSA and other neurodegenerative disorders faces challenges in delivery. Current care models are often plagued by a lack of patient-centered care with poor continuity and delayed or reactive care delivery [92, 93]. Two multidisciplinary models, the Canadian and Dutch models, have been evaluated for clinical effectiveness [94, 95]. Although both models show benefit, the Canadian model appears more robust, although no universal standard has yet been recognized [96]. More recent work has focused on expanding and standardizing the Canadian Chronic Care Model (CCM) for patients with chronic neurologic conditions including parkinsonian disorders for the purposes of objective evaluation and standardization, but this remains a developing area in need of further study [97,98,99,100]. It is important to note that much of this work has focused on dementia and Parkinson disease, with the presumption that atypical parkinsonism would similarly benefit. Such a presumption, however, remains an untested hypothesis.
Parkinsonism
Tremor, rigidity, bradykinesia, and postural instability are core features of MSA-P and are observed irrespective of subtype in nearly 90% of cases [2]. A poor or unstained response to levodopa is common and among the core diagnostic features of MSA, helping to differentiate MSA-P from Parkinson disease [35, 101]. Nearly a third of MSA-P patients benefit from levodopa therapy, but temporarily with mean duration of 3.5 years in one study [6]. Levodopa remains a first-line therapy with trial up to 2 g total daily dose of levodopa (titrated from 100 to 300 mg 3-4 times daily) recommended for at least 3 months [102]. Clinically significant improvement is characterized as 30% decrease in the Unified Multiple System Atrophy Rating Scale (UMSARS) [101]. Levodopa may worsen orthostatic hypotension or cause other more severe side effects less common among Parkinson disease patients. Reports of deterioration with withdrawal of the medication even in the context of apparent nonresponse are common and may justify continuation of treatment [4].
Dopamine agonists may be used as a second choice, but lack supporting evidence and are generally not preferred because of their increased side effect profile relative to levodopa [61, 102]. Amantadine, up to 300 mg in 3 divided doses, is a reasonable alternative treatment option (including as an add-on), though there are no controlled studies to date [2, 103, 104].
Nonpharmacological approaches such as physical, occupational, and speech therapy are both complementary and essential therapies in MSA. Randomized controlled trials support occupational therapy in mild to moderate MSA [105], and although there are no such trials supporting physical therapy in MSA specifically, given the strength of the evidence for the motor symptoms of Parkinson disease [106], the assumption is that parkinsonian MSA symptoms will similarly benefit.
Deep brain stimulation for parkinsonism in MSA is not recommended as symptoms often poorly respond and rapidly progress relative to Parkinson disease. Case studies and series suggest it is ineffective and, as a result, should not be considered a therapeutic option [107,108,109].
Dystonia and Spasticity
Focal dystonia such as anterocollis or blepharospasm is a common feature of MSA with prevalence reported between 12 and 46% [16, 41]. Medical therapies for dystonia including benzodiazepines and anticholinergics are not commonly used because of cognitive and respiratory risks. As is true with other causes of dystonia, botulinum toxin injections can be quite effective for dystonic symptoms in MSA, especially for blepharospasm and limb dystonia [110]. Treatment of cervical dystonia, in particular anterocollis, may confer substantial risk because of the potential for exacerbating underlying dysphagia often associated with disease, and abundant caution is advised [111].
Ataxia
There are currently no medications available that provide evidence-supported improvement in ataxic symptoms in MSA. Similar to parkinsonism, however, evidence supports intensive physical therapy for gait, balance, and overall coordination, as well as speech therapy for ataxic dysarthria [112,113,114,115,116].
Sleep Disturbance
Sleep disorders are common in MSA and include insomnia, daytime somnolence, restless legs syndrome (RLS), REM sleep behavior disorder (RBD), and sleep disordered breathing [117]. Up to 71% of MSA patients report insomnia and have problems falling or staying asleep, or early morning awakenings. The causes are multifactorial and include nocturnal discomfort (due to rigidity or dystonia), urinary dysfunction (e.g., nocturia), anxiety and depression, and other parasomnias [118]. Good sleep hygiene is important and include maintaining consistent bed and wake times (avoid also prolonged in-bed time) and avoiding caffeine and excessive daytime naps. For those who get out of bed or are unable to fall asleep, having a planned relaxing activity such as reading a book, listening to music, or a warm bath can help. Consider also a referral to psychology for brief behavioral therapy. Treat anxiety and depression, if present. If discomfort is causing sleep difficulty, inquire about specific symptoms such as rigidity, dystonia, RLS, or tremors and treat accordingly. Mild pain relievers may be necessary and appropriate for some patients.
Daytime sleepiness is a frequent complaint and may lead to naps or more problematic “sleep attacks” that interfere with meals. Factors that contribute to fatigue and excessive daytime somnolence include sleep deprivation (from insomnia), sleep disordered breathing, circadian rhythm disruption, and drug side effects (e.g., pain medications, benzodiazepines, and change in dopaminergic medications). A baseline sleep study can help check for other sleep disorders such sleep apnea, RBD, RLS, or periodic limb movements in sleep. RLS occurs more frequently in MSA-P than in MSA-C, and may be associated with iron deficiency, although studies are conflicting [45]. Physicians should review medications that may exacerbate RLS such anticholinergics and antihistamines, and consider typical therapies such as dopaminergic agents, gabapentin (regular or enacarbil forms), pregabalin, or low-dose benzodiazepines.
History of REM behavior disorder (RBD) is a common risk factor for synucleinopathies and almost ubiquitous in MSA [4, 66]. Symptoms should be elicited in clinical history with a referral to polysomnography for objective assessment. If RBD is present, safety is foremost and includes securing the bedroom environment and sleeping partner. Patients with RBD often sleep alone, in a separate bed, or with a divider in place. Consider also padding for hard edges, a railing, or putting the mattress directly on the floor to prevent falls and injury. Despite the frequency of RBD in MSA, no specific treatment trials are available to guide pharmacotherapy and general guidelines for RBD are usually followed. First-line therapy for RBD is clonazepam 0.5 to 2 mg at night with lower doses often quite effective [119]. Given the frequency of respiratory dysfunction among MSA patients in sleep, however, clonazepam should be avoided in the context of apnea. In these situations, melatonin at a starting dose of 3 mg is an effective second-line agent, but higher doses (10-20 mg) are often needed and well-tolerated. Additional agents such as gabapentin and pregabalin have been reported as potentially effective alternatives [120].
Sleep disordered breathing such as obstructive sleep apnea is common and occurs in 37 to 65% of MSA patients. Continuous positive airway pressure (CPAP) or bilevel positive airway pressure provides benefit [121,122,123], but some patients experience central sleep apnea and require AutoPAP, or positive airway pressure machines that can automatically sense snoring, apneas, and hypopneas, and increase pressure until these disturbances stop. Nocturnal stridor is a particularly concerning and common symptom, with up to 13% of patients affected in one report, and is associated with oxygen desaturations, respiratory failure, and sudden death during sleep [69, 124]. In small studies, both CPAP and noninvasive positive pressure ventilation (NPPV) have shown efficacy in eliminating oxygen desaturations and stridor [121, 122], with these treatments generally well tolerated [123]. In more severe cases, tracheostomy may be considered to address vocal cord paralysis but may worsen sleep disordered breathing [125]. Because of the absence of therapeutic trials for RBD in MSA, management relies on the standard of care for RBD with some consideration for MSA-specific medication side effects and risks. Although low-dose clonazepam is often trialed (0.5-2 mg) [126], comorbid obstructive respiratory symptom may preclude this therapy. Sustained-release melatonin is a common and likely effective alternative, with doses ranging from 3 to 15 mg nightly. Gabapentin, pregabalin, and sodium oxybate are additional second-line alternatives [120]. Although noninvasive continuous positive pressure ventilation has shown some benefit here as well, tracheostomy may be required in advanced MSA patients to prevent fatal obstruction from vocal cord paralysis [124, 127, 128].
Dysarthria, Dysphagia, and Aspiration Risk
No medical therapies have proven effective for dysarthria, dysphagia, or aspiration. Evaluation of speech and swallow by a speech–language pathologist should be considered at the initial clinical presentation and targeted therapy established as appropriate. Speech–language pathologists are an integral member of the multidisciplinary team and work closely with clinicians, patients, and their caregivers to communicate the importance of speech and swallow interventions and approaches to optimize long-term outcomes. Speech therapy can be helpful to improve both articulation and volume. Dysphagia occurs earlier in MSA than in Parkinson disease and is more severe [129, 130]. Although therapeutic swallow measures and changes in diet consistency can help, many patients aspirate (often silently) and are at risk for choking or pneumonia. Swallow difficulty, and poor appetite, can lead to failure to thrive. Percutaneous gastrostomy tube placement (PEG) and enteral feeds may be an option for patients who need supplemental nutrition, hydration, and medication delivery, but does not change disease outcome or fully protect against aspiration. Discussion of nutritional support via PEG should be done early as part of advanced directives and should involve a team approach.
Depression
Despite the frequency and severity of depression and anxiety in patients with MSA [53] with up to half of patients reporting symptoms and evidence they play a major role in quality of life, randomized controlled trials providing therapeutic guidance are lacking [50, 51]. Treatment is largely based on Parkinson disease and other parkinsonisms. Smaller studies have suggested that paroxetine may improve motor symptoms, but without affecting depressive symptoms [131]. The combination of selective serotonin reuptake inhibitors, which have a lower risk of hypotension as compared to other classes of antidepressants, with psychotherapy is a common clinical approach and recommended for MSA patients with depression or anxiety based on evidence in Parkinson disease [132].
Cognitive Impairment
Although historically thought to have limited cognitive impairment compared to other atypical syndromes and Parkinson disease, recent evidence indicates the presence of cognitive dysfunction and dementia estimated in 14 to 16% of MSA patients causing significant impact on quality of life [133]. Empiric therapy is based on data from other Parkinson disorders, and acetylcholinesterase inhibitors such as donepezil or rivastigmine are often employed with modest benefit [119].
Orthostatic Dizziness
Three of every 4 MSA patients will suffer from orthostatic dizziness and hypotension [7, 61]. The management of orthostatic hypotension begins with behavioral or lifestyle changes including increased fluid and salt intake (i.e., volume expansion), wearing compression stockings or an abdominal binder, and being mindful of exacerbating conditions such as activity in hot, humid weather. When these are not enough, pharmacological therapy is needed. Both midodrine, an α-1 adrenergic agonist, and the mineralocorticoid fludrocortisone have demonstrated efficacy for symptomatic orthostatic hypotension and are used with good effect in MSA [134,135,136]. Although ephedrine also functions as a sympathomimetic, evidence from placebo-controlled trials or head-to-head comparisons is lacking [137]. Droxidopa (l-threo-3,4-dihydroxyphenylserine or l-DOPS) is a synthetic amino acid precursor that is converted by dopa decarboxylase to norepinephrine, resulting in peripheral vasoconstriction [138]. The effect of droxidopa in symptomatic neurogenic orthostatic hypotension has been studied in multicenter trials involving patients with Parkinson disease and related disorders including MSA, pure autonomic failure, and nondiabetic autonomic neuropathy. An initial trial involving an open-label dose optimization phase, followed by 7-day washout, and 7-day double-blind, drug-versus-placebo phase demonstrated increased blood pressure and decreased orthostatic symptoms such as weakness, fatigue, dizziness, and vision disturbance with droxidopa [139]. Based on 2 subsequent multicenter trials over a 2-week treatment period, droxidopa received FDA approval for short-term treatment of neurogenic orthostatic hypotension [140,141,142]; however, long-term efficacy has not been shown [138, 143].
Additionally, there is emerging evidence supporting the use of atomoxetine, a norepinephrine transport inhibitor, for neurogenic orthostatic hypotension, especially in those with central autonomic failure [144,145,146]. Atomoxetine in one report showed comparable results (and safety) to midodrine for treatment of neurogenic orthostatic hypotension [147], suggesting potential use as an alternative agent. Ampreloxetine, another norepinephrine reuptake inhibitor (NRI), developed by Theravance Biopharma (South San Francisco, CA), is currently under investigation in a phase 3 trial for symptomatic orthostatic hypotension in subjects with MSA, PD, and pure autonomic failure (NCT03750552). CERC-301 (rislenemdaz; developed by Cerecor, Inc., Rockville, MD) is another investigational drug developed for the treatment of symptomatic orthostatic hypotension but targets the NMDA receptor 2B subunit.
Urinary Dysfunction
Urinary dysfunction is the most common autonomic symptom of MSA and is recognized as a diagnostic criterion. There are several types of dysfunction and associated medical therapies; therefore, patients benefit from care coordinated with a urologist familiar with MSA. Sphincter detrusor dyssynergy and detrusor hyperreflexia result in urge incontinence that may respond to antispasmodics, including anticholinergics [104, 116, 148]. Conservative dosing and close monitoring are important as MSA patients can easily develop urinary retention as well. Anticholinergics such as oxybutynin may also cause confusion or delirium; thus, newer agents such as solifenacin, trospium, or fesoterodine may be preferred, though definitive evidence of superiority compared to other drugs is lacking [149]. By contrast, mirabegron is an antispasmodic that stimulates β3-adrenoceptors resulting in detrusor muscle relaxation and decreased afferent bladder signaling, and has comparable safety and efficacy for control of urinary urgency and incontinence [150]. Hypertension is the most common side effect but comparable to anticholinergics, though antimuscarinic effects appear less common (2% vs 25%) [151, 152]. Detrusor muscle overactivity may also be addressed with botulinum injections in coordination with urology [153, 154]. In addition, excessive nocturnal polyuria (in which more than 33% of the 24-h urinary volume is secreted at night) is a common complaint among MSA patients and evidence supports the use of desmopressin (oral or intranasal spray) before bed without clear side effects in several case series [155, 156]. Finally, neurogenic urinary retention is seen in MSA and can lead to complicated or intractable urinary tract infections and impairment in kidney function. Alpha-adrenergic antagonist medications such as tamsulosin or prazosin may be helpful [58] but increase the risk of orthostatic hypotension. As a result, intermittent self-catheterization is thought to be the first-line therapy for urinary retention in MSA [157].
Erectile Dysfunction
Erectile dysfunction is a key criterion for diagnosis of probable MSA in men and, given the social stigma and patient embarrassment, is likely underreported. As a result, questions about sexual dysfunction are an essential component of the clinical evaluation in MSA. Randomized placebo-controlled, crossover studies support efficacy in sildenafil citrate (50 mg) [6, 158]. Treatment of sildenafil should be initiated with caution, however, because of the potential for sildenafil to exacerbate orthostatic hypotension [119].
Disease-Modifying Therapies and Trials
A relatively limited number of trials to date have failed to identify an effective disease-modifying therapy. This failure likely results from an incomplete understanding of the underlying pathophysiology as well as limitations in preclinical animal models of disease and, at clinical levels of research, a lack of early and accurate assessments of diagnosis and progression of disease. Table 3 summaries current and past efforts to develop disease-modifying therapies in MSA.
Targeting α-Synuclein
Although GCIs containing α-synuclein aggregates are associated with clinical and pathological MSA, oligodendrocytes do not produce synuclein, leaving obscure how a protein with a purported synaptic localization and function is found in oligodendrocytes. Research pursuing the possibility of oligodendrocyte expression has found little evidence to support glial expression [177,178,179,180]. Given the lack of endogenous oligodendroglial synuclein expression, the field has explored the hypothesis that there may be cell-to-cell transmission of synuclein from neurons to glia, with preclinical evidence demonstrating trans-synaptic transmission of α-synuclein between neurons, as well as transfer to glia specifically [181,182,183,184,185,186]. Clinical evidence supporting this hypothesis includes GCIs found in patients with SNCA triplication [187]. Synuclein expression in the brains of purportedly sporadic MSA patients, however, has failed to show increased neuronal SNCA gene expression [177, 179, 180]. Recent work suggests that α-synuclein can produce a variety of potentially pathogenic conformations or “strains” which may explain the variety of pathology and localization seen among α-synucleinopathies. In the case of MSA, it may be that a particular strain is predisposed to cell-to-cell transfer from neurons to oligodendrocytes [188, 189], although no such MSA-specific synuclein strain has yet been described. Impaired protein processing or degradation has also been implicated [12, 26]. In this model, either pathologic conformations of synuclein induce protein processing impairments in a feed forward mechanism or a primary impairment in autophagic or proteasomal dysfunction leads to synuclein accumulation [26, 190, 191]. Additional work has proposed not that pathologic synuclein itself is pathologic but that abnormal glial proteasomal function results in synuclein accumulation that glia bear the burden of [12, 192].
The focus on synuclein has resulted in several trials targeting synuclein pathology through inhibition of aggregation, enhancement of degradation, or immunotherapy. The European Artemis Consortium, a collaborative effort funded by the European Union to develop treatments for MSA named after the Greek goddess of the hunt, is evaluating α-synuclein therapies in several transgenic mouse models, including an increasing number of small molecule modulators of α-synuclein.
Anle138b is a small molecule that targets and inhibits α-synuclein oligomerization and aggregation in MSA mouse models, slowing of disease progression and cell death [162]. These data have motivated a phase I trial in MSA sponsored by MODAG neuroscience solutions (http://www.modag.net/images/pressrelease_modag_ series_a.pdf). An additional small molecule, CLR01, has been shown in preclinical studies to reduce the formation of synuclein oligomers [164]. The small molecule PBT434 has also shown potential to reduce glial aggregates in an MSA mouse model through modulation of reactive iron species, and a phase I trial is currently ongoing for this agent as well [193, 194]. Preclinical data suggests that PBT434 mediates transmembrane reactive iron distribution, sequestering these reactive species away from intracellular proteins and thus reducing aggregation and oxidative stress. Based on this evidence, a phase I trial (U1111-1211-0052) is ongoing. The small molecule NPT200-11 has similarly shown multiple synuclein-related benefits in animal models [195], including reduced synuclein pathology burden and astrogliosis, and improved motor function. Finally, the repurposed epilepsy drug and caspase-1 inhibitor VX-765 has own some potential to limit synuclein aggregation, progression of motor deficits, and neuronal death in MSA animal models [196]. Along with phase II tolerability data in patients with epilepsy, further clinical trials in MSA patients are currently being pursued for VX-765.
Despite promising preclinical data and ongoing clinical trials in this area, caution is warranted given previous failed trials of drugs claiming a similar ability to affect α-synuclein aggregation. Rifampicin showed similar promise in MSA mouse models [197] as well in reducing β-amyloid aggregation in Alzheimer disease [198, 199]. However, a large double-blind trial in MSA patients failed to show effects on progression of disease. In addition, polyphenol epigallocatechin-gallate (EGCG) found in green tea showed modulation of synuclein aggregation in in vitro and cell models, but phase III randomized studies (NCT02008721) and (2012-000928-18) were negative.
Further preclinical evidence has implicated impairment of protein processing and autophagy in the pathogenesis of α-synuclein aggregation, spurring several clinical trials. Lithium was shown to enhance autophagy in in vitro and in vivo models [200,201,202]. A randomized, double-blind, placebo-controlled study (NCT00997672) was terminated, however, because of adverse effects [159]. Another autophagy enhancer, sirolimus (or rapamycin), showed similar preclinical benefits, and a clinical trial is ongoing in MSA patients (NCT03589976). The c-Abl tyrosine kinase inhibitor nilotinib has also been proposed as an autophagy enhancer in α-synucleinopathies with promising evidence in animal models of Parkinsonism, but discouraging results thus far in clinical trials of PD and an unclear path forward in MSA patients. Finally, toll-like receptors and antisense oligonucleotides have been explored as possible additional therapies but remain in the preclinical phase.
Monophosphoryl lipid A (MPLA), a TLR4 selective agonist and inducer of phagocytosis, is thought to aid in the degradation of pathologic protein aggregates, reduction of GCIs, neuroprotection, and improvements in motor deficits in a mouse model of MSA [165]. In addition, TLR4 appears increased in MSA brains, suggesting it may participate in an endogenous reactive process of glial protein clearance in humans [203, 204], all of which support future testing in clinical trials.
Both passive and active (i.e., vaccines) immune therapies targeting α-synuclein with the goal of reduction or clearance are ongoing. Several studies in animal models have shown efficacy of both passive and active immunization against α-synuclein [167, 205,206,207,208,209,210]. AFFiRiS (Vienna, Austria) recently completed phase I studies (2018) on the active vaccine immunotherapies AFFITOPE® PD01A and PD03A that target α-synuclein in both MSA and PD, showing safety, tolerability, and immunogenicity. Using a passive immunotherapy approach, Prothena (South San Francisco, CA) designed a humanized monoclonal antibody against α-synuclein (PRX002) that in phase I studies resulted in over 96% reduction of serum α-synuclein levels and appeared safe and well-tolerated [161]. A phase 2 randomized and double-blind clinical trial, led by Roche, is now underway (NCT03100149). Similarly, Biogen (Cambridge, MA) is investigating passive immunotherapy with the antibody BIIB-054 directed against α-synuclein early PD (NCT03716570).
Antisense oligonucleotides (ASO) are increasingly being explored and used clinically to knockdown gene products in neurodegenerative diseases from Alzheimer disease to spinomuscular atrophy [211,212,213]. ASOs targeting the SCNA gene product in preclinical models of synucleinopathy have demonstrated some success [52, 214,215,216], suggesting they may have a role in treating MSA, although current work in this area remains in the preclinical phase. MicroRNA-101 has been found to upregulate oligodendroglial α-synuclein pathology in MSA mouse models and to be overexpressed in MSA brains [163]. Anti-MiR-101 is currently in preclinical evaluations as a potential disease-modifying therapy.
Neuroprotective Therapies
Glutamatergic excitotoxicity is an important contributor and a well-known trigger for neuronal death. Several glutamate antagonists have been studied in CNS disorders for their potential neuroprotective effect, in particular riluzole, currently approved for amyotrophic lateral sclerosis as a disease-modifying therapy. Riluzole has an indirect effect on glutamate receptors via blockade of sodium and potassium channels. Riluzole was shown to reduce behavioral deficits and striatal degeneration in a rat model of MSA-parkinsonism [217]. These promising findings spurred clinical trials of riluzole in MSA (and progressive supranuclear palsy). The NNIPPS trial compared riluzole to placebo for 3 years with survival as the primary outcome. Despite recruiting 400 patients to a trial of such a rare disease, survival was unchanged [168]. Other antiglutaminergic drugs have also been explored including estrogens, but with disappointing results [218]. Tllsh-2910, a specific NMDA receptor modulator, is one exception and is currently in phase III trials to evaluate efficacy for symptomatic management of ataxia in MSA (NCT03901638).
Rasagiline is a monoamine oxidase (MAO) B inhibitor with symptomatic benefit and suggested disease-modifying effect in PD [219]. The putative neuroprotective effects of rasagiline are thought to be related to mitochondrial metabolism rather than its MAOb effects [220]. A trial of rasagiline in a transgenic MSA mouse model showed evidence of decreased synuclein oligomers and improved motor function [221]. However, a subsequent phase II randomized, placebo-controlled trial in MSA patients failed to show benefit [173].
Neuroprotective strategies include also increasing levels of neurotrophic factors in the brain such as GDNF and BDNF. Both GDNF and neurturin have been explored as a potential therapeutic for PD [222, 223], but so far not in MSA. The SSRI fluoxetine has been shown to increase GDNF and BDNF and to rescue hippocampal neurogenesis in a transgenic mouse model of synucleinopathy [224, 225]. These preclinical findings spurred a clinical trial of fluoxetine in MSA (NCT01146548) that failed to show benefit.
Research has focused on the role of mitochondria in pathogenesis as well, with mitochondrial mutations and toxins playing a key role in striatonigral degeneration and parkinsonism in both animal models and patients [226,227,228,229,230]. Specific to MSA, both fibroblasts and induced pluripotent stem cells derived from MSA patients have shown a variety of mitochondrial impairments. Recent descriptions of mutations in the COQ2 gene, which contributes to the mitochondrial electron transport protein coenzyme Q10 (CoQ10) biosynthesis, among possible MSA patients, have focused the field on mitochondrial pathophysiology. Clinical trial testing of CoQ10, however, has not been successful [171].
Exendin-4, an FDA-approved antidiabetic drug, is a glucagon-like peptide-1 (GLP-1) analogue that has been shown to reduce dopaminergic cell loss and monomeric α-synuclein levels in a mouse model of MSA [172]. Additional clinical evidence in PD patients [172, 231] has motivated a phase I trial in MSA.
AZD3241 is an orally bioavailable inhibitor of myeloperoxidase, a key enzyme in the production of reactive oxygen species. Treatment with AZD3241 has shown neuroprotective efficacy in the MPTP model of Parkinsonism and the proteolipid protein (PLP)–α-synuclein transgenic mouse model of MSA, delaying progression of disease [232]. As a result, inhibition with AZD3241, verdiperstat, is being explored in clinical trials as a potential therapy, currently in phase III (NCT02388295), sponsored by Biohaven Pharmaceuticals (New Haven, CT) .
Some preclinical work has identified agents that have effects across mechanisms. For example a derivative of the FDA-approved multiple sclerosis immunosuppressive FTY720 has shown effects both in mitochondria and GDNF expression as well as synuclein pathology in MSA mouse models [176].
Recently, autologous mesenchymal stem cells (MSC) treatment has been examined as a possible neuroprotective therapy, driven by the hypothesis that these cells can differentiate into a variety of supportive cell types which secrete neuroprotective and immunomodulatory agents [233, 234]. Studies in animal models provide support for this hypothesis [235, 236], and early clinical trials showed evidence for delayed disease progression but raised concern about interarterial delivery and ischemic lesions [169, 237]. A more recent trial involving intrathecal MSC administration showed that it was safe and well-tolerated, but may be associated with implantation pain at higher doses [170]. Larger controlled trials are needed to assess the safety and efficacy of MSC treatment, as well as further elucidate the mechanism.
Reducing Neuroinflammation
Increasing evidence suggests that the inflammatory response to abnormal α-synuclein accumulation and aggregation plays a key role in the pathogenesis of MSA (and other synucleinopathies). This neuroinflammatory response is mediated by activated microglia that respond to neuronal damage by secreting proinflammatory cytokines, stimulating further immune reaction, and phagocytosis of injured/diseased cells. A consequence of chronic microglial activation in neurodegenerative disorders like MSA is the perpetuation of the neuroinflammatory response and release of cytotoxic molecules that may promote further neuronal injury. Suppression of microglia-associated inflammation has thus been proposed as a potential therapeutic strategy in MSA. Minocycline is a tetracycline antibiotic that crosses the blood–brain barrier and has additional effects including inhibition of microglia, apoptosis, and proteolysis [238]. Despite conflicting preclinical evidence [203, 239, 240], minocycline was pursued clinically through the MEMSA trial which randomized 63 MSA-P patients to minocycline or placebo with a small subgroup receiving PET scans to assess microglial activation. Despite evidence for decreased microglial activity, as measured by PET, after 1 year of treatment, the study failed to show clinical improvement and reported premature loss of 22 of 32 subjects in the minocycline group alone, primarily because of adverse events [174].
Although its mechanism remains poorly understood, intravenous immunoglobulins (IVIg) have been widely used in a number of immune-mediate neurological disorders. IVIg contains pooled antibodies from human plasma and is thought to reduce circulating inflammatory proteins that may mediate neurological disease. Based on this theory, IVIg was trialed in a single-center, 6-month, open-label pilot study in 7 MSA patients [175]. The study reported tolerability and improvement in clinical scores (both UMSARS I and II), but lacked controls, raising concern for potential placebo effects.
Conclusions
Treatment of MSA remains a challenge and requires a multidisciplinary approach including allied healthcare providers, rehabilitation, and supportive care. Although there are a number of effective symptomatic therapies, these treatments are limited by the multisystem nature of the disease, potential side effects, drug interactions, and relentless disease progression. These factors underscore the pressing need for disease-modifying therapies in MSA. A growing number of potentially disease-modifying agents are currently in preclinical development and include clinical trials focusing on α-synuclein modulation, neurotoxicity, neuroinflammation, trophic cell support, and even genetic modulation, among others. The success of these agents remains limited, however, by the number and validity of preclinical animal models, clinical diagnosis (especially early in disease), and the sensitivity and specificity of clinical fluid and imaging biomarkers to help with diagnosis and to monitor disease progression. In addition, an incomplete understanding of the underlying pathophysiologic mechanisms of MSA is a fundamental limitation. Specifically, the role of α-synuclein in oligodendrocyte-mediated neurodegeneration remains unclear. Despite these limitations, it is encouraging that recent research and clinical trials in MSA are increasing, raising hope for new discoveries and therapeutic developments for this devastating disease.
References
Gilman S, May SJ, Shults CW, et al. The North American Multiple System Atrophy Study Group. J Neural Transm (Vienna) 2005;112:1687-1694. https://doi.org/10.1007/s00702-005-0381-6.
Kollensperger M, Geser F, Ndayisaba JP, et al. Presentation, diagnosis, and management of multiple system atrophy in Europe: final analysis of the European multiple system atrophy registry. Mov Disord 2010;25:2604-2612. https://doi.org/10.1002/mds.23192.
Watanabe H, Saito Y, Terao S, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain 2002;125:1070-1083.
Fanciulli A, Wenning GK. Multiple-system atrophy. N Engl J Med 2015;372:249-263. https://doi.org/10.1056/NEJMra1311488.
Ben-Shlomo Y, Wenning GK, Tison F, Quinn NP. Survival of patients with pathologically proven multiple system atrophy: a meta-analysis. Neurology 1997;48:384-393.
Wenning GK, Geser F, Krismer F, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol 2013;12:264-274. https://doi.org/10.1016/S1474-4422(12)70327-7.
Low PA, Reich SG, Jankovic J, et al. Natural history of multiple system atrophy in the USA: a prospective cohort study. Lancet Neurol 2015;14:710-719. https://doi.org/10.1016/S1474-4422(15)00058-7.
Foubert-Samier A, Pavy-Le Traon A, Guillet F, et al. Disease progression and prognostic factors in multiple system atrophy: a prospective cohort study. Neurobiol Dis 2020;139:104813. https://doi.org/10.1016/j.nbd.2020.104813.
McFarland NR. Diagnostic approach to atypical parkinsonian syndromes. Continuum (Minneap Minn) 2016;22(4 Movement Disorders):1117-1142. https://doi.org/10.1212/CON.0000000000000348.
Cong S, Xiang C, Wang H, Cong S. Diagnostic utility of fluid biomarkers in multiple system atrophy: a systematic review and meta-analysis. J Neurol 2020. https://doi.org/10.1007/s00415-020-09781-9.
Chelban V, Bocchetta M, Hassanein S, Haridy NA, Houlden H, Rohrer JD. An update on advances in magnetic resonance imaging of multiple system atrophy. J Neurol 2019;266:1036-1045. https://doi.org/10.1007/s00415-018-9121-3.
Jellinger KA. Multiple system atrophy: an oligodendroglioneural synucleinopathy. J Alzheimers Dis 2018;62:1141-1179. https://doi.org/10.3233/JAD-170397.
Jellinger KA. Neuropathology of multiple system atrophy: new thoughts about pathogenesis. Mov Disord 2014;29:1720-1741. https://doi.org/10.1002/mds.26052.
Sato K, Kaji R, Matsumoto S, Nagahiro S, Goto S. Compartmental loss of striatal medium spiny neurons in multiple system atrophy of parkinsonian type. Mov Disord 2007;22:2365-2370. https://doi.org/10.1002/mds.21732.
Tsuchiya K, Ozawa E, Haga C, et al. Constant involvement of the Betz cells and pyramidal tract in multiple system atrophy: a clinicopathological study of seven autopsy cases. Acta Neuropathol 2000;99:628-636. https://doi.org/10.1007/s004010051173.
Wenning GK, Tison F, Ben Shlomo Y, Daniel SE, Quinn NP. Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 1997;12:133-147. https://doi.org/10.1002/mds.870120203.
Jellinger KA, Lantos PL. Papp-Lantos inclusions and the pathogenesis of multiple system atrophy: an update. Acta Neuropathol 2010;119:657-667. https://doi.org/10.1007/s00401-010-0672-3.
Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989;94:79-100.
Papp MI, Lantos PL. The distribution of oligodendroglial inclusions in multiple system atrophy and its relevance to clinical symptomatology. Brain 1994;117 235-243. https://doi.org/10.1093/brain/117.2.235.
Ubhi K, Low P, Masliah E. Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci 2011;34 581-590. https://doi.org/10.1016/j.tins.2011.08.003.
Nishie M, Mori F, Yoshimoto M, Takahashi H, Wakabayashi K. A quantitative investigation of neuronal cytoplasmic and intranuclear inclusions in the pontine and inferior olivary nuclei in multiple system atrophy. Neuropathol Appl Neurobiol 2004;30:546-554.https://doi.org/10.1111/j.1365-2990.2004.00564.x.
Wenning GK, Jellinger KA. The role of alpha-synuclein in the pathogenesis of multiple system atrophy. Acta Neuropathol 2005;109:129-140. https://doi.org/10.1007/s00401-004-0935-y.
Ahmed Z, Asi YT, Sailer A, et al. The neuropathology, pathophysiology and genetics of multiple system atrophy. Neuropathol Appl Neurobiol 2012;38:4-24. https://doi.org/10.1111/j.1365-2990.2011.01234.x.
Bendor JT, Logan TP, Edwards RH. The function of alpha-synuclein. Neuron 2013;79:1044-1066. https://doi.org/10.1016/j.neuron.2013.09.004.
Uchihara T, Giasson BI. Propagation of alpha-synuclein pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131 49-73. https://doi.org/10.1007/s00401-015-1485-1
Jellinger KA, Wenning GK. Multiple system atrophy: pathogenic mechanisms and biomarkers. J Neural Transm (Vienna). 2016;123:555-572. https://doi.org/10.1007/s00702-016-1545-2
Henderson MX, Trojanowski JQ, Lee VMY. alpha-Synuclein pathology in Parkinson’s disease and related alpha-synucleinopathies. Neurosci Lett. 2019;709. https://doi.org/10.1016/j.neulet.2019.134316.
Dhillon JS, Trejo-Lopez JA, Riffe C, et al. Dissecting alpha-synuclein inclusion pathology diversity in multiple system atrophy: implications for the prion-like transmission hypothesis. Lab Invest 2019;99:982-992. https://doi.org/10.1038/s41374-019-0198-9.
Lau A, So RWL, Lau HHC, et al. alpha-Synuclein strains target distinct brain regions and cell types. Nat Neurosci 2020;23:21-31. https://doi.org/10.1038/s41593-019-0541-x.
Holec SAM, Woerman AL. Evidence of distinct alpha-synuclein strains underlying disease heterogeneity. Acta Neuropathol 2020. https://doi.org/10.1007/s00401-020-02163-5.
Prusiner SB, Woerman AL, Mordes DA, et al. Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 2015. https://doi.org/10.1073/pnas.1514475112.
Woerman AL, Oehler A, Kazmi SA, et al. Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol. 2019;137:437-454. https://doi.org/10.1007/s00401-019-01959-4
Brundin P, Dave KD, Kordower JEH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 2017;298:225-235. https://doi.org/10.1016/j.expneurol.2017.10.003.
Gilman S, Low PA, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. J Neurol Sci 1999;163:94-98.
Gilman S, Wenning GK, Low PA, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology 2008;71 670-676. https://doi.org/10.1212/01.wnl.0000324625.00404.15.
Kollensperger M, Geser F, Seppi K, et al. Red flags for multiple system atrophy. Mov Disord 2008;23 1093-1099. https://doi.org/10.1002/mds.21992.
Kojovic M, Cordivari C, Bhatia K. Myoclonic disorders: a practical approach for diagnosis and treatment. Ther Adv Neurol Disord 2011;4:47-62. https://doi.org/10.1177/1756285610395653.
Koga S, Aoki N, Uitti RJ, et al. When DLB, PD, and PSP masquerade as MSA: an autopsy study of 134 patients. Neurology 2015;85:404-412. https://doi.org/10.1212/WNL.0000000000001807.
Joutsa J, Gardberg M, Roytta M, Kaasinen V. Diagnostic accuracy of parkinsonism syndromes by general neurologists. Parkinsonism Relat Disord 2014;20:840-844. https://doi.org/10.1016/j.parkreldis.2014.04.019
Jabbari E, Holland N, Chelban V, et al. Diagnosis across the spectrum of progressive supranuclear palsy and corticobasal syndrome. JAMA Neurol 2019. https://doi.org/10.1001/jamaneurol.2019.4347.
Boesch SM, Wenning GK, Ransmayr G, Poewe W. Dystonia in multiple system atrophy. J Neurol Neurosurg Psychiatry 2002;72:300-303. https://doi.org/10.1136/jnnp.72.3.300.
Azher SN, Jankovic J. Camptocormia: pathogenesis, classification, and response to therapy. Neurology 2005;65:355-359. https://doi.org/10.1212/01.wnl.0000171857.09079.9f.
Slawek J, Derejko M, Lass P, Dubaniewicz M. Camptocormia or Pisa syndrome in multiple system atrophy. Clin Neurol Neurosurg 2006;108:699-704.https://doi.org/10.1016/j.clineuro.2005.07.004.
Bhalsing K, Suresh K, Muthane UB, Pal PK. Prevalence and profile of Restless Legs Syndrome in Parkinson’s disease and other neurodegenerative disorders: a case-control study. Parkinsonism Relat Disord 2013;19 426-430. https://doi.org/10.1016/j.parkreldis.2012.12.005.
Ghorayeb I, Dupouy S, Tison F, Meissner WG. Restless legs syndrome in multiple system atrophy. J Neural Transm (Vienna) 2014;121:1523-1527. https://doi.org/10.1007/s00702-014-1232-0.
Kluin KJ, Gilman S, Lohman M, Junck L. Characteristics of the dysarthria of multiple system atrophy. Arch Neurol 1996;53 545-548. https://doi.org/10.1001/archneur.1996.00550060089021.
Rusz J, Tykalova T, Salerno G, Bancone S, Scarpelli J, Pellecchia MT. Distinctive speech signature in cerebellar and parkinsonian subtypes of multiple system atrophy. J Neurol 2019;266 1394-1404. https://doi.org/10.1007/s00415-019-09271-7.
Eschlbock S, Delazer M, Krismer F, et al. Cognition in multiple system atrophy: a single-center cohort study. Ann Clin Transl Neurol 2020;7:219-228. https://doi.org/10.1002/acn3.50987.
Stankovic I, Krismer F, Jesic A, et al. Cognitive impairment in multiple system atrophy: a position statement by the Neuropsychology Task Force of the MDS Multiple System Atrophy (MODIMSA) study group. Mov Disord 2014;29:857-867. https://doi.org/10.1002/mds.25880.
Benrud-Larson LM, Sandroni P, Schrag A, Low PA. Depressive symptoms and life satisfaction in patients with multiple system atrophy. Mov Disord 2005;20:951-957. https://doi.org/10.1002/mds.20450.
Schrag A, Geser F, Stampfer-Kountchev M, et al. Health-related quality of life in multiple system atrophy. Mov Disord 2006;21:809-815. https://doi.org/10.1002/mds.20808.
Zhang LY, Cao B, Zou YT, et al. Depression and anxiety in multiple system atrophy. Acta Neurol Scand 2018;137:33-37. https://doi.org/10.1111/ane.12804.
Almeida L, Ahmed B, Walz R, et al. Depressive symptoms are frequent in atypical Parkinsonian disorders. Mov Disord Clin Pract 2017;4 191-197. https://doi.org/10.1002/mdc3.12382.
Tison F, Yekhlef F, Chrysostome V. Depression and self-reported depressive symptoms in multiple system atrophy compared to Parkinson’s disease. Mov Disord 2006;21 1056-1057. https://doi.org/10.1002/mds.20891.
Parvizi J, Joseph J, Press DZ, Schmahmann JD. Pathological laughter and crying in patients with multiple system atrophy-cerebellar type. Mov Disord 2007;22 798-803. https://doi.org/10.1002/mds.21348.
Roncevic D, Palma JA, Martinez J, Goulding N, Norcliffe-Kaufmann L, Kaufmann H. Cerebellar and parkinsonian phenotypes in multiple system atrophy: similarities, differences and survival. J Neural Transm (Vienna) 2014;121 507-12. https://doi.org/10.1007/s00702-013-1133-7.
Iodice V, Lipp A, Ahlskog JE, et al. Autopsy confirmed multiple system atrophy cases: Mayo experience and role of autonomic function tests. J Neurol Neurosurg Psychiatry 2012;83:453-459. https://doi.org/10.1136/jnnp-2011-301068.
Sakakibara R, Hattori T, Uchiyama T, et al. Urinary dysfunction and orthostatic hypotension in multiple system atrophy: which is the more common and earlier manifestation? J Neurol Neurosurg Psychiatry 2000;68:65-69. https://doi.org/10.1136/jnnp.68.1.65.
Ito T, Sakakibara R, Yasuda K, et al. Incomplete emptying and urinary retention in multiple-system atrophy: when does it occur and how do we manage it? Mov Disord 2006;21:816-823. https://doi.org/10.1002/mds.20815.
Kirchhof K, Apostolidis AN, Mathias CJ, Fowler CJ. Erectile and urinary dysfunction may be the presenting features in patients with multiple system atrophy: a retrospective study. Int J Impot Res 2003;15:293-298. https://doi.org/10.1038/sj.ijir.3901014.
Wenning GK, Ben Shlomo Y, Magalhaes M, Daniel SE, Quinn NP. Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain. 1994;117:835-845.
Coon EA, Sletten DM, Suarez MD, et al. Clinical features and autonomic testing predict survival in multiple system atrophy. Brain 2015;138:3623-3631. https://doi.org/10.1093/brain/awv274.
Glasmacher SA, Leigh PN, Saha RA. Predictors of survival in progressive supranuclear palsy and multiple system atrophy: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2017;88:402-411. https://doi.org/10.1136/jnnp-2016-314956.
Xia C, Postuma RB. Diagnosing multiple system atrophy at the prodromal stage. Clin Auton Res 2020;30:197-205. https://doi.org/10.1007/s10286-020-00682-5.
Silveira-Moriyama L, Mathias C, Mason L, Best C, Quinn NP, Lees AJ. Hyposmia in pure autonomic failure. Neurology 2009;72:1677-1681. https://doi.org/10.1212/WNL.0b013e3181a55fd2.
Palma JA, Fernandez-Cordon C, Coon EA, et al. Prevalence of REM sleep behavior disorder in multiple system atrophy: a multicenter study and meta-analysis. Clin Auton Res 2015;25:69-75. https://doi.org/10.1007/s10286-015-0279-9.
Claassen DO, Josephs KA, Ahlskog JE, Silber MH, Tippmann-Peikert M, Boeve BF. REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology 2010;75:494-499. https://doi.org/10.1212/WNL.0b013e3181ec7fac.
Abbott SM, Videnovic A. Sleep disorders in atypical Parkinsonism. Mov Disord Clin Pract 2014;1:89-96. https://doi.org/10.1002/mdc3.12025.
Iranzo A. Sleep and breathing in multiple system atrophy. Curr Treat Options Neurol 2007;9:347-353.
Cortelli P, Calandra-Buonaura G, Benarroch EE, et al. Stridor in multiple system atrophy: consensus statement on diagnosis, prognosis, and treatment. Neurology 2019;93:630-639. https://doi.org/10.1212/WNL.0000000000008208.
Kaufmann H, Norcliffe-Kaufmann L, Palma JA, Autonomic Disorders C. Reply to “Pure autonomic failure vs. manifest CNS synucleinopathy: relevance of stridor and autonomic biomarkers”. Ann Neurol 2017;81:910-911. https://doi.org/10.1002/ana.24949.
Osaki Y, Ben-Shlomo Y, Lees AJ, Wenning GK, Quinn NP. A validation exercise on the new consensus criteria for multiple system atrophy. Mov Disord 2009;24:2272-2276. https://doi.org/10.1002/mds.22826.
Tha KK, Terae S, Tsukahara A, et al. Hyperintense putaminal rim at 1.5 T: prevalence in normal subjects and distinguishing features from multiple system atrophy. BMC Neurol 2012;12:39. https://doi.org/10.1186/1471-2377-12-39.
Planetta PJ, Kurani AS, Shukla P, et al. Functional and macrostructural MRI in Parkinson's disease and multiple system atrophy. Mov Disord 2014;29:S90.
Planetta PJ, Kurani AS, Shukla P, et al. Distinct functional and macrostructural brain changes in Parkinson’s disease and multiple system atrophy. Hum Brain Mapp 2014. https://doi.org/10.1002/hbm.22694.
Lee JH, Baik SK. Putaminal hypointensity in the parkinsonian variant of multiple system atrophy: simple visual assessment using susceptibility-weighted imaging. J Mov Disord. 2011;4:60-63. https://doi.org/10.14802/jmd.11012
Sugiyama A, Sato N, Kimura Y, et al. Quantifying iron deposition in the cerebellar subtype of multiple system atrophy and spinocerebellar ataxia type 6 by quantitative susceptibility mapping. J Neurol Sci 2019;407:116525.https://doi.org/10.1016/j.jns.2019.116525.
Juh R, Kim J, Moon D, Choe B, Suh T. Different metabolic patterns analysis of Parkinsonism on the 18F-FDG PET. Eur J Radiol 2004;51:223-233.https://doi.org/10.1016/S0720-048X(03)00214-6.
Perju-Dumbrava LD, Kovacs GG, Pirker S, et al. Dopamine transporter imaging in autopsy-confirmed Parkinson’s disease and multiple system atrophy. Mov Disord 2012;27:65-71. https://doi.org/10.1002/mds.24000.
Orimo S, Suzuki M, Inaba A, Mizusawa H. 123I-MIBG myocardial scintigraphy for differentiating Parkinson’s disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis. Parkinsonism Relat Disord 2012;18 494-500. https://doi.org/10.1016/j.parkreldis.2012.01.009.
Nagayama H, Ueda M, Yamazaki M, Nishiyama Y, Hamamoto M, Katayama Y. Abnormal cardiac [(123)I]-meta-iodobenzylguanidine uptake in multiple system atrophy. Mov Disord 2010;25 1744-1747. https://doi.org/10.1002/mds.23338.
Nagayama H, Yamazaki M, Ueda M, et al. Low myocardial MIBG uptake in multiple system atrophy with incidental Lewy body pathology: an autopsy case report. Mov Disord 2008;23:1055-1057. https://doi.org/10.1002/mds.22031.
Braune S. The role of cardiac metaiodobenzylguanidine uptake in the differential diagnosis of parkinsonian syndromes. Clin Auton Res 2001;11:351-355. https://doi.org/10.1007/bf02292766.
Goldstein DS. Dysautonomia in Parkinson disease. Compr Physiol 2014;4805-826. https://doi.org/10.1002/cphy.c130026.
Kaufmann H, Goldstein DS. Autonomic dysfunction in Parkinson disease. Handb Clin Neurol 2013;117:259-278. https://doi.org/10.1016/B978-0-444-53491-0.00021-3.
Orimo S, Oka T, Miura H, et al. Sympathetic cardiac denervation in Parkinson's disease and pure autonomic failure but not in multiple system atrophy. J Neurol Neurosurg Psychiatry 2002;73:776-777. https://doi.org/10.1136/jnnp.73.6.776.
Marques TM, Kuiperij HB, Bruinsma IB, et al. MicroRNAs in cerebrospinal fluid as potential biomarkers for Parkinson’s disease and multiple system atrophy. Mol Neurobiol 2017;54 7736-7745. https://doi.org/10.1007/s12035-016-0253-0.
Bougea A, Stefanis L, Paraskevas GP, Emmanouilidou E, Vekrelis K, Kapaki E. Plasma alpha-synuclein levels in patients with Parkinson’s disease: a systematic review and meta-analysis. Neurol Sci 2019;40:929-938. https://doi.org/10.1007/s10072-019-03738-1.
Kim JY, Illigens BM, McCormick MP, Wang N, Gibbons CH. Alpha-synuclein in skin nerve fibers as a biomarker for alpha-synucleinopathies. J Clin Neurol 2019;15 135-142. https://doi.org/10.3988/jcn.2019.15.2.135.
De Luca CMG, Elia AE, Portaleone SM, et al. Efficient RT-QuIC seeding activity for alpha-synuclein in olfactory mucosa samples of patients with Parkinson's disease and multiple system atrophy. Transl Neurodegener 2019;8:24. https://doi.org/10.1186/s40035-019-0164-x
Manne S, Kondru N, Jin H, et al. alpha-Synuclein real-time quaking-induced conversion in the submandibular glands of Parkinson’s disease patients. Mov Disord 2020;35:268-278. https://doi.org/10.1002/mds.27907.
Vlaanderen FP, Rompen L, Munneke M, Stoffer M, Bloem BR, Faber MJ. The voice of the Parkinson customer. J Parkinsons Dis 2019;9:197-201. https://doi.org/10.3233/JPD-181431.
van der Eijk M, Faber MJ, Al Shamma S, Munneke M, Bloem BR. Moving towards patient-centered healthcare for patients with Parkinson’s disease. Parkinsonism Relat Disord 2011;17:360-364. https://doi.org/10.1016/j.parkreldis.2011.02.012.
van der Marck MA, Bloem BR, Borm GF, Overeem S, Munneke M, Guttman M. Effectiveness of multidisciplinary care for Parkinson’s disease: a randomized, controlled trial. Mov Disord 2013;28:605-611. https://doi.org/10.1002/mds.25194.
van der Marck MA, Munneke M, Mulleners W, et al. Integrated multidisciplinary care in Parkinson’s disease: a non-randomised, controlled trial (IMPACT). Lancet Neurol 2013;12 947-956. https://doi.org/10.1016/S1474-4422(13)70196-0.
van der Marck MA, Bloem BR. How to organize multispecialty care for patients with Parkinson’s disease. Parkinsonism Relat Disord 2014;20 S167-S173. https://doi.org/10.1016/S1353-8020(13)70040-3.
Tenison E, Smink A, Redwood S, et al. Proactive and integrated management and empowerment in Parkinson’s disease: designing a new model of care. Parkinsons Dis 2020;2020:8673087. https://doi.org/10.1155/2020/8673087.
Jaglal SB, Guilcher SJ, Bereket T, et al. Development of a Chronic Care Model for Neurological Conditions (CCM-NC). BMC Health Serv Res 2014;14:409. https://doi.org/10.1186/1472-6963-14-409.
Ouwens M, Wollersheim H, Hermens R, Hulscher M, Grol R. Integrated care programmes for chronically ill patients: a review of systematic reviews. International J Qual Health Care 2005;17(2):141-146. https://doi.org/10.1093/intqhc/mzi016.
Coleman K, Austin BT, Brach C, Wagner EH. Evidence on the Chronic Care Model in the new millennium. Health Aff (Millwood) 2009;28 75-85. https://doi.org/10.1377/hlthaff.28.1.75.
Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol 2004;3 93-103.
Flabeau O, Meissner WG, Tison F. Multiple system atrophy: current and future approaches to management. Ther Adv Neurol Disord 2010;3 249-263. https://doi.org/10.1177/1756285610375328.
Rajrut AH, Uitti RJ, Fenton ME, George D. Amantadine effectiveness in multiple system atrophy and progressive supranuclear palsy. Parkinsonism Relat Disord 1997;3 211-214.
Wenning GK, Working Group on Atypical Parkinsonism of the Austrian Parkinson’s S. Placebo-controlled trial of amantadine in multiple-system atrophy. Clin Neuropharmacol 2005;28:225-227. https://doi.org/10.1097/01.wnf.0000183240.47960.f0.
Jain S, Dawson J, Quinn NP, Playford ED. Occupational therapy in multiple system atrophy: a pilot randomized controlled trial. Mov Disord 2004;19:1360-1364. https://doi.org/10.1002/mds.20211.
Tomlinson CL, Patel S, Meek C, et al. Physiotherapy versus placebo or no intervention in Parkinson’s disease. Cochrane Database Syst Rev 2013:CD002817. https://doi.org/10.1002/14651858.CD002817.pub4.
Chou KL, Forman MS, Trojanowski JQ, Hurtig HI, Baltuch GH. Subthalamic nucleus deep brain stimulation in a patient with levodopa-responsive multiple system atrophy. Case report. J Neurosurg 2004;100 553-556. https://doi.org/10.3171/jns.2004.100.3.0553.
Lezcano E, Gomez-Esteban JC, Zarranz JJ, et al. Parkinson's disease-like presentation of multiple system atrophy with poor response to STN stimulation: a clinicopathological case report. Mov Disord 2004;19 973-977. https://doi.org/10.1002/mds.20108.
Meissner WG, Laurencin C, Tranchant C, et al. Outcome of deep brain stimulation in slowly progressive multiple system atrophy: a clinico-pathological series and review of the literature. Parkinsonism Relat Disord 2016;24:69-75. https://doi.org/10.1016/j.parkreldis.2016.01.005.
Muller J, Wenning GK, Wissel J, Seppi K, Poewe W. Botulinum toxin treatment in atypical parkinsonian disorders associated with disabling focal dystonia. J Neurol 2002;249:300-304. https://doi.org/10.1007/s004150200009.
Thobois S, Broussolle E, Toureille L, Vial C. Severe dysphagia after botulinum toxin injection for cervical dystonia in multiple system atrophy. Mov Disord 2001;16:764-765. https://doi.org/10.1002/mds.1101.
Ilg W, Bastian AJ, Boesch S, et al. Consensus paper: management of degenerative cerebellar disorders. Cerebellum 2014;13 248-268. https://doi.org/10.1007/s12311-013-0531-6.
Ilg W, Synofzik M, Brotz D, Burkard S, Giese MA, Schols L. Intensive coordinative training improves motor performance in degenerative cerebellar disease. Neurology 2009;73:1823-1830. https://doi.org/10.1212/WNL.0b013e3181c33adf.
Landers M, Adams M, Acosta K, Fox A. Challenge-oriented gait and balance training in sporadic olivopontocerebellar atrophy: a case study. J Neurol Phys Ther 2009;33 160-168. https://doi.org/10.1097/NPT.0b013e3181b511f4.
Wedge F. The impact of resistance training on balance and functional ability of a patient with multiple system atrophy. J Geriatr Phys Ther 2008;31:79-83. https://doi.org/10.1519/00139143-200831020-00007.
Colosimo C, Tiple D, Wenning GK. Management of multiple system atrophy: state of the art. J Neural Transm (Vienna) 2005;112:1695-704. https://doi.org/10.1007/s00702-005-0379-0.
Cochen De Cock V. Sleep abnormalities in multiplesystem atrophy. Curr Treat Options Neurol. 2018;20:16. https://doi.org/10.1007/s11940-018-0503-8.
Videnovic A. Management of sleep disorders in Parkinson’s disease and multiple system atrophy. Mov Disord 2017;32 659-668. https://doi.org/10.1002/mds.26918.
Rohrer G, Hoglinger GU, Levin J. Symptomatic therapy of multiple system atrophy. Auton Neurosci 2018;211:26-30. https://doi.org/10.1016/j.autneu.2017.10.006.
Escriba J, Hoyo B. Alternatives to clonazepam in REM behavior disorder treatment. J Clin Sleep Med 2016;12 1193. https://doi.org/10.5664/jcsm.6068.
Nonaka M, Imai T, Shintani T, Kawamata M, Chiba S, Matsumoto H. Non-invasive positive pressure ventilation for laryngeal contraction disorder during sleep in multiple system atrophy. J Neurol Sci 2006;247:53-58. https://doi.org/10.1016/j.jns.2006.03.008.
Iranzo A, Santamaria J, Tolosa E, et al. Long-term effect of CPAP in the treatment of nocturnal stridor in multiple system atrophy. Neurology 2004;63:930-932. https://doi.org/10.1212/01.wnl.0000137043.76383.a4.
Ghorayeb I, Bioulac B, Tison F. Sleep disorders in multiple system atrophy. J Neural Transm (Vienna) 2005;112:1669-1675. https://doi.org/10.1007/s00702-005-0348-7.
Silber MH, Levine S. Stridor and death in multiple system atrophy. Mov Disord 2000;15 699-704. https://doi.org/10.1002/1531-8257(200007)15:4<699::aid-mds1015>3.0.co;2-l.
Garcia-Sanchez A, Fernandez-Navarro I, Garcia-Rio F. Central apneas and REM sleep behavior disorder as an initial presentation of multiple system atrophy. J Clin Sleep Med 2016;12:267-270. https://doi.org/10.5664/jcsm.5500.
St Louis EK, Boeve AR, Boeve BF. REM sleep behavior disorder in Parkinson’s disease and other synucleinopathies. Mov Disord 2017;32:645-658. https://doi.org/10.1002/mds.27018.
Tada M, Onodera O, Tada M, et al. Early development of autonomic dysfunction may predict poor prognosis in patients with multiple system atrophy. Arch Neurol 2007;64:256-260. https://doi.org/10.1001/archneur.64.2.256.
Giannini G, Calandra-Buonaura G, Mastrolilli F, et al. Early stridor onset and stridor treatment predict survival in 136 patients with MSA. Neurology 2016;87 1375-1383. https://doi.org/10.1212/WNL.0000000000003156.
Lee HH, Seo HG, Kim KD, et al. Characteristics of early oropharyngeal dysphagia in patients with multiple system atrophy. Neurodegener Dis 2018;18:84-90. https://doi.org/10.1159/000487800
Do HJ, Seo HG, Lee HH, et al. Progression of oropharyngeal dysphagia in patients with multiple system atrophy. Dysphagia. 2020;35:24-31. https://doi.org/10.1007/s00455-019-09990-z.
Friess E, Kuempfel T, Modell S, et al. Paroxetine treatment improves motor symptoms in patients with multiple system atrophy. Parkinsonism Relat Disord 2006;12:432-437. https://doi.org/10.1016/j.parkreldis.2006.04.002.
Zhang Q, Yang X, Song H, Jin Y. Cognitive behavioral therapy for depression and anxiety of Parkinson’s disease: a systematic review and meta-analysis. Complement Ther Clin Pract 2020;39:101111. https://doi.org/10.1016/j.ctcp.2020.101111.
Brown RG, Lacomblez L, Landwehrmeyer BG, et al. Cognitive impairment in patients with multiple system atrophy and progressive supranuclear palsy. Brain 2010;133:2382-2393. https://doi.org/10.1093/brain/awq158
Jankovic J, Gilden JL, Hiner BC, et al. Neurogenic orthostatic hypotension: a double-blind, placebo-controlled study with midodrine. Am J Med 1993;95:38-48. https://doi.org/10.1016/0002-9343(93)90230-m.
Ten Harkel AD, Van Lieshout JJ, Wieling W. Treatment of orthostatic hypotension with sleeping in the head-up tilt position, alone and in combination with fludrocortisone. J Intern Med 1992;232139-145. https://doi.org/10.1111/j.1365-2796.1992.tb00563.x.
Schoffer KL, Henderson RD, O'Maley K, O’Sullivan JD. Nonpharmacological treatment, fludrocortisone, and domperidone for orthostatic hypotension in Parkinson's disease. Mov Disord 2007;22:1543-1549. https://doi.org/10.1002/mds.21428.
Brooks DJ, Redmond S, Mathias CJ, Bannister R, Symon L. The effect of orthostatic hypotension on cerebral blood flow and middle cerebral artery velocity in autonomic failure, with observations on the action of ephedrine. J Neurol Neurosurg Psychiatry 1989;52 962-966. https://doi.org/10.1136/jnnp.52.8.962.
Elgebaly A, Abdelazeim B, Mattar O, Gadelkarim M, Salah R, Negida A. Meta-analysis of the safety and efficacy of droxidopa for neurogenic orthostatic hypotension. Clin Auton Res 2016;26:171-180. https://doi.org/10.1007/s10286-016-0349-7.
Kaufmann H, Freeman R, Biaggioni I, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology 2014;83 328-335. https://doi.org/10.1212/WNL.0000000000000615.
Biaggioni I, Freeman R, Mathias CJ, et al. Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension 2015;65:101-107. https://doi.org/10.1161/HYPERTENSIONAHA.114.04035.
Hauser RA, Isaacson S, Lisk JP, Hewitt LA, Rowse G. Droxidopa for the short-term treatment of symptomatic neurogenic orthostatic hypotension in Parkinson’s disease (nOH306B). Mov Disord 2015;30:646-654. https://doi.org/10.1002/mds.26086.
Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin Neurol 2003;23:351-363. https://doi.org/10.1055/s-2004-817719.
Biaggioni I, Arthur Hewitt L, Rowse GJ, Kaufmann H. Integrated analysis of droxidopa trials for neurogenic orthostatic hypotension. BMC Neurol 2017;17:90. https://doi.org/10.1186/s12883-017-0867-5.
Patel H, Simpson A, Palevoda G, Hale GM. Evaluating the effectiveness of atomoxetine for the treatment of primary orthostatic hypotension in adults. J Clin Hypertens (Greenwich) 2018;20 794-797. https://doi.org/10.1111/jch.13260.
Hale GM, Brenner M. Atomoxetine for orthostatic hypotension in an elderly patient over 10 weeks: a case report. Pharmacotherapy 2015;35:e141-e148. https://doi.org/10.1002/phar.1635.
Ramirez CE, Okamoto LE, Arnold AC, et al. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension. 2014;64:1235-1240. https://doi.org/10.1161/HYPERTENSIONAHA.114.04225
Byun JI, Kim DY, Moon J, et al. Efficacy of atomoxetine versus midodrine for neurogenic orthostatic hypotension. Ann Clin Transl Neurol 2020;7:112-120. https://doi.org/10.1002/acn3.50968.
Wenning GK, Geser F, Poewe W. Therapeutic strategies in multiple system atrophy. Mov Disord 2005;20 S67-S76. https://doi.org/10.1002/mds.20543.
Nygaard I. Clinical practice. Idiopathic urgency urinary incontinence. N Engl J Med 2010;363:1156-1162. https://doi.org/10.1056/NEJMcp1003849.
Peyronnet B, Vurture G, Palma JA, et al. Mirabegron in patients with Parkinson disease and overactive bladder symptoms: a retrospective cohort. Parkinsonism Relat Disord 2018;57:22-26. https://doi.org/10.1016/j.parkreldis.2018.07.005.
Gubbiotti M, Conte A, Di Stasi SM, Tambasco N, Giannantoni A. Feasibility of mirabegron in the treatment of overactive bladder in patients affected by Parkinson’s disease: a pilot study. Ther Adv Neurol Disord 2019;12:1756286419843458. https://doi.org/10.1177/1756286419843458
Athanasiou S, Pitsouni E, Grigoriadis T, Zacharakis D, Salvatore S, Serati M. Mirabegron in female patients with overactive bladder syndrome: what’s new? A systematic review and meta-analysis. Eur J Obstet Gynecol Reprod Biol 2020;251:73-82. https://doi.org/10.1016/j.ejogrb.2020.05.018.
Giannantoni A, Rossi A, Mearini E, Del Zingaro M, Porena M, Berardelli A. Botulinum toxin A for overactive bladder and detrusor muscle overactivity in patients with Parkinson's disease and multiple system atrophy. J Urol 2009;182 1453-1457. https://doi.org/10.1016/j.juro.2009.06.023.
Knuepfer S, Juenemann KP. Experience with botulinum toxin type A in the treatment of neurogenic detrusor overactivity in clinical practice. Ther Adv Urol 2014;6:34-42. https://doi.org/10.1177/1756287213510962.
Sakakibara R, Uchiyama T, Asahina M, Yoshiyama M, Yamanishi T, Hattori T. Amezinium metilsulfate, a sympathomimetic agent, may increase the risk of urinary retention in multiple system atrophy. Clin Auton Res 2003;13 51-53. https://doi.org/10.1007/s10286-003-0034-5.
Mathias CJ, Fosbraey P, da Costa DF, Thornley A, Bannister R. The effect of desmopressin on nocturnal polyuria, overnight weight loss, and morning postural hypotension in patients with autonomic failure. Br Med J (Clin Res Ed) 1986;293:353-354. https://doi.org/10.1136/bmj.293.6543.353.
Fowler CJ, O’Malley KJ. Investigation and management of neurogenic bladder dysfunction. J Neurol Neurosurg Psychiatry. 2003;74 iv27-iv31. https://doi.org/10.1136/jnnp.74.suppl_4.iv27.
Hussain IF, Brady CM, Swinn MJ, Mathias CJ, Fowler CJ. Treatment of erectile dysfunction with sildenafil citrate (Viagra) in parkinsonism due to Parkinson’s disease or multiple system atrophy with observations on orthostatic hypotension. J Neurol Neurosurg Psychiatry 2001;71:371-374. https://doi.org/10.1136/jnnp.71.3.371.
Sacca F, Marsili A, Quarantelli M, et al. A randomized clinical trial of lithium in multiple system atrophy. J Neurol 2013;260:458-461. https://doi.org/10.1007/s00415-012-6655-7.
Wenning GK, Krismer F, Poewe W. Rifampicin for multiple system atrophy. Lancet Neurol 2014;13 237-239. https://doi.org/10.1016/S1474-4422(14)70022-5.
Schenk DB, Koller M, Ness DK, et al. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 2017;32:211-218. https://doi.org/10.1002/mds.26878.
Heras-Garvin A, Weckbecker D, Ryazanov S, et al. Anle138b modulates alpha-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov Disord 2019;34:255-263. https://doi.org/10.1002/mds.27562.
Valera E, Spencer B, Mott J, et al. MicroRNA-101 modulates autophagy and oligodendroglial alpha-synuclein accumulation in multiple system atrophy. Front Mol Neurosci 2017;10:329. https://doi.org/10.3389/fnmol.2017.00329.
Herrera-Vaquero M, Bouquio D, Kallab M, et al. The molecular tweezer CLR01 reduces aggregated, pathologic, and seeding-competent alpha-synuclein in experimental multiple system atrophy. Biochim Biophys Acta Mol Basis Dis 2019;1865:165513. https://doi.org/10.1016/j.bbadis.2019.07.007.
Venezia S, Refolo V, Polissidis A, Stefanis L, Wenning GK, Stefanova N. Toll-like receptor 4 stimulation with monophosphoryl lipid A ameliorates motor deficits and nigral neurodegeneration triggered by extraneuronal alpha-synucleinopathy. Mol Neurodegener 2017;12 52. https://doi.org/10.1186/s13024-017-0195-7.
Bassil F, Fernagut PO, Bezard E, et al. Reducing C-terminal truncation mitigates synucleinopathy and neurodegeneration in a transgenic model of multiple system atrophy. Proc Natl Acad Sci U S A 2016;113:9593-9598. https://doi.org/10.1073/pnas.1609291113
Kallab M, Herrera-Vaquero M, Johannesson M, et al. Region-specific effects of immunotherapy with antibodies targeting alpha-synuclein in a transgenic model of synucleinopathy. Front Neurosci 2018;12:452. https://doi.org/10.3389/fnins.2018.00452.
Bensimon G, Ludolph A, Agid Y, Vidailhet M, Payan C, Leigh PN. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain 2009;132 156-171.
Lee PH, Lee JE, Kim HS, et al. A randomized trial of mesenchymal stem cells in multiple system atrophy. Ann Neurol 2012;72:32-40. https://doi.org/10.1002/ana.23612.
Singer W, Dietz AB, Zeller AD, et al. Intrathecal administration of autologous mesenchymal stem cells in multiple system atrophy. Neurology 2019;93 e77-e87. https://doi.org/10.1212/WNL.0000000000007720
Mitsui J, Koguchi K, Momose T, et al. Three-year follow-up of high-dose ubiquinol supplementation in a case of familial multiple system atrophy with compound heterozygous COQ2 mutations. Cerebellum. 2017;16 664-672. https://doi.org/10.1007/s12311-017-0846-9.
Bassil F, Canron MH, Vital A, et al. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain 2017;140 1420-1436. https://doi.org/10.1093/brain/awx044
Poewe W, Seppi K, Fitzer-Attas CJ, et al. Efficacy of rasagiline in patients with the parkinsonian variant of multiple system atrophy: a randomised, placebo-controlled trial. Lancet Neurol 2015;14:145-152. https://doi.org/10.1016/S1474-4422(14)70288-1.
Dodel R, Spottke A, Gerhard A, et al. Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord 2010;25 97-107. https://doi.org/10.1002/mds.22732.
Novak P, Williams A, Ravin P, Zurkiya O, Abduljalil A, Novak V. Treatment of multiple system atrophy using intravenous immunoglobulin. BMC Neurol 2012;12:131. https://doi.org/10.1186/1471-2377-12-131.
Vidal-Martinez G, Segura-Ulate I, Yang B, et al. FTY720-Mitoxy reduces synucleinopathy and neuroinflammation, restores behavior and mitochondria function, and increases GDNF expression in Multiple System Atrophy mouse models. Exp Neurol 2020;325:113120. https://doi.org/10.1016/j.expneurol.2019.113120.
Langerveld AJ, Mihalko D, DeLong C, Walburn J, Ide CF. Gene expression changes in postmortem tissue from the rostral pons of multiple system atrophy patients. Mov Disord 2007;22:766-777. https://doi.org/10.1002/mds.21259.
Miller DW, Johnson JM, Solano SM, Hollingsworth ZR, Standaert DG, Young AB. Absence of alpha-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Transm (Vienna) 2005;112:1613-1624. https://doi.org/10.1007/s00702-005-0378-1.
Ozawa T, Okuizumi K, Ikeuchi T, Wakabayashi K, Takahashi H, Tsuji S. Analysis of the expression level of alpha-synuclein mRNA using postmortem brain samples from pathologically confirmed cases of multiple system atrophy. Acta Neuropathol 2001;102:188-190. https://doi.org/10.1007/s004010100367.
Jin H, Ishikawa K, Tsunemi T, Ishiguro T, Amino T, Mizusawa H. Analyses of copy number and mRNA expression level of the alpha-synuclein gene in multiple system atrophy. J Med Dent Sci 2008;55:145-153.
Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 2009;106:13010-13015. https://doi.org/10.1073/pnas.0903691106.
Hansen C, Angot E, Bergstrom AL, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest 2011;121:715-725. https://doi.org/10.1172/JCI43366.
Kisos H, Pukass K, Ben-Hur T, Richter-Landsberg C, Sharon R. Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS One 2012;7:e46817. https://doi.org/10.1371/journal.pone.0046817.
Konno M, Hasegawa T, Baba T, et al. Suppression of dynamin GTPase decreases alpha-synuclein uptake by neuronal and oligodendroglial cells: a potent therapeutic target for synucleinopathy. Mol Neurodegener 2012;7:38. https://doi.org/10.1186/1750-1326-7-38.
Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 2012;209 975-986. https://doi.org/10.1084/jem.20112457.
Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014;62:387-398. https://doi.org/10.1002/glia.22611.
Gwinn-Hardy K, Mehta ND, Farrer M, et al. Distinctive neuropathology revealed by alpha-synuclein antibodies in hereditary parkinsonism and dementia linked to chromosome 4p. Acta Neuropathol 2000;99:663-672. https://doi.org/10.1007/s004010051177.
Peelaerts W, Bousset L, Van der Perren A, et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015;522:340-344. https://doi.org/10.1038/nature14547.
Peng C, Gathagan RJ, Covell DJ, et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018;557 558-563. https://doi.org/10.1038/s41586-018-0104-4.
Schwarz L, Goldbaum O, Bergmann M, Probst-Cousin S, Richter-Landsberg C. Involvement of macroautophagy in multiple system atrophy and protein aggregate formation in oligodendrocytes. J Mol Neurosci 2012;47:256-266. https://doi.org/10.1007/s12031-012-9733-5.
Tanji K, Odagiri S, Maruyama A, et al. Alteration of autophagosomal proteins in the brain of multiple system atrophy. Neurobiol Dis 2013;49:190-198. https://doi.org/10.1016/j.nbd.2012.08.017
Hasegawa T, Baba T, Kobayashi M, et al. Role of TPPP/p25 on alpha-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy. Neurochem Int 2010;57:857-866. https://doi.org/10.1016/j.neuint.2010.09.002
Stamler D, Margaret B, Wong CH, Offman E. A first in human study of PBT434, a novel small molecule inhibitor of α-synuclein aggregation [abstract]. Mov Disord. 2019;34.
Finkelstein DI, Billings JL, Adlard PA, et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson's disease. Acta Neuropathol Commun 2017;5 53. https://doi.org/10.1186/s40478-017-0456-2.
Price DL, Koike MA, Khan A, et al. The small molecule alpha-synuclein misfolding inhibitor, NPT200-11, produces multiple benefits in an animal model of Parkinson’s disease. Sci Rep 2018;8 16165. https://doi.org/10.1038/s41598-018-34490-9.
Wang W, Nguyen LT, Burlak C, et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein alpha-synuclein. Proc Natl Acad Sci U S A 2016;113:9587-9592. https://doi.org/10.1073/pnas.1610099113.
Ubhi K, Rockenstein E, Mante M, et al. Rifampicin reduces alpha-synuclein in a transgenic mouse model of multiple system atrophy. Neuroreport 2008;19 1271-1276. https://doi.org/10.1097/WNR.0b013e32830b3661.
Chui DH, Tabira T, Izumi S, Koya G, Ogata J. Decreased beta-amyloid and increased abnormal Tau deposition in the brain of aged patients with leprosy. Am J Pathol 1994;145:771-775.
Tomiyama T, Shoji A, Kataoka K, et al. Inhibition of amyloid beta protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. J Biol Chem 1996;271 6839-6844. https://doi.org/10.1074/jbc.271.12.6839.
Sarkar S, Floto RA, Berger Z, et al. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 2005;170:1101-1111. https://doi.org/10.1083/jcb.200504035.
Lu SZ, Guo YS, Liang PZ, et al. Suppression of astrocytic autophagy by alphaB-crystallin contributes to alpha-synuclein inclusion formation. Transl Neurodegener 2019;8:3. https://doi.org/10.1186/s40035-018-0143-7.
Li XZ, Chen XP, Zhao K, Bai LM, Zhang H, Zhou XP. Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced parkinsonism in mice: possible mediation through enhanced autophagy. Int J Neurosci 2013;123 73-79. https://doi.org/10.3109/00207454.2012.729234.
Stefanova N, Reindl M, Neumann M, Kahle PJ, Poewe W, Wenning GK. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov Disord 2007;22:2196-2203. https://doi.org/10.1002/mds.21671.
Bruck D, Wenning GK, Stefanova N, Fellner L. Glia and alpha-synuclein in neurodegeneration: a complex interaction. Neurobiol Dis 2016;85:262-274. https://doi.org/10.1016/j.nbd.2015.03.003.
Mandler M, Valera E, Rockenstein E, et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol Neurodegener 2015;10:10. https://doi.org/10.1186/s13024-015-0008-9.
Mandler M, Valera E, Rockenstein E, et al. Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol 2014;127:861-879. https://doi.org/10.1007/s00401-014-1256-4.
Schneeberger A, Tierney L, Mandler M. Active immunization therapies for Parkinson’s disease and multiple system atrophy. Mov Disord 2016;31:214-224. https://doi.org/10.1002/mds.26377.
Bae EJ, Lee HJ, Rockenstein E, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454-13469. https://doi.org/10.1523/JNEUROSCI.1292-12.2012.
Masliah E, Rockenstein E, Mante M, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 2011;6:e19338. https://doi.org/10.1371/journal.pone.0019338.
Games D, Valera E, Spencer B, et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 2014;34 9441-9454. https://doi.org/10.1523/JNEUROSCI.5314-13.2014.
Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 2017;377 1723-1732. https://doi.org/10.1056/NEJMoa1702752.
DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9 https://doi.org/10.1126/scitranslmed.aag04819/374/eaag0481.
DeVos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics 2013;10 486-97. https://doi.org/10.1007/s13311-013-0194-5.
Zhao HT, John N, Delic V, et al. LRRK2 antisense oligonucleotides ameliorate alpha-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol Ther Nucleic Acids 2017;8:508-519. https://doi.org/10.1016/j.omtn.2017.08.002.
Alarcon-Aris D, Recasens A, Galofre M, et al. Selective alpha-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: potential therapy for Parkinson’s disease. Mol Ther 2018;26:550-567. https://doi.org/10.1016/j.ymthe.2017.11.015.
Scoles DR, Pulst SM. Antisense therapies for movement disorders. Mov Disord 2019;34:1112-1119. https://doi.org/10.1002/mds.27782.
Scherfler C, Sather T, Diguet E, et al. Riluzole improves motor deficits and attenuates loss of striatal neurons in a sequential double lesion rat model of striatonigral degeneration (parkinson variant of multiple system atrophy). J Neural Transm (Vienna) 2005;112 1025-1033. https://doi.org/10.1007/s00702-004-0245-5.
Heo JH, Lee ST, Chu K, Kim M. The efficacy of combined estrogen and buspirone treatment in olivopontocerebellar atrophy. J Neurol Sci 2008;271:87-90. https://doi.org/10.1016/j.jns.2008.03.016
Olanow CW, Rascol O, Hauser R, et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009;361:1268-1278. https://doi.org/10.1056/NEJMoa0809335.
Jenner P, Langston JW. Explaining ADAGIO: a critical review of the biological basis for the clinical effects of rasagiline. Mov Disord 2011;26:2316-23. https://doi.org/10.1002/mds.23926.
Stefanova N, Poewe W, Wenning GK. Rasagiline is neuroprotective in a transgenic model of multiple system atrophy. Exp Neurol 2008;210:421-427. https://doi.org/10.1016/j.expneurol.2007.11.022
Gasmi M, Herzog CD, Brandon EP, et al. Striatal delivery of neurturin by CERE-120, an AAV2 vector for the treatment of dopaminergic neuron degeneration in Parkinson’s disease. Mol Ther 2007;15 62-68. https://doi.org/10.1038/sj.mt.6300010
Johnston LC, Eberling J, Pivirotto P, et al. Clinically relevant effects of convection-enhanced delivery of AAV2-GDNF on the dopaminergic nigrostriatal pathway in aged rhesus monkeys. Hum Gene Ther. 2009;20 (5):497-510. https://doi.org/10.1089/hum.2008.137
Kohl Z, Winner B, Ubhi K, et al. Fluoxetine rescues impaired hippocampal neurogenesis in a transgenic A53T synuclein mouse model. Eur J Neurosci 2012;35 10-19. https://doi.org/10.1111/j.1460-9568.2011.07933.x.
Ubhi K, Inglis C, Mante M, et al. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp Neurol 2012;234 405-416. https://doi.org/10.1016/j.expneurol.2012.01.008
Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006;38 515-517. https://doi.org/10.1038/ng1769.
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 2000;3 1301-1306. https://doi.org/10.1038/81834.
Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 2008;7 97-109. https://doi.org/10.1016/S1474-4422(07)70327-7.
Schapira AH. Mitochondrial myopathies. BMJ 1989;298:1127-1128. https://doi.org/10.1136/bmj.298.6681.1127.
Singer TP, Salach JI, Castagnoli N, Jr., Trevor A. Interactions of the neurotoxic amine 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine with monoamine oxidases. Biochem J 1986;235 785-789. https://doi.org/10.1042/bj2350785.
Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 2017;390 1664-1675. https://doi.org/10.1016/S0140-6736(17)31585-4.
Stefanova N, Georgievska B, Eriksson H, Poewe W, Wenning GK. Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox Res 2012;21 393-404. https://doi.org/10.1007/s12640-011-9294-3.
Dezawa M, Kanno H, Hoshino M, et al. Specific induction of neuronal cells from bone marrow stromal cells and application for autologous transplantation. J Clin Invest 2004;113:1701-1710. https://doi.org/10.1172/JCI20935.
Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. J Cell Biochem 2006;98:1076-1084. https://doi.org/10.1002/jcb.20886.
Park HJ, Bang G, Lee BR, Kim HO, Lee PH. Neuroprotective effect of human mesenchymal stem cells in an animal model of double toxin-induced multiple system atrophy parkinsonism. Cell Transplant 2011;20:827-835. https://doi.org/10.3727/096368910X540630.
Stemberger S, Jamnig A, Stefanova N, Lepperdinger G, Reindl M, Wenning GK. Mesenchymal stem cells in a transgenic mouse model of multiple system atrophy: immunomodulation and neuroprotection. PLoS One 2011;6:e19808. https://doi.org/10.1371/journal.pone.0019808.
Lee PH, An YS, Yong SW, Yoon SN. Cortical metabolic changes in the cerebellar variant of multiple system atrophy: a voxel-based FDG-PET study in 41 patients. Neuroimage 2008;40:796-801. https://doi.org/10.1016/j.neuroimage.2007.11.055.
Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol 2013;169 337-52. https://doi.org/10.1111/bph.12139.
Diguet E, Gross CE, Tison F, Bezard E. Rise and fall of minocycline in neuroprotection: need to promote publication of negative results. Exp Neurol 2004;189 1-4. https://doi.org/10.1016/j.expneurol.2004.05.016.
Stefanova N, Mitschnigg M, Ghorayeb I, et al. Failure of neuronal protection by inhibition of glial activation in a rat model of striatonigral degeneration. J Neurosci Res 2004;78 87-91. https://doi.org/10.1002/jnr.20233.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 1225 kb)
Rights and permissions
About this article

Cite this article
Burns, M.R., McFarland, N.R. Current Management and Emerging Therapies in Multiple System Atrophy. Neurotherapeutics 17, 1582–1602 (2020). https://doi.org/10.1007/s13311-020-00890-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00890-x