High-Density SNP-Based Association Mapping of Seed Traits in Fenugreek Reveals Homology with Clover


 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
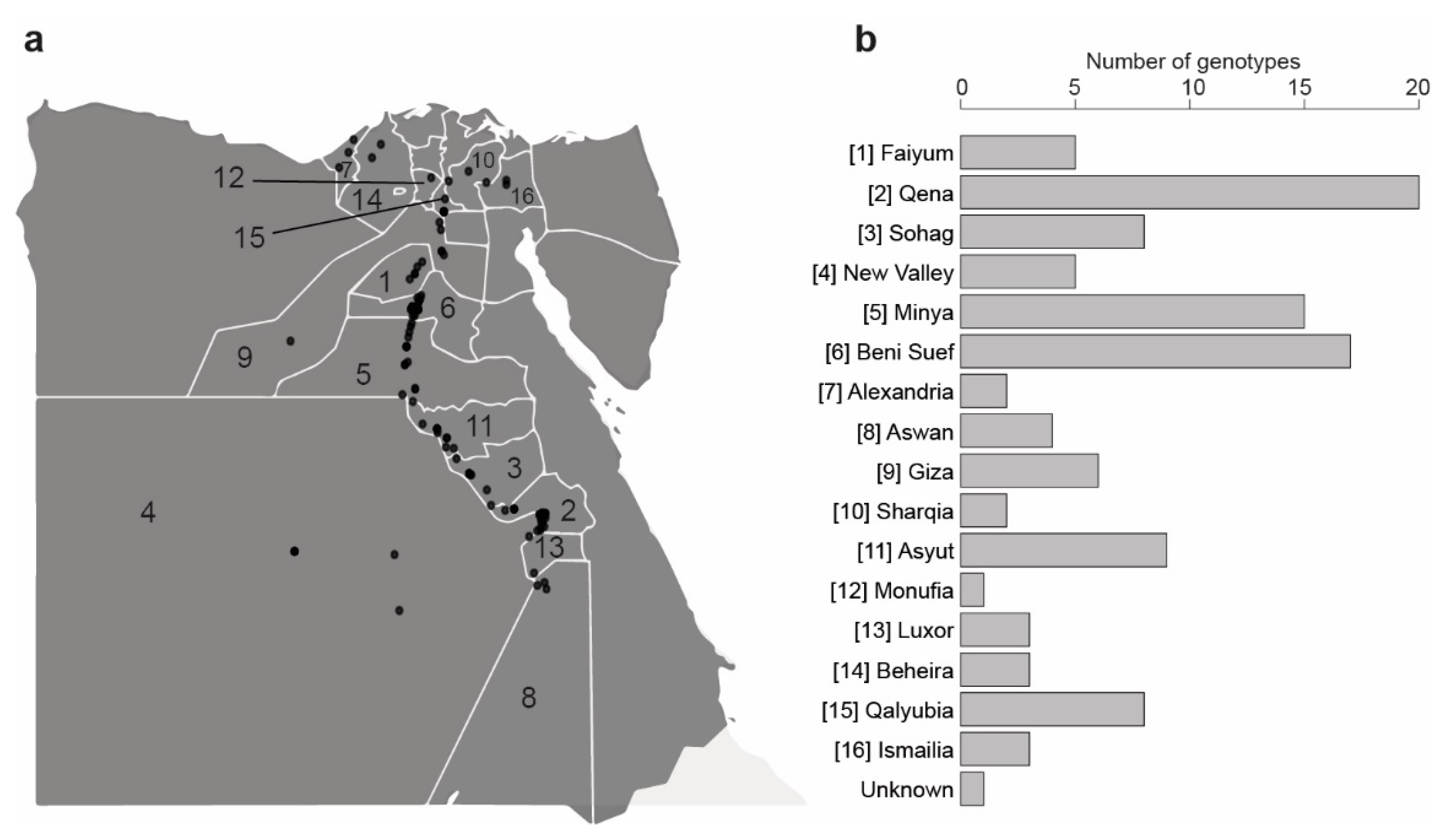
2.1. Fenugreek Genotypes Collection
2.2. DNA Extraction and Library Preparation for Sequencing
2.3. Bioinformatics and Statistical Analyses
2.4. Population Structure Estimation
2.5. Phenotyping and Association Mapping
2.6. Homology Analysis to Predict Candidate Genes
3. Results
3.1. Sequencing Quality
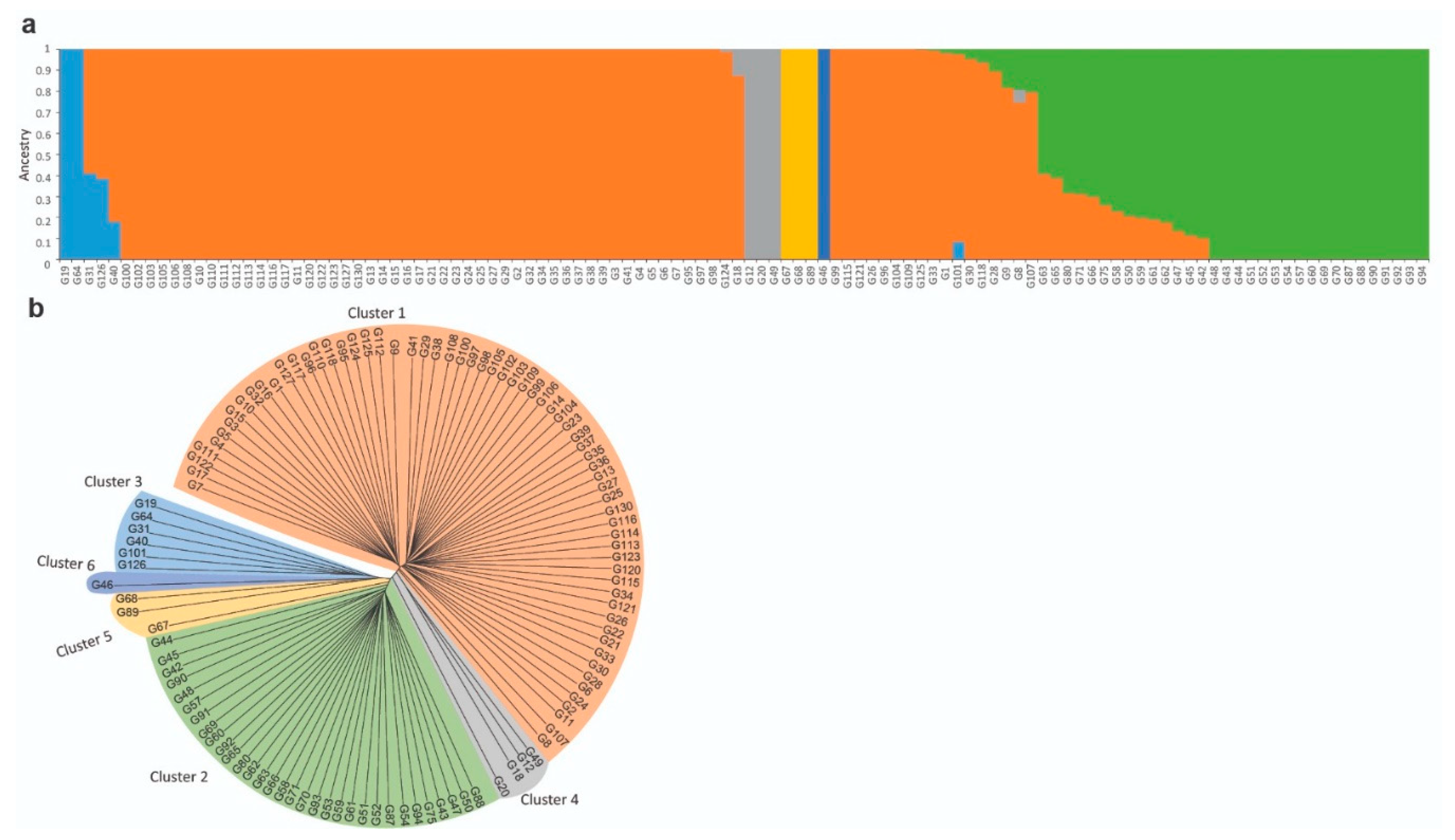
3.2. Population Structure and Genetic Diversity
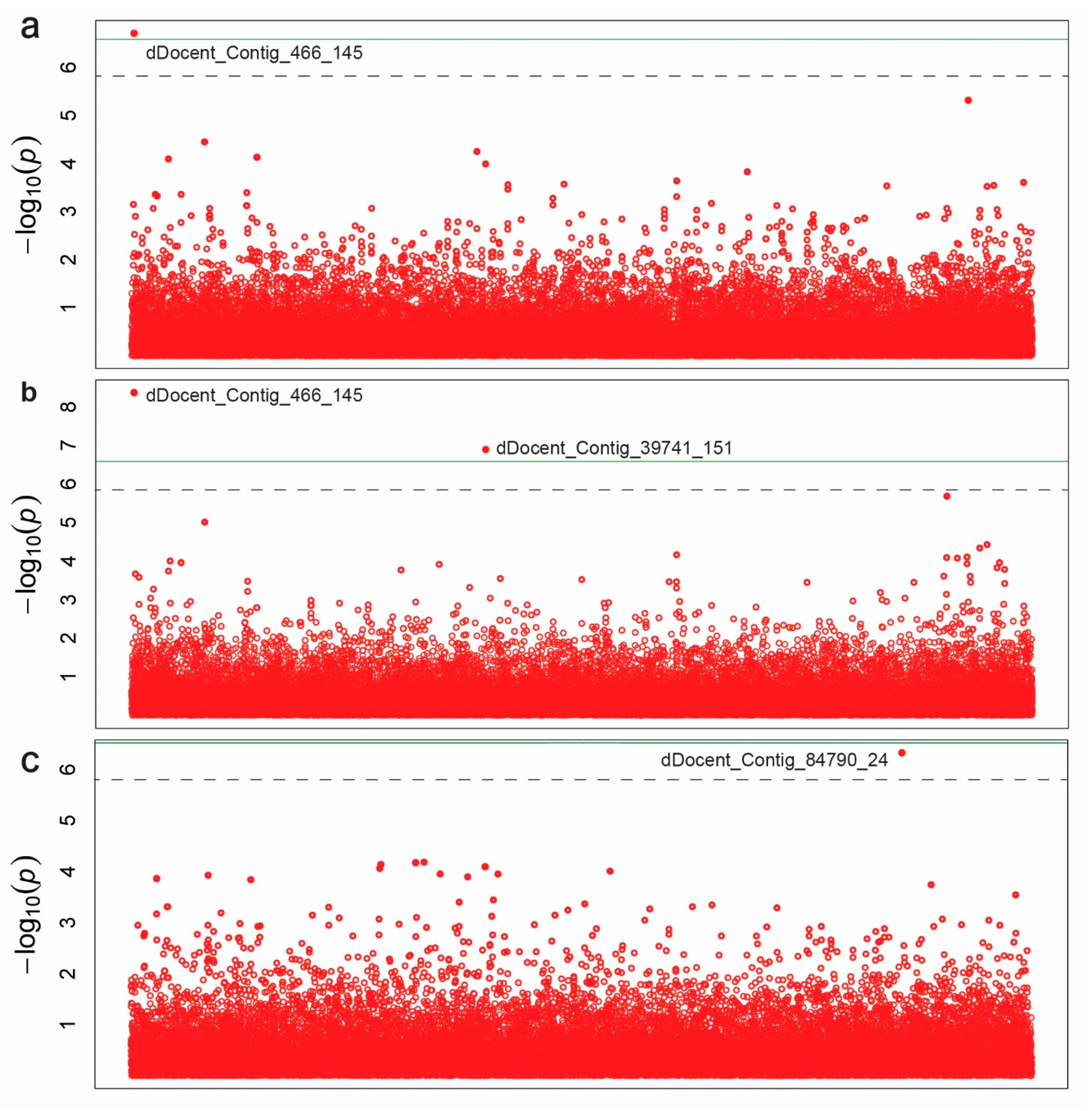
3.3. Association Mapping
3.4. Homology Analysis to Predict Candidate Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Small, E.; Lassen, P.; Brookes, B.S. An expanded circumscription of medicago (Leguminosae, Trifolieae) based on explosive flower tripping. Willdenowia 1987, 16, 415–437. [Google Scholar]
- Bena, G. Molecular phylogeny supports the morphologically based taxonomic transfer of the “medicagoid”Trigonella species to the genus Medicago L. Plant Syst. Evol. 2001, 229, 217–236. [Google Scholar] [CrossRef]
- Wani, S.A.; Kumar, P. Fenugreek: A review on its nutraceutical properties and utilization in various food products. J. Saudi Soc. Agric. Sci. 2016, 17, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Alghamdi, S.S.; Mahmood, K.; Afzal, M. Fenugreek a multipurpose crop: Potentialities and improvements. Saudi J. Biol. Sci. 2016, 23, 300–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, W.G. Understanding and using quantitative genetic variation. Philos. Trans. Royal Soc. Lond. Ser. B. Biol. Sci. 2010, 365, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedoruk, M.J.; Vandenberg, A.; Bett, K.E. Quantitative trait loci analysis of seed quality characteristics in lentil using single nucleotide polymorphism markers. Plant Genome 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Cober, E.R.; Voldeng, H.D.; Frégeau-Reid, J.A. Heritability of seed shape and seed size in soybean. Crop Sci. 1997, 37. [Google Scholar] [CrossRef]
- Drabo, I.; Redden, R.; Smithson, J.B.; Aggarwal, V.D. Inheritance of seed size in cowpea (Vigna unguiculata (L.) Walp.). Euphytica 1984, 33, 929–934. [Google Scholar] [CrossRef]
- Altuntaş, E.; Özgöz, E.; Taşer, Ö.F. Some physical properties of fenugreek (Trigonella foenum-graceum L.) seeds. J. Food Eng. 2005, 71, 37–43. [Google Scholar] [CrossRef]
- Aasim, M.; Baloch, F.S.; Nadeem, M.A.; Bakhsh, A.; Sameeullah, M.; Day, S. Fenugreek (Trigonella foenum-graecum L.): An underutilized edible plant of modern world. In Global Perspectives on Underutilized Crops; Ozturk, M., Hakeem, K.R., Ashraf, M., Ahmad, M.S.A., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 381–408. [Google Scholar] [CrossRef]
- McCormick, K.M.; Norton, R.M.; Eagles, H.A. Phenotypic variation within a fenugreek (Trigonella foenum-graecum L.) germplasm collection. I. Description of the collection. Genet. Resour. Crop Evol. 2009, 56, 639–649. [Google Scholar] [CrossRef]
- Dangi, R.S.; Lagu, M.D.; Choudhary, L.B.; Ranjekar, P.K.; Gupta, V.S. Assessment of genetic diversity in Trigonella foenum-graecum and Trigonella caeruleausing ISSR and RAPD markers. BMC Plant Biol. 2004, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindhu, A.; Tehlan, S.K.; Chaudhury, A. Analysis of genetic diversity among medicinal therapist Trigonella foenum-graecum L. genotypes through RAPD and SSR markers. Acta Physiol. Plant. 2017, 39, 100. [Google Scholar] [CrossRef]
- Amiriyan, M.; Shojaeiyan, A.; Yadollahi, A.; Maleki, M.; Bahari, Z. Genetic diversity analysis and population structure of some Iranian Fenugreek (Trigonella foenum-graecum L.) landraces using SRAP Markers. Mol. Biol. Res. Commun. 2019, 8, 181–190. [Google Scholar] [PubMed]
- Jamann, T.M.; Balint-Kurti, P.J.; Holland, J.B. QTL mapping using high-throughput sequencing. In Plant Functional Genomics: Methods and Protocols; Alonso, J.M., Stepanova, A.N., Eds.; Springer: New York, NY, USA, 2015; pp. 257–285. [Google Scholar] [CrossRef]
- Burghardt, L.T.; Young, N.D.; Tiffin, P. A guide to genome-wide association mapping in plants. Curr. Protoc. Plant Biol. 2017, 2, 22–38. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, K.; Ghosh, A.; Kumar, V.; Chaudhary, S.; Srivastava, N.; Katudia, K.; Tiwari, T.; Chikara, S.K. De novo transcriptome sequencing in Trigonella foenum-graecum L. to identify genes involved in the biosynthesis of diosgenin. Plant Genome 2013, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Ciura, J.; Szeliga, M.; Grzesik, M.; Tyrka, M. Next-generation sequencing of representational difference analysis products for identification of genes involved in diosgenin biosynthesis in fenugreek (Trigonella foenum-graecum). Planta 2017, 245, 977–991. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Li, X.; Zhou, Z.; Li, C.; Zhang, Y. Comparative transcriptome analysis identifies genes involved in diosgenin biosynthesis in Trigonella foenum-graecum L. Molecules 2019, 24, 140. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Mashayekh, T.; Rashidi-Monfared, S.; Ebrahimi, A.; Abedini, D. New insights into diosgenin biosynthesis pathway and its regulation in Trigonella foenum-graecum L. Phytochem. Anal. 2020, 31, 229–241. [Google Scholar] [CrossRef]
- George, J.; Sawbridge, T.I.; Cogan, N.O.; Gendall, A.R.; Smith, K.F.; Spangenberg, G.C.; Forster, J.W. Comparison of genome structure between white clover and Medicago truncatula supports homoeologous group nomenclature based on conserved synteny. Genome 2008, 51, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Dluhošová, J.; Ištvánek, J.; Nedělník, J.; Řepková, J. Red clover (Trifolium pratense) and zigzag clover (T. medium)—A picture of genomic similarities and differences. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puritz, J.B.; Hollenbeck, C.M.; Gold, J.R. dDocent: A RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ 2014, 2, e431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F. Trim galore. A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. Bioinformatics 2015, 516, 517. [Google Scholar]
- Chong, Z.; Ruan, J.; Wu, C.-I. Rainbow: An integrated tool for efficient clustering and assembling RAD-seq reads. Bioinformatics 2012, 28, 2732–2737. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [Green Version]
- Gupta, G.S.; Bailey, D. Fast image capture and vision processing for robotic applications. In Sensors: Advancements in Modeling, Design Issues, Fabrication and Practical Applications; Mukhopadhyay, S.C., Huang, R.Y.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 329–352. [Google Scholar] [CrossRef]
- Lipka, A.E.; Tian, F.; Wang, Q.; Peiffer, J.; Li, M.; Bradbury, P.J.; Gore, M.A.; Buckler, E.S.; Zhang, Z. GAPIT: Genome association and prediction integrated tool. Bioinformatics 2012, 28, 2397–2399. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ersoz, E.; Lai, C.-Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.M.; Zaitlen, N.A.; Wade, C.M.; Kirby, A.; Heckerman, D.; Daly, M.J.; Eskin, E. Efficient control of population structure in model organism association mapping. Genetics 2008, 178, 1709–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Clarke, G.M.; Anderson, C.A.; Pettersson, F.H.; Cardon, L.R.; Morris, A.P.; Zondervan, K.T. Basic statistical analysis in genetic case-control studies. Nat. Protoc. 2011, 6, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakis, D.; Kanterakis, A.; Manousaki, T.; Tsakogiannis, A.; Tsagris, M.; Tsamardinos, I.; Papaharisis, L.; Chatziplis, D.; Potamias, G.; Tsigenopoulos, C.S. Scanning of genetic variants and genetic mapping of phenotypic traits in gilthead sea bream through ddRAD sequencing. Front. Genet. 2019, 10, 675. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Chaparro, C.; Gayraud, T.; de Souza, R.F.; Domingues, D.S.; Akaffou, S.; Laforga Vanzela, A.L.; Kochko, A.; Rigoreau, M.; Crouzillat, D.; Hamon, S.; et al. Terminal-repeat retrotransposons with GAG domain in plant genomes: A new testimony on the complex world of transposable elements. Genome Biol. Evol. 2015, 7, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Brock, J.; Dyer, J.M.; Kutchan, T.; Schachtman, D.; Augustin, M.; Ge, Y.; Fahlgren, N.; Abdel-Haleem, H. Genetic diversity and population structure of a camelina sativa spring panel. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Niu, S.; Song, Q.; Koiwa, H.; Qiao, D.; Zhao, D.; Chen, Z.; Liu, X.; Wen, X. Genetic diversity, linkage disequilibrium, and population structure analysis of the tea plant (Camellia sinensis) from an origin center, Guizhou plateau, using genome-wide SNPs developed by genotyping-by-sequencing. BMC Plant Biol. 2019, 19, 328. [Google Scholar] [CrossRef] [PubMed]
- Alemu, A.; Feyissa, T.; Tuberosa, R.; Maccaferri, M.; Sciara, G.; Letta, T.; Abeyo, B. Genome-wide association mapping for grain shape and color traits in Ethiopian durum wheat (Triticum turgidum ssp. durum). Crop J. 2020. [Google Scholar] [CrossRef]
- Gao, L.; Turner, M.K.; Chao, S.; Kolmer, J.; Anderson, J.A. Genome wide association study of seedling and adult plant leaf rust resistance in elite spring wheat breeding lines. PLoS ONE 2016, 11, e0148671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaler, A.S.; Purcell, L.C. Estimation of a significance threshold for genome-wide association studies. BMC Genom. 2019, 20, 618. [Google Scholar] [CrossRef] [Green Version]
- Kaler, A.S.; Abdel-Haleem, H.; Fritschi, F.B.; Gillman, J.D.; Ray, J.D.; Smith, J.R.; Purcell, L.C. Genome-wide association mapping of dark green color index using a diverse panel of soybean accessions. Sci. Rep. 2020, 10, 5166. [Google Scholar] [CrossRef] [Green Version]
- Dundas, I.S.; Nair, R.M.; Verlin, D.C. First report of meiotic chromosome number and karyotype analysis of an accession of Trigonella balansae (Leguminosae). N. Z. J. Agric. Res. 2006, 49, 55–58. [Google Scholar] [CrossRef]
- Ahmad, F.; Acharya, S.N.; Mir, Z.; Mir, P.S. Localization and activity of rRNA genes on fenugreek (Trigonella foenum-graecum L.) chromosomes by fluorescent in situ hybridization and silver staining. Theor. Appl. Genet. 1999, 98, 179–185. [Google Scholar] [CrossRef]
- Istvanek, J.; Jaros, M.; Krenek, A.; Repkova, J. Genome assembly and annotation for red clover (Trifolium pratense; Fabaceae). Am. J. Bot. 2014, 101, 327–337. [Google Scholar] [CrossRef]
- McClean, P.E.; Bett, K.E.; Stonehouse, R.; Lee, R.; Pflieger, S.; Moghaddam, S.M.; Geffroy, V.; Miklas, P.; Mamidi, S. White seed color in common bean (Phaseolus vulgaris) results from convergent evolution in the P (pigment) gene. New Phytol. 2018, 219, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Murube, E.; Campa, A.; Song, Q.; McClean, P.; Ferreira, J.J. Toward validation of QTLs associated with pod and seed size in common bean using two nested recombinant inbred line populations. Mol. Breed. 2019, 40, 7. [Google Scholar] [CrossRef]
- Radkova, M.; Revalska, M.; Kertikova, D.; Iantcheva, A. Zinc finger CCHC-type protein related with seed size in model legume species Medicago truncatula. Biotechnol. Biotechnol. Equip. 2019, 33, 278–285. [Google Scholar] [CrossRef] [Green Version]
- De Vega, J.J.; Ayling, S.; Hegarty, M.; Kudrna, D.; Goicoechea, J.L.; Ergon, Å.; Rognli, O.A.; Jones, C.; Swain, M.; Geurts, R. Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Sci. Rep. 2015, 5, 17394. [Google Scholar] [CrossRef] [PubMed]
- Cannon, S.B.; Sterck, L.; Rombauts, S.; Sato, S.; Cheung, F.; Gouzy, J.; Wang, X.; Mudge, J.; Vasdewani, J.; Schiex, T.; et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc. Natl. Acad. Sci. USA 2006, 103, 14959–14964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hougaard, B.K.; Madsen, L.H.; Sandal, N.; de Carvalho Moretzsohn, M.; Fredslund, J.; Schauser, L.; Nielsen, A.M.; Rohde, T.; Sato, S.; Tabata, S.; et al. Legume anchor markers link syntenic regions between Phaseolus vulgaris, Lotus japonicus, Medicago truncatula and Arachis. Genetics 2008, 179, 2299–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gujaria-Verma, N.; Vail, S.L.; Carrasquilla-Garcia, N.; Penmetsa, R.V.; Cook, D.R.; Farmer, A.D.; Vandenberg, A.; Bett, K.E. Genetic mapping of legume orthologs reveals high conservation of synteny between lentil species and the sequenced genomes of Medicago and chickpea. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Webb, A.; Cottage, A.; Wood, T.; Khamassi, K.; Hobbs, D.; Gostkiewicz, K.; White, M.; Khazaei, H.; Ali, M.; Street, D.; et al. A SNP-based consensus genetic map for synteny-based trait targeting in faba bean (Vicia faba L.). Plant Biotechnol. J. 2016, 14, 177–185. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | SNP | −LOG10 (P) | MAF | R2 | FDR | Sequences of ddRAD Primers |
|---|---|---|---|---|---|---|
| Seed Length | dDocent_Contig_466_145 | 6.71 | 0.36 | 0.39 | 0.007 | GAGACTGCTGAATTTTCCAAGTGTATTAAGTTTGAGAATGGTCTGCGTGC[T]GAGATTAAGTGGGCCATTGGGTACCAGAAGATCNNNNNNNNNNTAATTCT |
| Seed Width | dDocent_Contig_466_145 | 8.36 | 0.36 | 0.46 | 0.000 | |
| dDocent_Contig_39741_151 | 6.88 | 0.28 | 0.38 | 0.002 | TTGAAGGTTGCTAAGGAGGGCGCTGGCTCGGCAGGTCCGAAGGAGACTGC[T]GAGATTGCCAGCCTCAGTCGCGCAGAGTTGATCNNNNNNNNNNAATTCTG | |
| Seed Color | dDocent_Contig_84790_24 | 6.32 | 0.05 | 0.28 | 0.016 | NAATTCTAACTCTTCCCGTAGTG[C]TGGCCCCCGTTCTCCAACTGAGTACGTTCATCTCGATTGGGATGACGGCC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abd El-Wahab, M.M.H.; Aljabri, M.; Sarhan, M.S.; Osman, G.; Wang, S.; Mabrouk, M.; El-Shabrawi, H.M.; Gabr, A.M.M.; Abd El-Haliem, A.M.; O'Sullivan, D.M.; et al. High-Density SNP-Based Association Mapping of Seed Traits in Fenugreek Reveals Homology with Clover. Genes 2020, 11, 893. https://doi.org/10.3390/genes11080893
Abd El-Wahab MMH, Aljabri M, Sarhan MS, Osman G, Wang S, Mabrouk M, El-Shabrawi HM, Gabr AMM, Abd El-Haliem AM, O'Sullivan DM, et al. High-Density SNP-Based Association Mapping of Seed Traits in Fenugreek Reveals Homology with Clover. Genes. 2020; 11(8):893. https://doi.org/10.3390/genes11080893
Chicago/Turabian StyleAbd El-Wahab, Mustafa M. H., Maha Aljabri, Mohamed S. Sarhan, Gamal Osman, Shichen Wang, Mahmoud Mabrouk, Hattem M. El-Shabrawi, Ahmed M. M. Gabr, Ahmed M. Abd El-Haliem, Donal M. O'Sullivan, and et al. 2020. "High-Density SNP-Based Association Mapping of Seed Traits in Fenugreek Reveals Homology with Clover" Genes 11, no. 8: 893. https://doi.org/10.3390/genes11080893