DNA6mA-MINT: DNA-6mA Modification Identification Neural Tool



1
Department of Electronics and Information Engineering, Jeonbuk National University, Jeonju 54896, Korea
2
Department of Avionics Engineering, Air University, Islamabad 44000, Pakistan
3
Advanced Electronics and Information Research Center, Jeonbuk National University, Jeonju 54896, Korea
*
Author to whom correspondence should be addressed.
Genes 2020, 11(8), 898; https://doi.org/10.3390/genes11080898
Submission received: 3 July 2020
/
Revised: 28 July 2020
/
Accepted: 28 July 2020
/
Published: 5 August 2020
(This article belongs to the Special Issue Computational Methods for the Analysis of Genomic Data and Biological Processes)
Abstract
:DNA N-methyladenine (6mA) is part of numerous biological processes including DNA repair, DNA replication, and DNA transcription. The 6mA modification sites hold a great impact when their biological function is under consideration. Research in biochemical experiments for this purpose is carried out and they have demonstrated good results. However, they proved not to be a practical solution when accessed under cost and time parameters. This led researchers to develop computational models to fulfill the requirement of modification identification. In consensus, we have developed a computational model recommended by Chou’s 5-steps rule. The Neural Network (NN) model uses convolution layers to extract the high-level features from the encoded binary sequence. These extracted features were given an optimal interpretation by using a Long Short-Term Memory (LSTM) layer. The proposed architecture showed higher performance compared to state-of-the-art techniques. The proposed model is evaluated on Mus musculus, Rice, and “Combined-species” genomes with 5- and 10-fold cross-validation. Further, with access to a user-friendly web server, publicly available can be accessed freely.
1. Introduction
In genomes of distinct species, DNA N-methyladenine (6mA) illustrates a crucial epigenetic transformation [1,2]. DNA 6mA is a non-canonical process that modifies the catalyzed adenine ring of DNA methyltransferases [3]. Alteration occurs at the sixth position of the adenine ring where a methyl group is additionally introduced. DNA 6mA holds a vital role in numerous biological processes, which includes DNA replication [4], DNA repair [5], DNA transcription [6], and others. Recent research established that uneven 6mA modification has a role in different diseases such as cancer [7], immune systems, and others. Therefore, this makes it necessary to identify a 6mA position in the genome sites. Mammalian 6mA largely originates from the genomic incorporation mediated by DNA polymerase, while the methylase-generated 6mA in mice remains elusive [8].
Silico prediction is considered to be a principal approach to encounter the aforementioned problem, while N-methyladenine prediction is its alternative. Intensive labor with extravagant experiments and expenses limits the use of silico prediction, making 6mA prediction an ideal solution for tracking modifications in the genome. For the identification of 6mA, diversified techniques can be found in the literature. Initially, ultraviolet absorption spectra, paper chromatographic movement, and electrophoretic mobility were combined to represent a complete mechanism. Although this method was not efficacious enough to be used for detecting 6mA transformations in animals [9], this led to an introduction of another technique for identifying 6mA modification using a restriction enzyme, but this approach was only capable of identifying transformed adenines that are present in the target motifs [10].
For the detection of 6mA sites in prokaryotes and eukaryotes, numerous techniques were proposed such as single molecule real-time (SMRT) sequencing [11], methylated DNA immunoprecipitation sequencing [12], ultra-high performance liquid chromatography with mass spectrometry [1], and metabolically generated stable isotope-labeled deoxynucleoside code [13]. Chlamydomonas genes carry 84% N-methyladenine modifications, which was identified after 6mA an immunoprecipitation sequencing experiment [14]. SMRT sequencing found out that adenines of methylated sites carry 2.8% of initial-diverged fungi [15]. Utilization of SMRT, 6mA immunoprecipitation, and mass spectrometry result in 0.2% of adenines being methylated [16].
The experimental techniques proved to be expensive and prolonged processes, therefore researchers tried to come up with computational techniques for prediction of DNA 6mA modifications. For this purpose, numerous prediction tools were proposed in the literature. iDNA6mA-PseKNC was the first ever N-methyladenine modification prediction tool for the Mus musculus genome [17]. iDNA6mA-PseKNC proposed sequence sample formulation for feature extraction and employed six different classifiers to identify the modification. csDMA is another reported tool that predicts the modification in N-adenine methylation, which used K-mer pattern, KSNPF frequency, nucleic shift density, binary code, and motif score matrix for extraction of the feature vector of the sequence [18]. Further, they deployed five different classifiers to evaluate the performance of the extracted feature set. Recently, 6mA-Finder was introduced as an online tool for predicting 6mA modification [19]. 6mA-Finder engaged seven sequence encoding schemes to get three types of physico-chemical features encoded. These encoded features were then embedded in seven different classifiers to evaluate the performance of encoded features. The i6mA-Pred is an identification tool for N-methyladenine modification in the rice genome [20].
FastFeatGen is another tool present in the literature that predicts DNA N methyladenine sites [21]. FastFeatGen has used a parallel feature extraction technique followed by an exploratory feature selection algorithm to get the most relevant features. These features are then fed to Extra-Tree Classifier (ETC) for the prediction. Liang et al. proposed the i6mA-DNCP tool for the identification of 6mA sites [22]. i6mA-DNCP used optimized dinucleotide-based features with bagging classifier for the prediction model. Undoubtedly machine learning has illustrated high performance for many research problems, but the neural network has its benefits that need to be investigated for every research problem.
In recent years, Neural Network (NN)-based techniques, especially Convolution Neural Network (CNN), have shown tremendous improvement in many different research problems, e.g., in medical imaging [23,24] and bio-informatics [25,26,27], while the use of CNN for DNA-6mA modification identification is still in the infancy. Recently, a technique called iIM-CNN was reported by Wahab et al., which uses a CNN-based model for the N6-adenine methylation modification identification in genomes of different species [28]. The proposed CNN model in iIM-CNN carries two convolution layers with two max-pooling layers and a set of fully connected layers. iIM-CN showed high performance in prediction of N-methyladenine modification, somehow still, a research space is available where many aspects of CNN can be explored more.
This article aims to provide a CNN and Long Short-Term Memory (LSTM)-based efficient tool named DNA6mA-MINT, for DNA 6mA modification identification. The proposed model uses CNN for feature extraction while LSTM gives optimal interpretation to those features. The proposed architecture demonstrates higher performance than the existing state-of-the-art techniques on the “combined-species”, M. musculus genome, and rice genome benchmark datasets. For better comparative analysis between DNA6mA-MINT and existing techniques, we have carried out performance analysis on 5- and 10-fold cross-validation. When compared with respective models available in the literature, Matthews Correlation Coefficient (MCC) for the “combined-species” benchmark dataset is noted with an increase of 20.83% for 5-fold cross-validation. The five steps are construction of dataset, encoding samples, constructing prediction model, evaluation of the proposed model, and establishing an online server. For the development of a useful and effective biological predictor, Chou’s 5-steps rule needs to be followed [29,30]. These steps were followed by the previous researchers as well [17,18,19,20,28]. This research article follows Chou’s 5-steps rule.
2. Benchmark Dataset
In this work, we used three datasets. The M. musculus genome database for DNA 6mA was proposed in 2018 by Feng et al. [17]. The dataset consists of 1934 samples for each positive and negative case. The 6mA sites available in the mouse genome were collected from MethSMRT database [31] with Gene Expression Omnibus (GEO) accession number GSE71866. Another dataset was on the rice genome, which was presented in 2019 by Chen et al. [20]. This dataset consists of 880 samples for each positive and negative case. The 6mA sites in rice genomes were provided by Zhou et al. [16] with GEO accession number GSE103145. Combining both aforementioned databases, a “combined-species” dataset is generated which contains 2768 samples for the positive cases and 2716 for negative cases. While the “combined-species” dataset did not contain sequence redundancy, which is eliminated by CD-HIT software [32], the rigorous sequence identity threshold was 0.80. Further, the dataset for training comprises 2214 positive samples and 2214 negative samples, while for the purpose of independent training 554 positive samples and 502 negative samples are taken into account. The length of all sequences in the datasets are 41 bp centered with the 6mA and non-6mA site.
3. Methodology
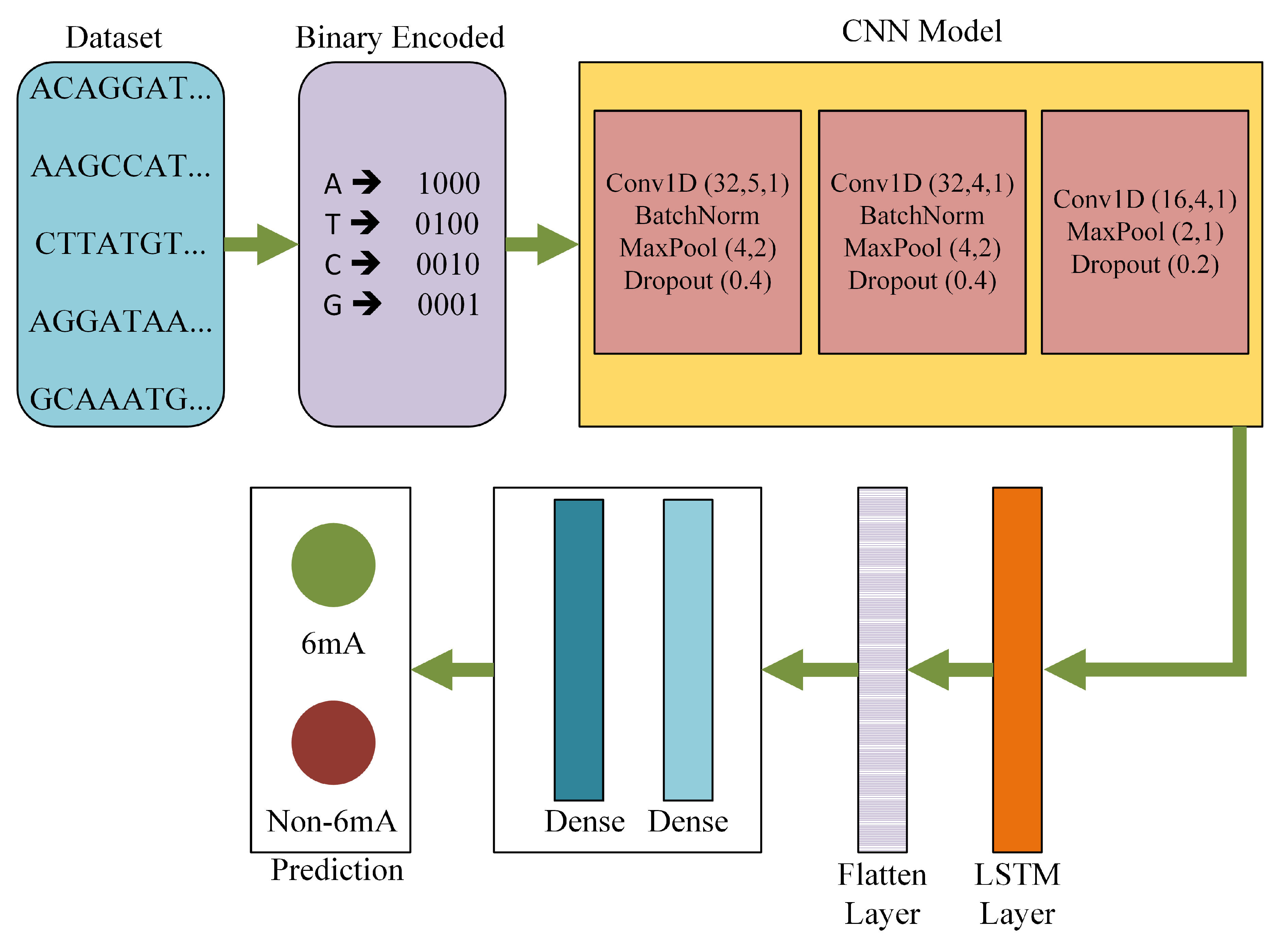
The proposed architecture was an efficient deep learning-based model comprised of several convolution layers, hidden layers, LSTM layers, and dense layers. Figure 1 is a visual representation of DNA6mA-MINT. This model holds the capability of extracting critical features from the input raw sequence, which are then used to carry prediction. The input sequence carries a combination of 4 nucleotides, A, T, C, and G, as can be seen in the dataset block of Figure 1. The NNs work on the numerical data only, therefore an encoding scheme is required here which can effectively convert the sequence-based data to a numerical representation. For the said purpose, binary encoding was taken into account. Where A, T, C, and G are represented as (1, 0, 0, 0), (0, 1, 0, 0), (0, 0, 1, 0), (0, 0, 0, 1), respectively.
Table 1 shows the architecture details of DNA6mA-MINT. The DNA6mA-MINT includes three convolution layers that use different parameters to extract the features from the input binary encoded sequence. The first convolution layer uses 32 filters with a filter size of five, followed by another convolution layer which uses 32 different filters with a filter size of four. The last convolution layer uses 16 filters of size four. Features extracted by the first two convolution layers undergo Batch normalization, Max-pooling layer, and a dropout layer discarding 40% of features, while the features extracted by the last convolution undergo Max-pooling and dropout of 20%. The number of filters for the convolution layer with their filter size, Stride length, pool-size, and the dropout ratio is decided after hyperparameter tuning. Therefore, the selected values of the parameters were capable of giving the best performance from the model.
In CNN models a greater number of convolution layers represents the extraction of deeper features, but for the research problem under consideration, we cannot use more number of convolution layers, as by further increasing the convolution layers, the overfitting problem is observed. Using three convolution layers was an ideal solution to classify the input data we have, as this leads us to a high-performance architecture. All the convolution layers used ReLU as an activation function which eases the training process. At this stage, sigmoid or tanh are not used as an activation function, the reason being their vanishing gradient problem. The vanishing gradient problem makes the training process difficult, where ReLU solves this problem due to its unbounded nature.
The set of features extracted from the CNN model was fed into LSTM, which is a recurrent neural network (RNN). Here, the LSTM supports the sequence prediction. Therefore, the proposed model consists of two sub-models: the feature extractor which is the CNN model and the feature interpreter, which is the LSTM layer. In the proposed model, LSTM is used with a filter size of four, which is selected after hyperparameter tuning. The optimally interpreted feature set was converted to a single feature column by using a flattened layer. A single column feature set undergoes two dense layers with 32 and 1 neurons respectively to give the final classification output. The first dense layer uses the ReLU activation function while the second dense layer uses the sigmoid activation function. Sigmoid activation function makes the output range between 0 and 1 which is required for a binary classification problem. Below are the equations for ReLU and sigmoid functions.
DNA6mA-MINT is implemented on the Keras framework [33]. The output of the sigmoid activation function will be an input to the objective function. Binary cross-entropy is used as an objective function [34] and its equation is as follows,
where is the label for class sample. The loss can also be expressed as
Stochastic gradient descent is used for optimizing the objective function. The equation below is used for calculating stochastic gradient descent,
where is the current estimation of at iteration ‘’, is the learning rate, and is computed gradient of the loss function.
Stochastic gradient descent reduces the computational complexity by achieving faster iterations [35]. In the optimization process, the learning rate and momentum were set to 0.004 and 0.9 respectively.
4. Figure of Merits
Evaluation of the DNA6mA-MINT is carried out using k-fold cross-validation where the value of k in our case is kept five and ten. In both cases, the whole dataset was divided into k subset. A single subset is chosen iteratively for the testing purpose where remaining subsets are used for training purposes. For the final performance estimation of the model, an average of k-trials is taken.
The figure of merits used in recent publications are listed with equations below,
where
TP = True Positive = 6mA correctly identified as 6mA
FP = False Positive = Non 6mA incorrectly identified as 6mA
TN = True Negative = Non 6mA correctly identified as Non 6mA
FN = False Negative = 6mA incorrectly identified as Non 6mA
Sensitivity, also known as True Positive Rate (TPR), is a statistical measure which calculates the ratio of positive samples identified as positive samples by the model. Specificity, also known as True Negative Rate (TNR), is also a statistical measure which calculates the ratio of negative samples identified as negative samples by the model. Accuracy measures the closeness of the model to the idle situation. While the Matthews correlation coefficient (MCC) depicts the quality of the model as a binary classifier, another figure of merit used in this study is the area under Receiver Operating Characteristics (auROC). It measures the performance of the model at various thresholds. The auROC indicates the capability of the model to distinguish two classes from each other.
5. Results and Discussion
The proposed model was evaluated on three datasets: M. musculus genome, rice genome, and “Combined-species”. The state-of-the-art techniques in the literature carried out their results either using 5-fold cross-validation or 10-fold cross-validation. Therefore, we validated DNA6mA-MINT by using both numbers of folds so that a better comparative analysis can be derived. Therefore, it is important to compare 5-fold cross-validation results with the models that have reported their results on 5-fold cross-validation. Similarly, 10-fold results should be compared with the 10-fold cross-validated model in the literature. A greater number of folds depicts higher performance, the reason being that by increasing the number of folds, the training dataset gets a higher ratio of the data which increases the model performance.
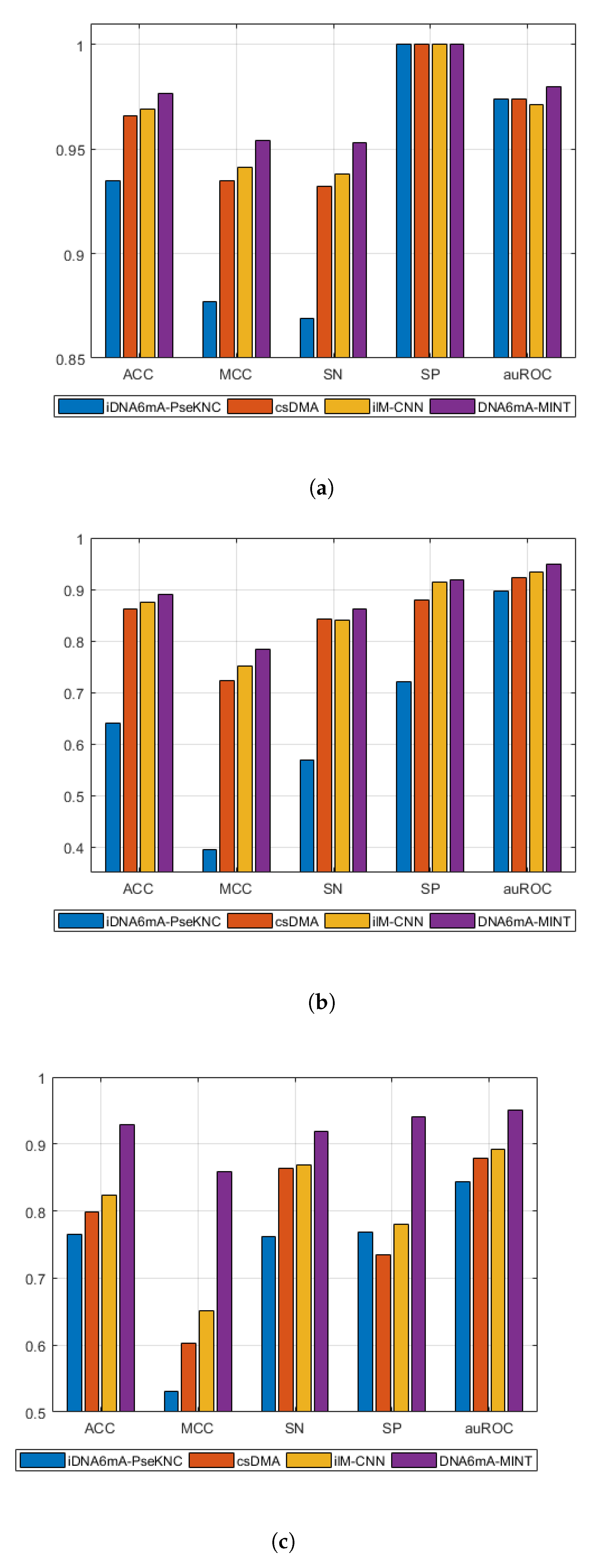
Table 2 shows a comparison of the proposed model with existing techniques, while Figure 2 shows the graphical visualization of performance differences between existing techniques and the proposed technique in this study. In the case of M. musculus genomes, the DNA6mA-MINT achieved high results in all figures of merit when compared with models validated on 5-fold cross-validation. On the other hand, compared on 10-fold cross-validation, the 6mA-Finder exhibits higher auROC then the proposed model. However, in all other figures of merit the proposed model remains higher in performance.
For Rice genomes with 5-fold cross-validation, the DNA6mA-MINT depicts an increase in all figures of merit, while in 10-fold cross-validation, 6mA-Finder has not reported results for all figures of merit, but the reported auROC achieved by 6mA-Finder is lower than that achieved by the proposed model in 10-fold cross-validation.
“Combined-species” is another benchmark dataset for the evaluation of the proposed model. In "combined-species", the proposed model has shown a tremendous increase in performance when compared with existing techniques. In 5-fold cross-validated models, the DNA6mA-MINT increased the sensitivity, specificity, accuracy, MCC, and AuROC by 4.92%, 16.09%, 10.55%, 20.83%, and 5.8%, respectively. For 10-fold cross-validation, the proposed model illustrated an increase of 3.93% in auROC when compared with 6mA-Finder. The sharp increase in MCC depicts the higher quality of the DNA6mA-MINT in comparison to existing state-of-the-art tools.
Figure 3 shows the auROC curves for three species. As can be determined by the curves, the proposed model curves are approaching the ideal scenario. Especially in the case of M. musculus, which is almost near to ideal. Upon evaluation of DNA6mA-MINT on the “combined-species” independent dataset with 10-fold cross-validation, a massive increase of 8.99% is observed in auROC. The 6mA Finder has reported 87.01% auROC while the proposed model has achieved 96% auROC for “combined-species” independent dataset. The high performance shown by the DNA6mA-MINT depicts the reliability of the proposed tool.
For functional genomics, such an architecture should be used which can effectively model the DNA motifs with some insertion/deletion (indels). Keeping it in mind to unfold the quality of DNA6mA-MINT, the silico mutagenesis method is adopted. Nucleotides in the benchmark dataset are computationally mutated. The effect of this mutation in model prediction is studied. One by one the data at position “1-41” is mutated and the corresponding absolute difference is stored. Last, the averaged predicted score for all the mutations over all the sequences in the benchmark dataset is computed to construct the heat map. Figure 4 represents the constructed heat map illustrating the important position of the input sequence. As can be seen, the final prediction is more affected by the mutations occurring at the center of the sequence than the mutations happening on both sides of the sequence.
In order to study the generalization of DNA6mA-MINT we have prepared additional dataset for Rice genome (which is a part of our future work) from the NCBI Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE103145. We have prepared from this repository 10,000 positive sequences and 10,000 negative sequences that are not 6mA. Obtained values for sensitivity, specificity, and accuracy are 84.77, 82.78, and 83.76, respectively. The obtained results show that proposed model generalizes well to the new sequences.
6. Conclusions
DNA modification results in presiding form which is DNA N-methyladenine (6mA). DNA-6mA identification is necessary to explore different biological functions. This study proposed an effective computational tool for the identification of DNA-6mA using a Neural Network framework. The proposed model uses a CNN for feature extraction followed by the LSTM layer, which gives interpretation of the high-dimensional feature vector so that they can be optimally utilized for classification of methylated or non-methylated sites. For comparison purpose results are computed on five and ten folds for three datasets. The proposed model outperformed the results achieved by existing state-of-the-art models in the case of all the datasets. The aim to introduce this model is to utilize it for different research fields working in the development of medicine and bioinformatics. For the said reason, a web server is created which is publicly available at: http://home.jbnu.ac.kr/NSCL/DNA6mA-MINT.htm.
Author Contributions
Conceptualization, M.U.R. and K.T.C.; methodology, M.U.R.; software, M.U.R.; validation, M.U.R. and K.T.C.; investigation, M.U.R. and K.T.C.; writing—original draft preparation: M.U.R.; writing—review and editing, M.U.R. and K.T.C.; supervision, K.T.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A2C2005612) and in part by the Brain Research Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (No. NRF-2017M3C7A1044816).
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizábal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA methylation on N6-adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Huang, H.; Liu, D.; Cheng, Y.; Liu, X.; Zhang, W.; Yin, R.; Zhang, D.; Zhang, P.; Liu, J.; et al. N6-methyladenine DNA modification in Drosophila. Cell 2015, 161, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.Z.; He, C. DNA N6-methyladenine in metazoans: Functional epigenetic mark or bystander? Nat. Struct. Mol. Biol. 2017, 24, 503–506. [Google Scholar] [CrossRef]
- Campbell, J.L.; Kleckner, N. E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell 1990, 62, 967–979. [Google Scholar] [CrossRef]
- Pukkila, P.J.; Peterson, J.; Herman, G.; Modrich, P.; Meselson, M. Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics 1983, 104, 571–582. [Google Scholar] [PubMed]
- Robbins-Manke, J.L.; Zdraveski, Z.Z.; Marinus, M.; Essigmann, J.M. Analysis of global gene expression and double-strand-break formation in DNA adenine methyltransferase-and mismatch repair-deficient Escherichia coli. J. Bacteriol. 2005, 187, 7027–7037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.L.; Zhu, S.; He, M.; Chen, D.; Zhang, Q.; Chen, Y.; Yu, G.; Liu, J.; Xie, S.Q.; Luo, F.; et al. N6-methyladenine DNA modification in the human genome. Mol. Cell 2018, 71, 306–318. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Lai, W.; Li, Y.; Chen, S.; Liu, B.; Zhang, N.; Mo, J.; Lyu, C.; Zheng, J.; Du, Y.R.; et al. N6-methyladenine is incorporated into mammalian genome by DNA polymerase. Cell Res. 2020. [Google Scholar] [CrossRef]
- Dunn, D.; Smith, J. Occurrence of a new base in the deoxyribonucleic acid of a strain of Bacterium coli. Nature 1955, 175, 336–337. [Google Scholar] [CrossRef]
- Bird, A.P. Use of restriction enzymes to study eukaryotic DNA methylation: II. The symmetry of methylated sites supports semi-conservative copying of the methylation pattern. J. Mol. Biol. 1978, 118, 49–60. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomraning, K.R.; Smith, K.M.; Freitag, M. Genome-wide high throughput analysis of DNA methylation in eukaryotes. Methods 2009, 47, 142–150. [Google Scholar] [CrossRef]
- Liu, B.; Liu, X.; Lai, W.; Wang, H. Metabolically generated stable isotope-labeled deoxynucleoside code for tracing DNA N6-Methyladenine in human cells. Anal. Chem. 2017, 89, 6202–6209. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Luo, G.Z.; Chen, K.; Deng, X.; Yu, M.; Han, D.; Hao, Z.; Liu, J.; Lu, X.; Doré, L.C.; et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 2015, 161, 879–892. [Google Scholar] [CrossRef] [Green Version]
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; LaButti, K.; Haridas, S.; Kuo, A.; Salamov, A.; Ahrendt, S.R.; et al. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017, 49, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Wang, C.; Liu, H.; Zhou, Q.; Liu, Q.; Guo, Y.; Peng, T.; Song, J.; Zhang, J.; Chen, L.; et al. Identification and analysis of adenine N6-methylation sites in the rice genome. Nat. Plants 2018, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Yang, H.; Ding, H.; Lin, H.; Chen, W.; Chou, K.C. iDNA6mA-PseKNC: Identifying DNA N6-methyladenosine sites by incorporating nucleotide physicochemical properties into PseKNC. Genomics 2019, 111, 96–102. [Google Scholar] [CrossRef]
- Liu, Z.; Dong, W.; Jiang, W.; He, Z. csDMA: An improved bioinformatics tool for identifying DNA 6mA modifications via Chou’s 5-step rule. Sci. Rep. 2019, 9, 1–9. [Google Scholar]
- Xu, H.; Hu, R.; Jia, P.; Zhao, Z. 6mA-Finder: A novel online tool for predicting DNA N6-methyladenine sites in genomes. Bioinformatics 2020, 36, 3257–3259. [Google Scholar] [CrossRef]
- Chen, W.; Lv, H.; Nie, F.; Lin, H. i6mA-Pred: Identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics 2019, 35, 2796–2800. [Google Scholar] [CrossRef]
- Rahman, M.K. FastFeatGen: Faster Parallel Feature Extraction from Genome Sequences and Efficient Prediction of DNA N6-Methyladenine Sites. In Proceedings of the International Conference on Computational Advances in Bio and Medical Sciences, Miami, FL, USA, 15–17 November 2019; pp. 52–64. [Google Scholar]
- Kong, L.; Zhang, L. i6mA-DNCP: Computational identification of DNA N6-methyladenine sites in the rice genome using optimized dinucleotide-based features. Genes 2019, 10, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, M.U.; Khan, S.H.; Abbas, Z.; Danish Rizvi, S.M. Classification of Diabetic Retinopathy Images Based on Customised CNN Architecture. In Proceedings of the 2019 Amity International Conference on Artificial Intelligence (AICAI), Dubai, UAE, 4–6 February 2019; pp. 244–248. [Google Scholar]
- Rehman, M.U.; Khan, S.H.; Rizvi, S.D.; Abbas, Z.; Zafar, A. Classification of skin lesion by interference of segmentation and convolotion neural network. In Proceedings of the 2018 2nd International Conference on Engineering Innovation (ICEI), Bangkok, Thailand, 5–6 July 2018; pp. 81–85. [Google Scholar]
- Mahmoudi, O.; Wahab, A.; Chong, K.T. iMethyl-Deep: N6 methyladenosine identification of yeast genome with automatic feature extraction technique by using deep learning algorithm. Genes 2020, 11, 529. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.; Mahmoudi, O.; Kim, J.; Chong, K.T. DNC4mC-Deep: Identification and analysis of DNA N4-methylcytosine sites based on different encoding schemes by using deep learning. Cells 2020, 9, 1756. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Wahab, A.; Nazari, I.; Ryu, J.H.; Chong, K.T. i6mA-DNC: Prediction of DNA N6-Methyladenosine sites in rice genome based on dinucleotide representation using deep learning. Chemom. Intell. Lab. Syst. 2020, 204, 104102. [Google Scholar] [CrossRef]
- Wahab, A.; Ali, S.D.; Tayara, H.; Chong, K.T. iIM-CNN: Intelligent identifier of 6mA sites on different species by using convolution neural network. IEEE Access 2019, 7, 178577–178583. [Google Scholar] [CrossRef]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Chou, K.C. Advances in predicting subcellular localization of multi-label proteins and its implication for developing multi-target drugs. Curr. Med. Chem. 2019, 26, 4918–4943. [Google Scholar] [CrossRef]
- Ye, P.; Luan, Y.; Chen, K.; Liu, Y.; Xiao, C.; Xie, Z. MethSMRT: An integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res. 2016, gkw950. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Chollet, F.; Keras Special Interest Group. Keras: Deep Learning Library for Theano and Tensorflow. 2015, Volume 7. p. T1. Available online: https://keras.io/ (accessed on 1 August 2020).
- De Boer, P.T.; Kroese, D.P.; Mannor, S.; Rubinstein, R.Y. A tutorial on the cross-entropy method. Ann. Oper. Res. 2005, 134, 19–67. [Google Scholar] [CrossRef]
- Bottou, L.; Bousquet, O. The tradeoffs of large scale learning. In Advances in Neural Information Processing Systems; Neural Information Processing Systems Foundation (NIPS): Vancouver, BC, Canada, 2008; pp. 161–168. [Google Scholar]
Figure 1.
DNA6mA-MINT architecture for identification of DNA 6mA modification. Acronyms: Convolution 1 Dimension (Conv1D), BatchNormalization (BatchNorm), MaxPool (Max Pooling), Convolution Neural Network (CNN), Conv1d (number of filters, size of the filters, number of strides), MaxPool (pool size, number of strides), Dropout (ratio of features which needs to be discarded), and Long Short-Term Memory (LSTM).
Figure 1.
DNA6mA-MINT architecture for identification of DNA 6mA modification. Acronyms: Convolution 1 Dimension (Conv1D), BatchNormalization (BatchNorm), MaxPool (Max Pooling), Convolution Neural Network (CNN), Conv1d (number of filters, size of the filters, number of strides), MaxPool (pool size, number of strides), Dropout (ratio of features which needs to be discarded), and Long Short-Term Memory (LSTM).

Figure 2.
Graphical comparison of DNA6mA-MINT with state-of-the-art tools using five fold cross validation on different species. (a) Mus musculus, (b) Rice, (c) “Combined-species”. Acronyms are Sensitivity (SN), Specificity (SP), Accuracy (ACC), Matthews Correlation Coefficient (MCC), and area under the Receiver Operating Characteristics (auROC).
Figure 2.
Graphical comparison of DNA6mA-MINT with state-of-the-art tools using five fold cross validation on different species. (a) Mus musculus, (b) Rice, (c) “Combined-species”. Acronyms are Sensitivity (SN), Specificity (SP), Accuracy (ACC), Matthews Correlation Coefficient (MCC), and area under the Receiver Operating Characteristics (auROC).

Figure 3.
AuROC for M. musculus, Rice, and “Combined-species” genomes.

Figure 4.
Heat Map to study the effect of mutation in model prediction.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Architecture details of DNA6mA-MINT.
| Layer | Output Shape | Number of Parameters |
|---|---|---|
| Input | (41,4) | - |
| Conv1D (32,5,1) | (37,32) | 672 |
| Batch Normalization | (37,32) | 128 |
| Max Pooling (4,2) | (17,32) | 0 |
| Dropout (0.4) | (17,32) | 0 |
| Conv1D (32,4,1) | (14,32) | 4128 |
| Batch Normalization | (14,32) | 128 |
| Max Pooling (4,2) | (6,32) | 0 |
| Dropout (0.4) | (6,32) | 0 |
| Conv1D (16,4,1) | (3,16) | 2064 |
| Max Pooling (2,1) | (1,16) | 0 |
| Dropout (0.2) | (1,16) | 0 |
| LSTM | (1,4) | 336 |
| Flatten | 4 | 0 |
| Dense | 32 | 160 |
| Dense | 1 | 33 |
Table 2.
Performance comparison of DNA6mA-MINT with existing techniques on different species with 5- and 10-fold cross-validation.
Table 2.
Performance comparison of DNA6mA-MINT with existing techniques on different species with 5- and 10-fold cross-validation.
| Model | Species | Folds | SN | SP | ACC | MCC | auROC |
|---|---|---|---|---|---|---|---|
| iDNA6mA-PseKNC | M. musculus | 5 | 0.869 | 1 | 0.935 | 0.877 | 0.974 |
| Rice | 5 | 0.569 | 0.721 | 0.641 | 0.394 | 0.896 | |
| Combined-species | 5 | 0.762 | 0.769 | 0.765 | 0.531 | 0.844 | |
| csDMA | M. musculus | 5 | 0.932 | 1 | 0.966 | 0.935 | 0.974 |
| Rice | 5 | 0.842 | 0.880 | 0.861 | 0.723 | 0.923 | |
| Combined-species | 5 | 0.863 | 0.735 | 0.799 | 0.603 | 0.879 | |
| ilM-CNN | M. musculus | 5 | 0.938 | 1 | 0.969 | 0.941 | 0.971 |
| Rice | 5 | 0.841 | 0.914 | 0.875 | 0.752 | 0.934 | |
| Combined-species | 5 | 0.869 | 0.780 | 0.824 | 0.651 | 0.892 | |
| 6mA-Finder | M. musculus | 10 | 0.9349 | 1 | 0.9674 | 0.935 | 0.9954 |
| Rice | 10 | - | - | - | - | 0.9394 | |
| Combined-species | 10 | - | - | - | - | 0.9207 | |
| DNA6mA-MINT | M. musculus | 5 | 0.9531 | 1 | 0.9766 | 0.9543 | 0.980 |
| Rice | 5 | 0.8621 | 0.9195 | 0.8908 | 0.7829 | 0.950 | |
| Combined-species | 5 | 0.9182 | 0.9409 | 0.9295 | 0.8593 | 0.950 | |
| DNA6mA-MINT | M. musculus | 10 | 0.9427 | 1 | 0.9714 | 0.9444 | 0.98 |
| Rice | 10 | 0.9425 | 0.908 | 0.9253 | 0.8511 | 0.950 | |
| Combined-species | 10 | 0.9318 | 0.9321 | 0.932 | 0.8639 | 0.960 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rehman, M.U.; Chong, K.T. DNA6mA-MINT: DNA-6mA Modification Identification Neural Tool. Genes 2020, 11, 898. https://doi.org/10.3390/genes11080898
AMA Style
Rehman MU, Chong KT. DNA6mA-MINT: DNA-6mA Modification Identification Neural Tool. Genes. 2020; 11(8):898. https://doi.org/10.3390/genes11080898
Chicago/Turabian StyleRehman, Mobeen Ur, and Kil To Chong. 2020. "DNA6mA-MINT: DNA-6mA Modification Identification Neural Tool" Genes 11, no. 8: 898. https://doi.org/10.3390/genes11080898
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.