Solid Lipid Nanoparticles for Duodenum Targeted Oral Delivery of Tilmicosin

 ,
,
Abstract
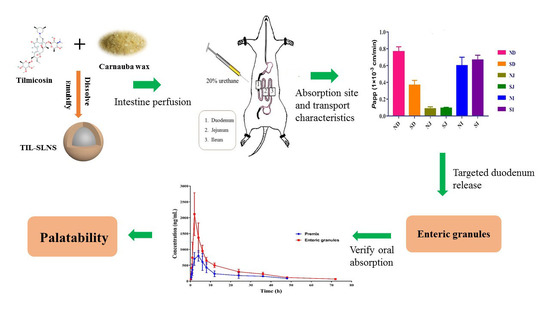
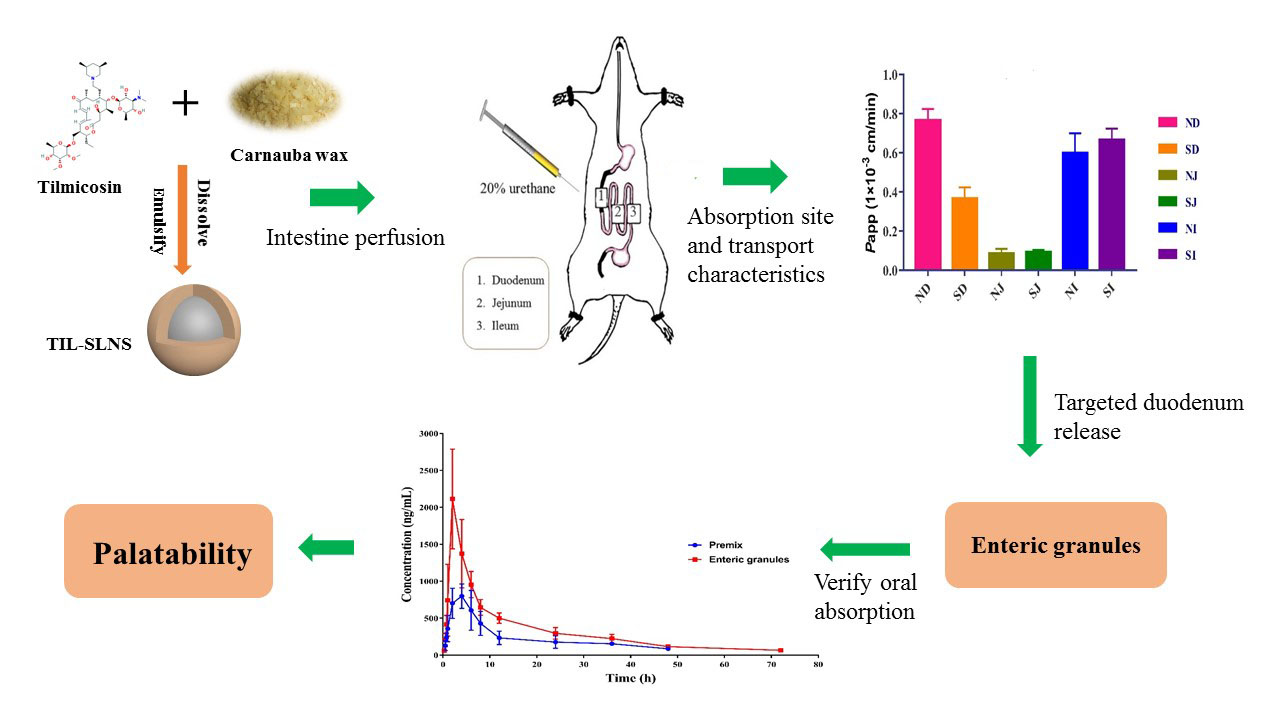
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
2.3. Preparation of TIL-SLNs Suspensions
2.4. Characterization of TIL-SLNs
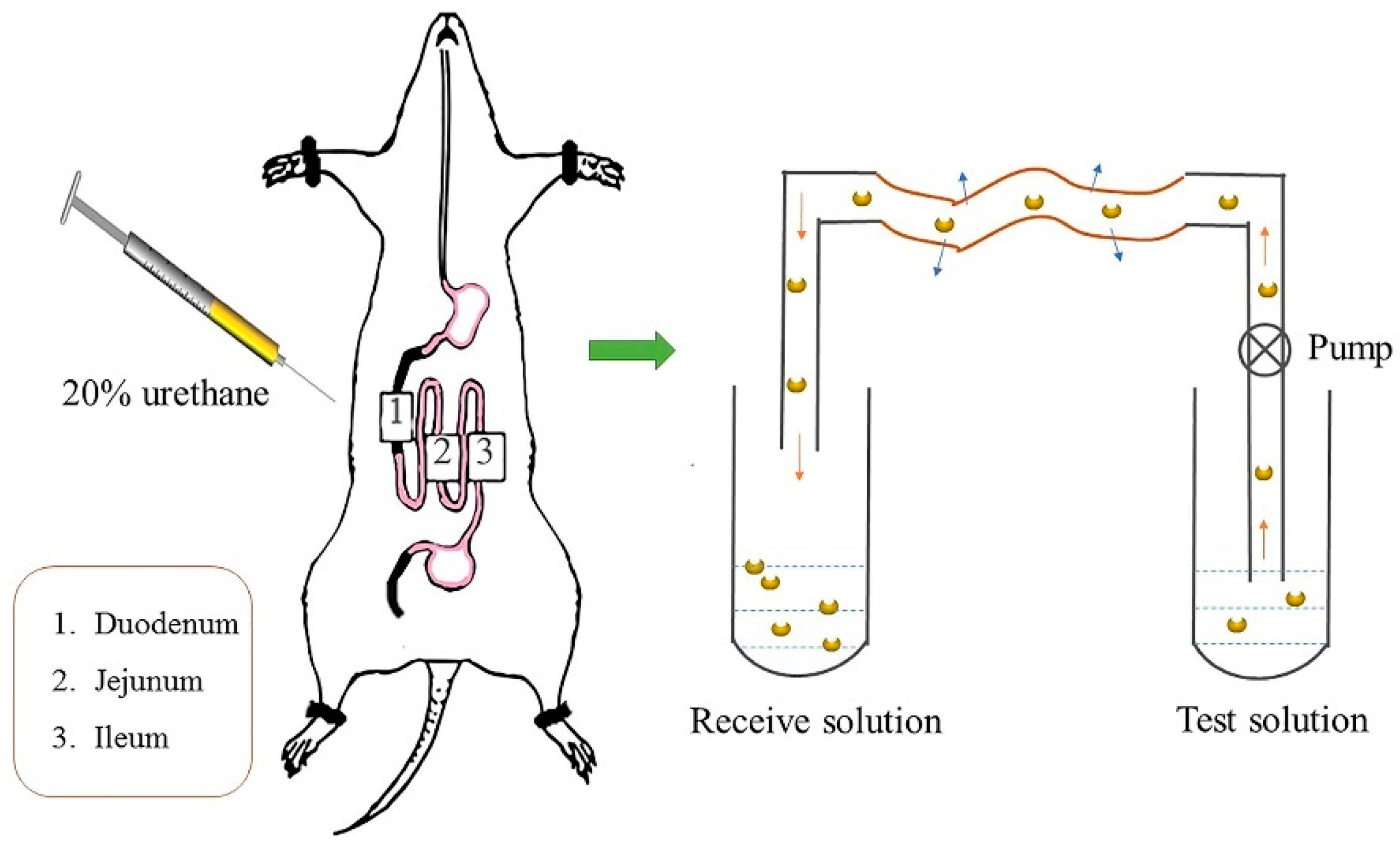
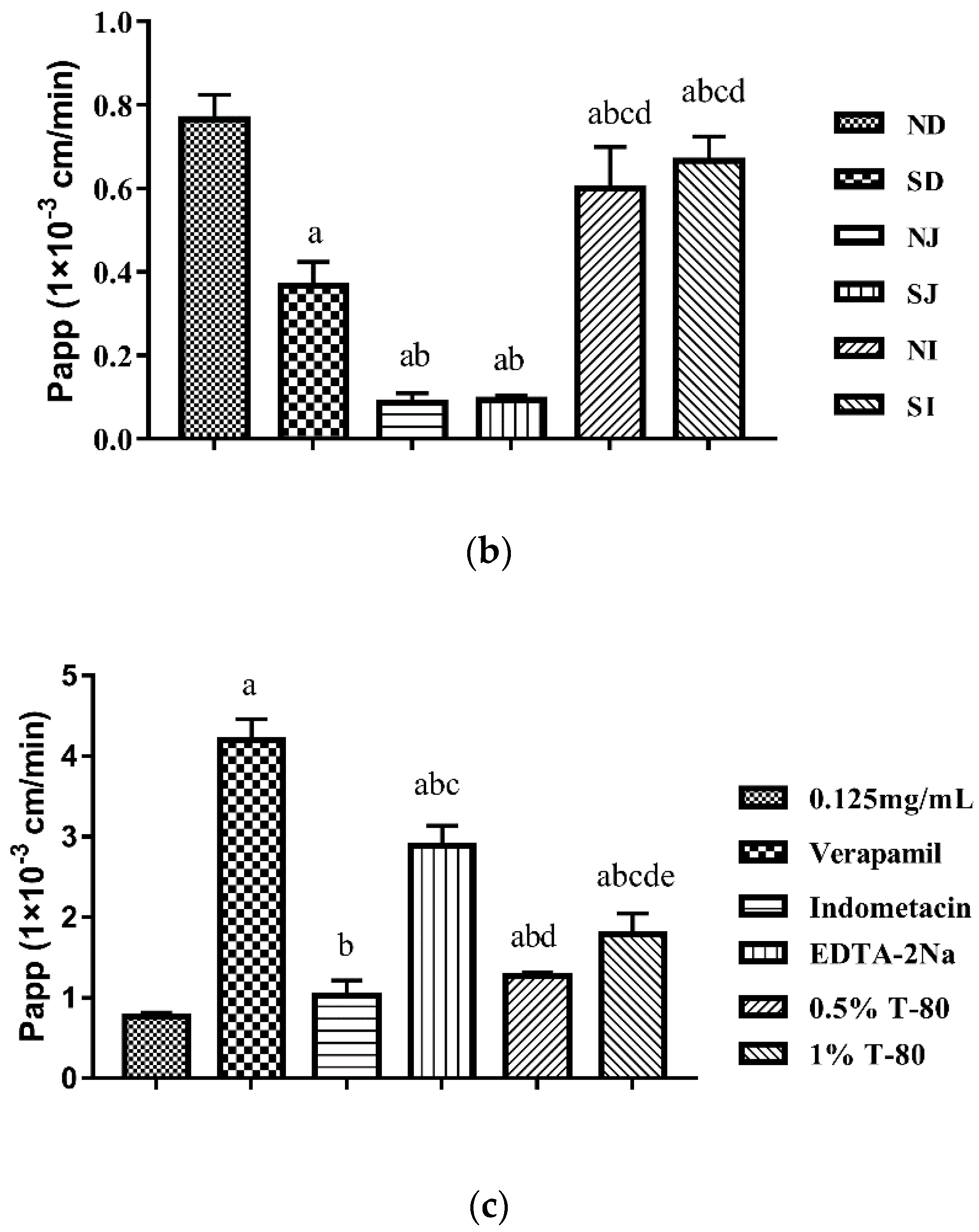
2.5. Single-Pass Intestine Perfusion in Situ
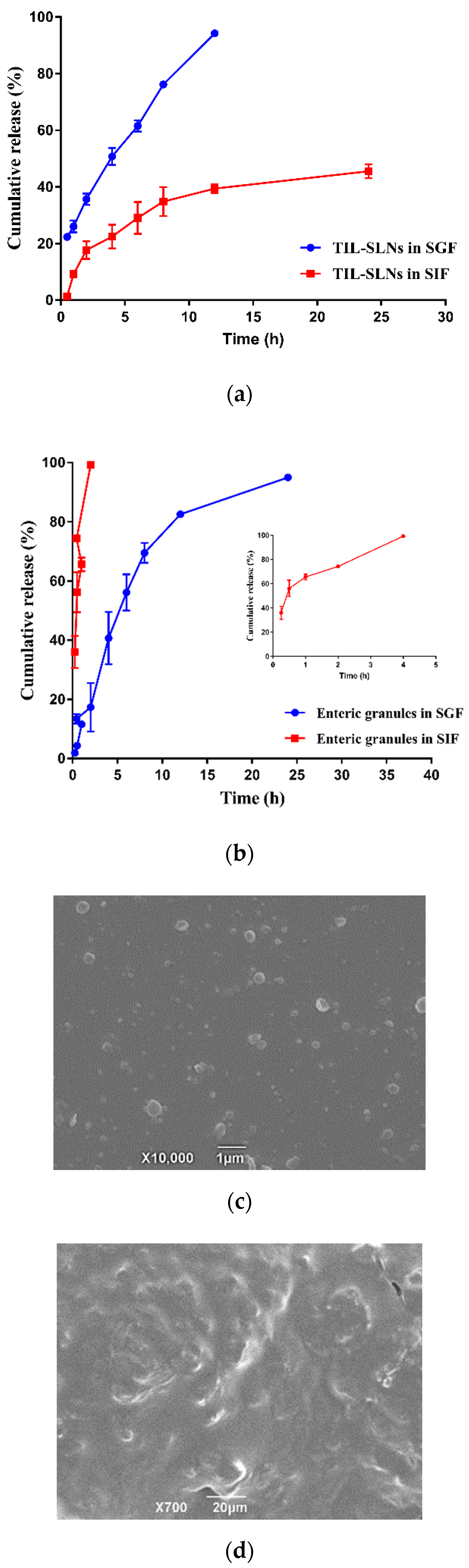
2.6. Preparation and Characteristics of Enteric Granules Used TIL-SLNs as Kernel
2.7. In Vitro Release and Release Mechanism
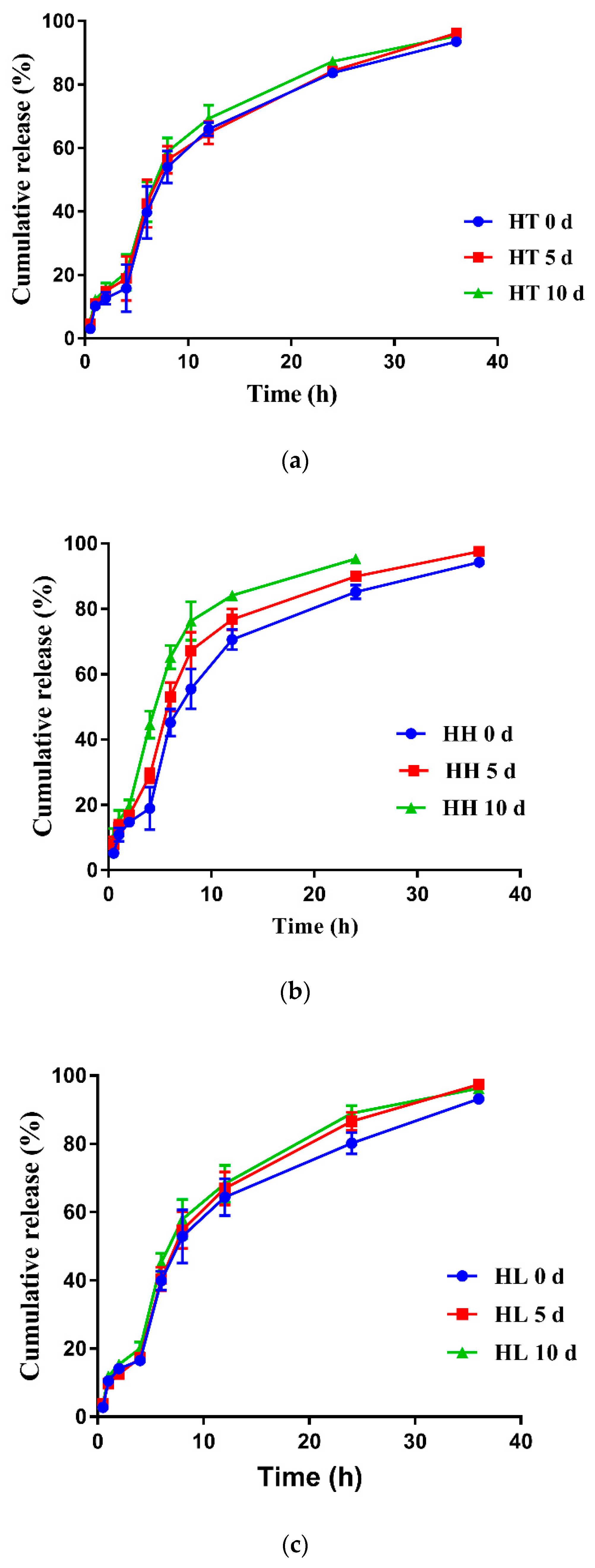
2.8. Stability of Enteric Granules
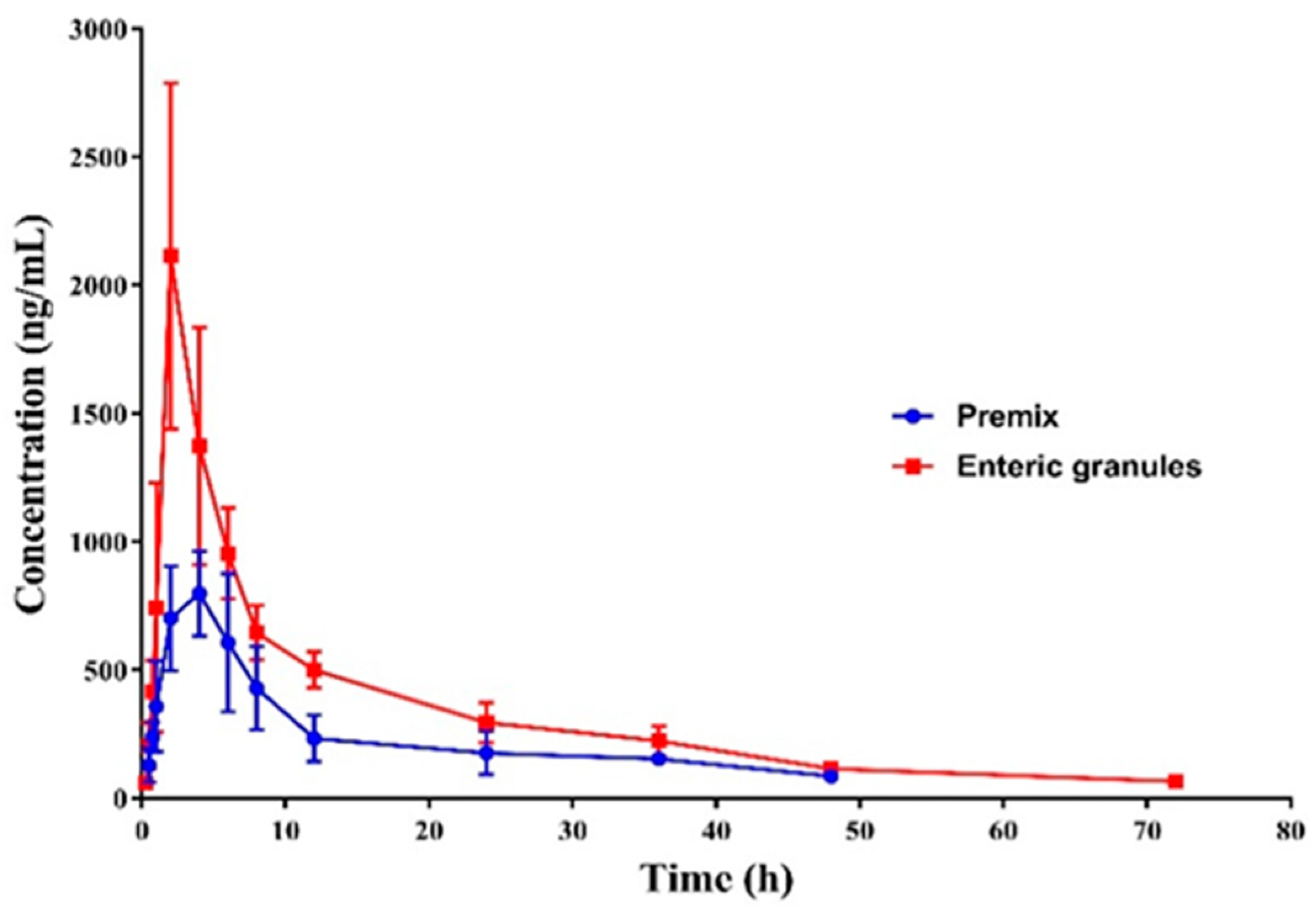
2.9. Pharmacokinetics of Enteric Granules
2.10. Quantitative Measurement of Tilmicosin
2.11. Palatability Experiment
3. Results and Discussions
3.1. Optimization of TIL-SLNs
3.2. Physicochemical Properties of TIL-SLNs
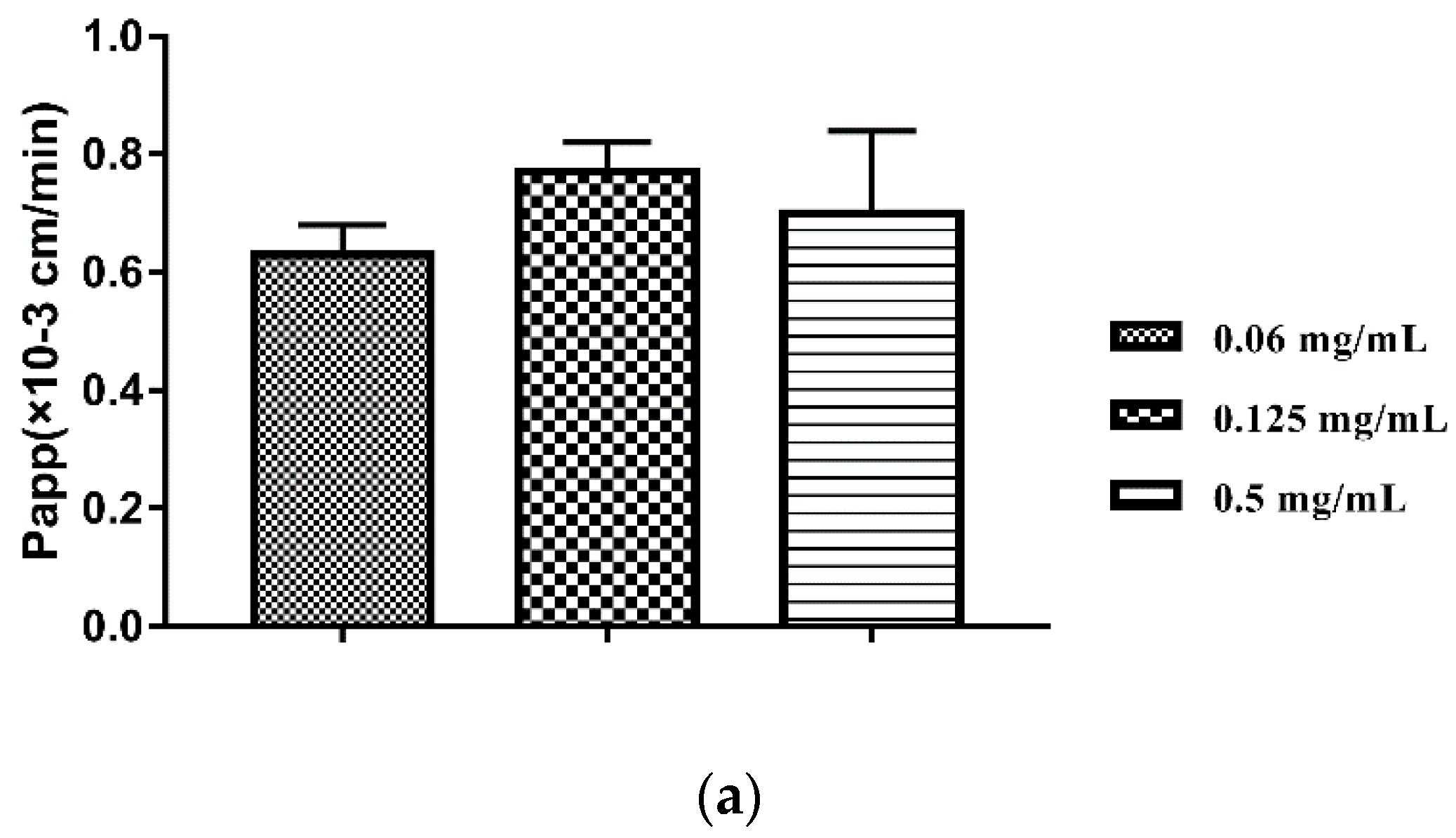
3.3. Absorption Site and Transport Characteristics of TIL-SLNs
3.4. Optimization and Characteristics of Enteric Granules
3.5. In Vitro Release and Release Mechanisms
3.6. Stability
3.7. Pharmacokinetics
3.8. Palatability of Enteric Granules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mole, B. MRSA: Farming up trouble. Nature 2013, 499, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Ferri, M.; Ranucci, E.; Romagnoli, P.; Giaccone, V. Antimicrobial resistance: A global emerging threat to public health systems. Crit. Rev. Food Sci. Nutr. 2017, 57, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.A.; Kullar, R.; Gilchrist, M.; File, T.M., Jr. Antibiotics and adverse events: The role of antimicrobial stewardship programs in doing no harm. Curr. Opin. Infect. Dis. 2019, 32, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; Toit, L.D.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. BioTechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-emulsifying oral lipid drug delivery systems: Advances and challenges. AAPS PharmSciTech 2019, 20, 129. [Google Scholar] [CrossRef]
- Raza, A.; Sime, F.B.; Cabot, P.J.; Maqbool, F.; Maqbool, F.; Roberts, J.A.; Falconer, J.R. Solid nanoparticles for oral antimicrobial drug delivery: A review. Drug Discov. Today 2019, 24, 858–866. [Google Scholar] [CrossRef]
- Sarmah, A.K.; Meyer, M.T.; Boxall, A.B. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 2006, 65, 725–759. [Google Scholar] [CrossRef]
- Xie, S.Y.; Tao, Y.F.; Pan, Y.H.; Qu, W.; Cheng, G.Y.; Huang, L.L.; Yuan, Z.H. Biodegradable nanoparticles for intracellular delivery of antimicrobial agents. J. Control. Release 2014, 187, 101–117. [Google Scholar] [CrossRef]
- Wang, F.; Cao, J.; Hao, J.; Liu, K. Pharmacokinetics, tissue distribution and relative bioavailability of geniposide-solid lipid nanoparticles following oral administration. J. Microencapsul. 2014, 31, 382–389. [Google Scholar] [CrossRef]
- Du, Y.; Ling, L.; Ismail, M.; He, W.; Xia, Q. Redox sensitive lipid-camptothecin conjugate encapsulated solid lipid nanoparticles for oral delivery. Int. J. Pharm. 2018, 549, 352–362. [Google Scholar] [CrossRef]
- Tao, Y.F.; Yang, F.; Meng, K.; Zhou, K.; Luo, W.; Qu, W.; Pan, Y.; Yuan, Z.; Xie, S. Exploitation of enrofloxacin-loaded docosanoic acid solid lipid nanoparticle suspension as oral and intramuscular sustained release formulations for pig. Drug Deliv. 2019, 26, 273–280. [Google Scholar] [CrossRef]
- Patel, M.H.; Mundada, V.P.; Sawant, K.K. Fabrication of solid lipid nanoparticles of lurasidone HCl for oral delivery: Optimization, in vitro characterization, cell line studies and in vivo efficacy in schizophrenia. Drug Dev. Ind. Pharm. 2019, 45, 1242–1257. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Singh, S.K. Biological Voyage of solid lipid nanoparticles: A proficient carrier in nanomedicine. Ther. Deliv. 2016, 7, 691–709. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, X.; Ma, Y.; Zhai, G.; Li, L.; Lou, H. Enhancement of gastrointestinal absorption of quercetin by solid lipid nanoparticles. J. Control. Release 2009, 133, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Ping, Q.; Huang, G.; Han, X.; Cheng, Y.; Xu, W. Transport characteristics of wheat germ agglutinin-modified insulin-liposomes and solid lipid nanoparticles in a perfused rat intestinal model. J. Nanosci. Nanotechnol. 2006, 6, 2959–2966. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ibáñez, G.; del Val Bermejo-Sanz, M.; Rius-Alarcó, F.; Martín-Villodre, A. Experimental studies on the influence of surfactants on intestinal absorption of drugs. Cefadroxil as model drug and sodium taurocholate as natural model surfactant: Studies in rat colon and in rat duodenum. Arzneimittelforschung 1999, 49, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gao, F.; Bu, H.; Xiao, J.; Li, Y. Solid lipid nanoparticles loading candesartan cilexetil enhance oral bioavailability: In vitro characteristics and absorption mechanism in rats. Nanomedicine 2012, 8, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, L.; Wu, L.; Du, Y.; Sun, J.; Wang, Y.; Fu, Q.; Zhang, P.; He, Z. Development of novel self-assembled ES-PLGA hybrid nanoparticles for improving oral absorption of doxorubicin hydrochloride by P-gp inhibition: In vitro and in vivo evaluation. Eur. J. Pharm. Sci. 2017, 99, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Cornaire, G.; Woodley, J.; Hermann, P.; Cloarec, A.; Arellano, C.; Houin, G. Impact of excipients on the absorption of P-glycoprotein substrates in vitro and in vivo. Int. J. Pharm. 2004, 278, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, K.; Chen, D.; Xu, W.; Tao, Y.; Pan, Y.; Meng, K.; Shabbir, M.A.B.; Liu, Q.; Huang, L.; et al. Solid lipid nanoparticles with enteric coating for improving stability, palatability, and oral bioavailability of enrofloxacin. Int. J. Nanomed. 2019, 14, 1619–1631. [Google Scholar] [CrossRef] [Green Version]
- Bargoni, A.; Cavalli, R.; Caputo, O.; Fundarò, A.; Gasco, M.R.; Zara, G.P. Solid lipid nanoparticles in lymph and plasma after duodenal administration to rats. Pharm. Res. 1998, 15, 745–750. [Google Scholar] [CrossRef]
- Cavalli, R.; Bargoni, A.; Podio, V.; Muntoni, E.; Zara, G.P.; Gasco, M.R. Duodenal administration of solid lipid nanoparticles loaded with different percentages of tobramycin. J. Pharm. Sci. 2003, 92, 1085–1094. [Google Scholar] [CrossRef]
- Manjunath, K.; Venkateswarlu, V. Pharmacokinetics, tissue distribution and bioavailability of clozapine solid lipid nanoparticles after intravenous and intraduodenal administration. J. Control. Release 2005, 107, 215–228. [Google Scholar] [CrossRef]
- Jordan, W.H.; Byrd, R.A.; Cochrane, R.L.; Hanasono, G.K.; Hoyt, J.A.; Main, B.W.; Meyerhoff, R.D.; Sarazan, R.D. A review of the toxicology of the antibiotic MICOTIL 300. Vet. Hum. Toxicol. 1993, 35, 151–158. [Google Scholar] [PubMed]
- Ramadan, A. Pharmacokinetics of tilmicosin in serum and milk of goats. Res. Vet. Sci. 1997, 62, 48–50. [Google Scholar] [CrossRef]
- Abdel-Daim, M.M.; Ghazy, E.W.; Fayez, M. Synergistic protective role of mirazid (commiphora molmol) and ascorbic acid against tilmicosin-induced cardiotoxicity in mice. Can. J. Physiol. Pharmaco. 2015, 93, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Gong, S.Y.; Zhou, X.Z.; Yang, Y.J.; Li, J.Y.; Wei, X.J.; Cheng, F.S.; Niu, J.R.; Liu, X.W.; Zhang, J.Y. Determination of antibacterial agent tilmicosin in pig plasma by LC/MS/MS and its application to pharmacokinetics. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef]
- Zhang, P.; Hao, H.; Li, J.; Cheng, G.; Chen, D.; Tao, Y.; Huang, L.; Wang, Y.; Dai, M.; Liu, Z.; et al. The epidemiologic and pharmacodynamic cutoff values of tilmicosin against haemophilus parasui. Front. Microbiol. 2016, 7, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Yun, H. Postantibiotic effects and postantibiotic sub-mic effects of erythromycin, roxithromycin, tilmicosin, and tylosin on pasteurella multocida. Int. J. Antimicrob. Agents 2001, 17, 471–476. [Google Scholar] [CrossRef]
- Zhou, K.; Wang, X.; Chen, D.; Yuan, Y.; Wang, S.; Li, C.; Yan, Y.; Liu, Q.; Shao, L.; Huang, L.; et al. Enhanced treatment effects of tilmicosin against staphylococcus aureus cow mastitis by self-assembly sodium alginate-chitosan nanogel. Pharmaceutics 2019, 11, 524. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhai, B.; Fan, Y.; Wang, J.; Sun, J.; Shi, Y.; Guo, D. Intestinal absorption mechanisms of araloside A in situ single-pass intestinal perfusion and in vitro Caco-2 cell model. Biomed. Pharmacother. 2018, 106, 1563–1569. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, J.; Wu, L.; Wu, H.; Dai, M. Paeonol nanoemulsion for enhanced oral bioavailability: Optimization and mechanism. Nanomedicine 2018, 13, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Dubbelboer, I.R.; Dahlgren, D.; Sjögren, E.; Lennernäs, H. Rat intestinal drug permeability: A status report and summary of repeated determinations. Eur. J. Pharm. Biopharm. 2019, 142, 364–376. [Google Scholar] [CrossRef]
- Dahlgren, D.; Roos, C.; Peters, K.; Lundqvist, A.; Tannergren, C.; Sjögren, E.; Sjöblom, M.; Lennernäs, H. Evaluation of drug permeability calculation based on luminal disappearance and plasma appearance in the rat single-pass intestinal perfusion model. Eur. J. Pharm. Biopharm. 2019, 142, 31–37. [Google Scholar] [CrossRef]
- Zhang, B.; Lu, Y.; Li, P.; Wen, X.; Yang, J. Study on the absorption of corosolic acid in the gastrointestinal tract and its metabolites in rats. Toxicol. Appl. Pharmacol. 2019, 378, 114600. [Google Scholar] [CrossRef]
- He, X.; Song, Z.J.; Jiang, C.P.; Zhang, C.F. Absorption properties of luteolin and apigenin in genkwa flos using in situ single-pass intestinal perfusion system in the rat. Am. J. Chin. Med. 2017, 45, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Medarević, D.P.; Kachrimanis, K.; Mitrić, M.; Djuriš, J.; Djurić, Z.; Ibrić, S. Dissolution rate enhancement and physicochemical characterization of carbamazepine-poloxamer solid dispersions. Pharm. Dev. Technol. 2016, 21, 268–276. [Google Scholar] [CrossRef]
- Hu, X.Y.; Lou, H.; Hageman, M.J. Preparation of lapatinib ditosylate solid dispersions using solvent rotary evaporation and hot melt extrusion for solubility and dissolution enhancement. Int. J. Pharm. 2018, 552, 154–163. [Google Scholar] [CrossRef]
- Ali, S.F.B.; Afrooz, H.; Hampel, R.; Mohamed, E.M.; Bhattacharya, R.; Cook, P.; Khan, M.A.; Rahman, Z. Blend of cellulose ester and enteric polymers for delayed and enteric coating of core tablets of hydrophilic and hydrophobic drugs. Int. J. Pharm. 2019, 567, 118462. [Google Scholar] [CrossRef]
- Horster, L.; Bernhardt, A.; Kiehm, K.; Langer, K. Conversion of PLGA nanoparticle suspensions into solid dosage forms via fluid bed granulation and tableting. Eur. J. Pharm. Biopharm. 2019, 134, 77–87. [Google Scholar] [CrossRef]
- Wang, X.F.; Zhang, S.L.; Zhu, L.Y.; Xie, S.Y.; Dong, Z.; Wang, Y.; Zhou, W.Z. Enhancement of antibacterial activity of tilmicosin against Staphylococcus aureus by solid lipid nanoparticles in vitro and in vivo. Vet. J. 2012, 191, 115–120. [Google Scholar] [CrossRef] [PubMed]
- zur Mühlen, A.; Schwarz, C.; Mehnert, W. Solid lipid nanoparticles (SLN) for controlled drug delivery--drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998, 45, 149–155. [Google Scholar] [CrossRef]
- Müller, R.H.; Mäder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery-a review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Ling, Z.; Yonghong, L.; Junfeng, L.; Li, Z.; Xianqiang, L. Tilmicosin- and florfenicol-loaded hydrogenated castor oil-solid lipid nanoparticles to pigs: Combined antibacterial activities and pharmacokinetics. J. Vet. Pharmacol. Ther. 2018, 41, 307–313. [Google Scholar] [CrossRef]
- Kreizinger, Z.; Grózner, D.; Sulyok, K.M.; Nilsson, K.; Hrivnák, V.; Benčina, D.; Gyuranecz, M. Antibiotic susceptibility profiles of mycoplasma synoviae strains originating from central and eastern Europe. BMC Vet. Res. 2017, 13, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Huang, J.; Liu, Y.; Guo, T.; Wang, L. Using the lentiviral vector system to stably express chicken P-gp and BCRP in MDCK cells for screening the substrates and studying the interplay of both transporters. Arch. Toxicol. 2018, 92, 2027–2042. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, L.; Huang, J.; Sun, Y.; He, F.; Zloh, M.; Wang, L. Inhibitory Effect of Berberine on Broiler P-glycoprotein Expression and Function: In Situ and In Vitro Studies. Int. J. Mol. Sci. 2019, 20, 1966. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Level | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| Type (A) | PVA (Polyvinyl alcohol) | PVP (Poly-vinyl pyrrolidone) | Poloxamer188 |
| Concentration (B) | 1% | 2% | 3% |
| Volume (C) | 15 mL | 20 mL | 25 mL |
| Sample | Type (A) | Concentration (B) | Volume (C) | LC (%) | Size (μm) | PDI |
|---|---|---|---|---|---|---|
| 1 | 2 | 2 | 3 | 16.02 ± 0.85 | 1.34 ± 0.01 | >0.50 |
| 2 | 3 | 1 | 3 | 16.52 ± 0.70 | 1.68 ± 0.01 | >0.50 |
| 3 | 2 | 3 | 1 | 18.99 ± 1.97 | 5.85 ± 0.07 | >0.50 |
| 4 | 1 | 3 | 3 | 20.90 ± 0.65 | 0.34 ± 0.01 | 0.37 ± 0.02 |
| 5 | 3 | 3 | 2 | 17.61 ± 0.95 | 5.35 ± 0.04 | >0.50 |
| 6 | 3 | 2 | 1 | 17.04 ± 1.65 | 2.45 ± 0.01 | >0.50 |
| 7 | 1 | 1 | 1 | 17.19 ± 0.98 | 0.48 ± 0.01 | 0.42 ± 0.03 |
| 8 | 1 | 2 | 2 | 15.47 ± 0.98 | 0.31 ± 0.01 | 0.31 ± 0.02 |
| 9 | 2 | 1 | 2 | 15.91 ± 1.23 | 1.42 ± 0.01 | >0.50 |
| k1 | 17.85 | 16.54 | 17.74 | |||
| k2 | 16.64 | 15.84 | 16.33 | |||
| k3 | 17.05 | 19.16 | 17.81 | |||
| R | 1.21 | 3.32 | 1.48 | |||
| Optimum | A1 | B3 | C3 |
| Time | Factors | Appearance | Content (%) | Unqualified Granularity (%) |
|---|---|---|---|---|
| 0th d | High temperature | Yellowish | 9.95 ± 0.14 | 8.28 ± 0.77 |
| High humidity | Yellowish | 9.95 ± 0.24 | 8.28 ± 0.77 | |
| High light | Yellowish | 9.95 ± 0.15 | 8.28 ± 0.77 | |
| 5th d | High temperature | Yellowish | 9.40 ± 0.14 | 8.02 ± 0.52 |
| High humidity | Yellowish | 9.62 ± 0.21 | 8.45 ± 0.64 | |
| High light | Yellowish | 9.59 ± 0.18 | 8.18 ± 0.89 | |
| 10th d | High temperature | Yellowish | 9.24 ± 0.13 | 7.94 ± 0.43 |
| High humidity | Yellowish | 9.44 ± 0.24 | 8.89 ± 1.01 | |
| High light | Yellowish | 9.36 ± 0.10 | 8.05 ± 0.61 |
| Parameter | Unit | Enteric Granules | Premix |
|---|---|---|---|
| Vz F | mL/kg | 234,913.52 ± 50216.91 | 212,708.7 ± 54,024.98 |
| MRT | h | 13.13 ± 4.78 | 10.73 ± 5.79 |
| AUC0-last | µg·h·mL−1 | 23.17 ± 4.12 | 8.50 ± 3.23 |
| T1/2β | h | 9.10 ± 3.31 | 7.44 ± 4.02 |
| CL | L/h/kg | 8203.84 ± 1374.71 | 23,895.02 ± 8991.91 |
| Tmax | h | 2 ± 0 | 4.0 ± 1.26 |
| Cmax | µg/mL | 2.11 ± 0.62 | 0.92 ± 0.04 |
| F (%) | / | 272.46% | / |
| Groups | Before Experiment (kg/group) | During Experiment (kg/group) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1d | 2d | 3d | x ± SD | 1d | 2d | 3d | 4d | 5d | x ± SD | |
| Control | 1.30 | 1.28 | 1.35 | 1.32 ± 0.02 | 1.35 | 1.32 | 1.40 | 1.38 | 1.43 | 1.38 ± 0.04 |
| Mixed Tilmicosin | 1.24 | 1.22 | 1.28 | 1.25 ± 0.02 | 0.95 | 0.96 | 0.88 | 0.97 | 0.77 | 0.91 ± 0.07a |
| Premix | 1.26 | 1.20 | 1.23 | 1.23 ± 0.02 | 0.74 | 0.80 | 1.05 | 1.38 | 1.46 | 1.09 ± 0.29 |
| Enteric Granules | 1.39 | 1.33 | 1.35 | 1.36 ± 0.02 | 1.10 | 1.25 | 1.32 | 1.59 | 1.58 | 1.37 ± 0.19b |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, K.; Yan, Y.; Chen, D.; Huang, L.; Li, C.; Meng, K.; Wang, S.; Algharib, S.A.; Yuan, Z.; Xie, S. Solid Lipid Nanoparticles for Duodenum Targeted Oral Delivery of Tilmicosin. Pharmaceutics 2020, 12, 731. https://doi.org/10.3390/pharmaceutics12080731
Zhou K, Yan Y, Chen D, Huang L, Li C, Meng K, Wang S, Algharib SA, Yuan Z, Xie S. Solid Lipid Nanoparticles for Duodenum Targeted Oral Delivery of Tilmicosin. Pharmaceutics. 2020; 12(8):731. https://doi.org/10.3390/pharmaceutics12080731
Chicago/Turabian StyleZhou, Kaixiang, Yuanyuan Yan, Dongmei Chen, Lingli Huang, Chao Li, Kuiyu Meng, Shuge Wang, Samah Attia Algharib, Zonghui Yuan, and Shuyu Xie. 2020. "Solid Lipid Nanoparticles for Duodenum Targeted Oral Delivery of Tilmicosin" Pharmaceutics 12, no. 8: 731. https://doi.org/10.3390/pharmaceutics12080731