Characterization of Single Gene Deletion Mutants Affecting Alternative Oxidase Production in Neurospora crassa: Role of the yvh1 Gene

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Growth and Handling of N. crassa
2.2. Measurement of Growth Rates
2.3. Construction of Plasmids Used for Gene Rescue and Isolation of Transformants
2.4. Construction of Plasmids Encoding Mutant Versions of the YVH1 Protein
2.5. Isolation of Mitochondria, Cytosolic, and Post-Mitochondrial Pellet (PMP) Fractions
2.6. Isolation of Nuclei
2.7. Whole Cell Extracts
2.8. Quantitative Real Time Polymerase Chain Reaction (qPCR)
2.9. Spectral Analysis
2.10. General Procedures and Other Techniques
2.11. Antibody to S. cerevisiae RPL3
3. Results
3.1. Attempts to Rescue Single Deletion Mutant Strains by Transformation
3.2. The NCU08158 Gene Encodes a YVH1 Protein
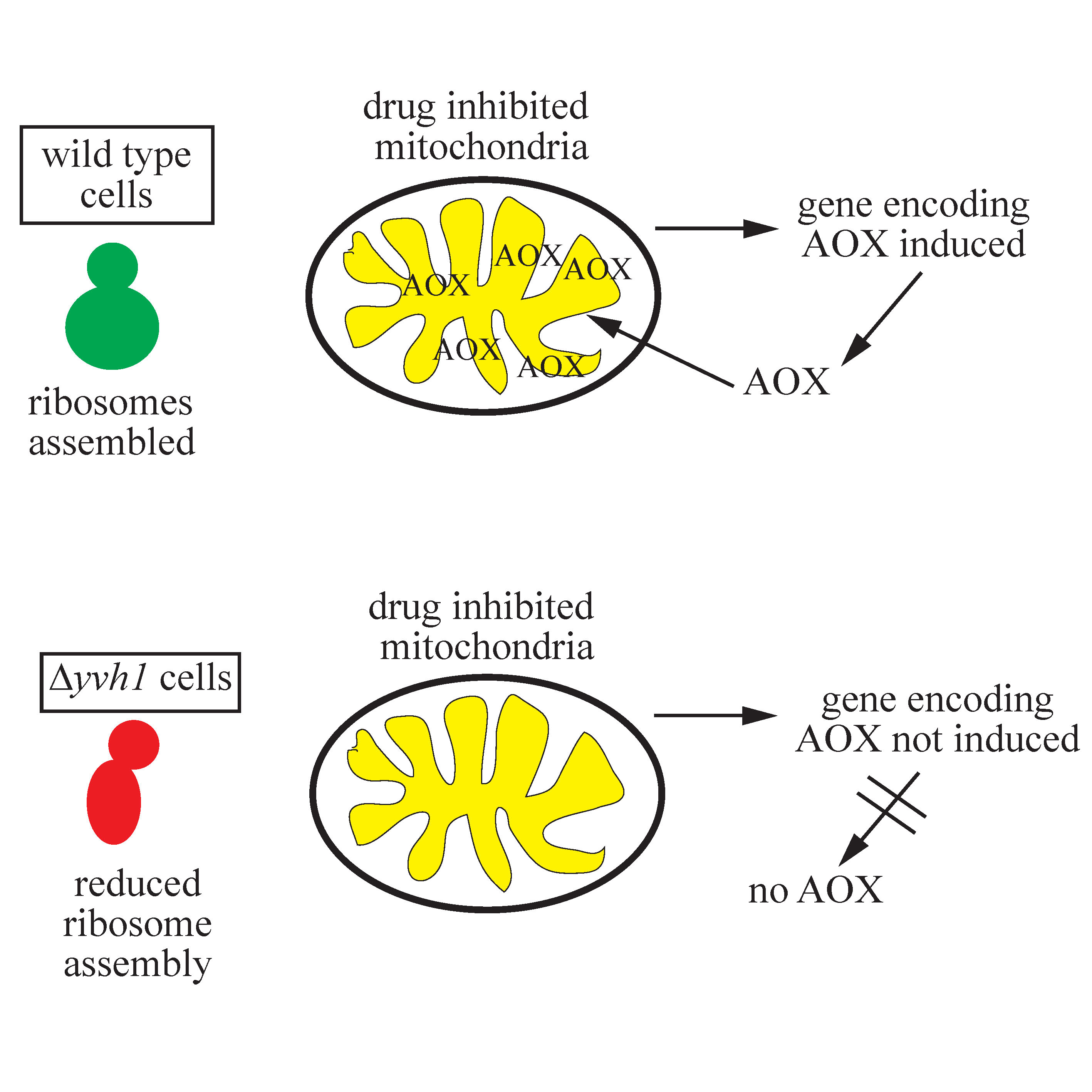
3.3. Temporal Expression of AOD1 in the Δyvh1 Mutant
3.4. Reduced Transcription of aod-1 in the Δyvh1 Mutant
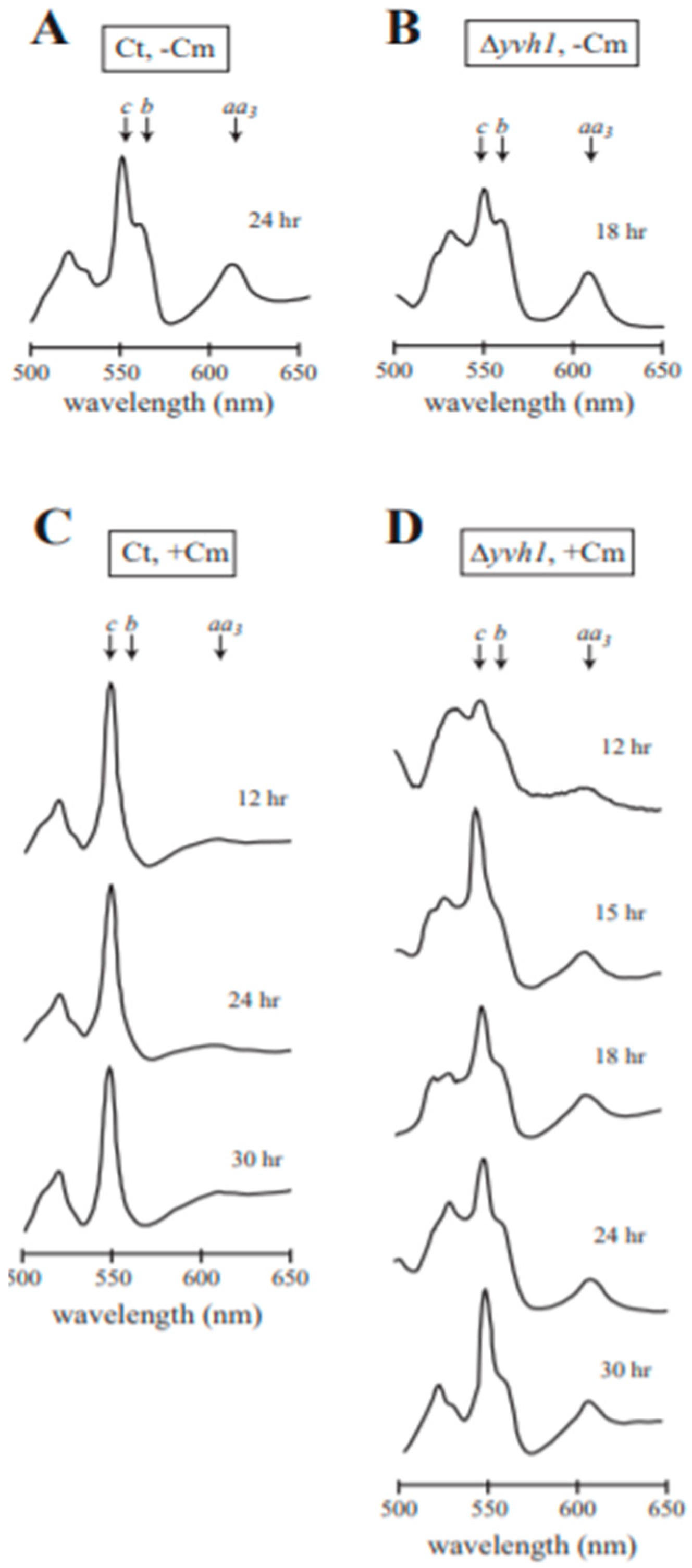
3.5. Analysis of Cytochrome Spectra for Efficiency of Cm Inhibition
3.6. Characterization of Δyvh1 Transformants
3.7. AOX Expression Requires a Functional Zinc Binding Domain but Not the Phosphatase Domain of YVH1
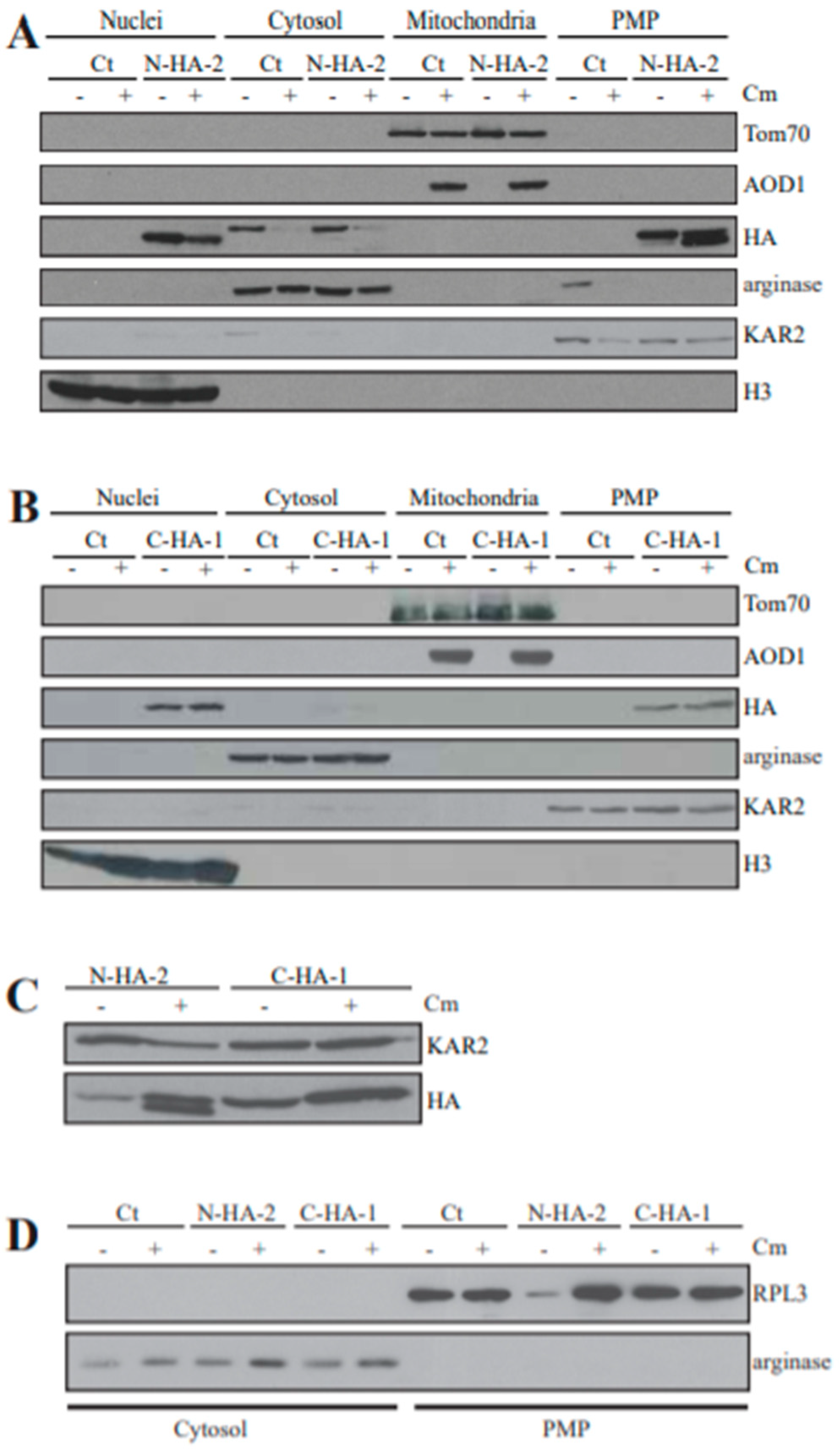
3.8. Localization of Mutant YVH1 Proteins
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, Z.; Butow, R.A. Mitochondrial retrograde regulation. Annu. Rev. Genet. 2006, 40, 159–185. [Google Scholar] [CrossRef] [PubMed]
- Jazwinski, S.M. The retrograde response: When mitochondrial quality control is not enough. Biochim. Biophys. Acta 2013, 1833, 400–409. [Google Scholar] [CrossRef] [Green Version]
- da Cunha, F.M.; Torelli, N.Q.; Kowaltowski, A.J. Mitochondrial retrograde signaling: Triggers, pathways, and outcomes. Oxid. Med. Cell. Longev. 2015, 2015, 482582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardamone, M.D.; Tanasa, B.; Cederquist, C.T.; Huang, J.; Mahdaviani, K.; Li, W.; Rosenfeld, M.G.; Liesa, M.; Perissi, V. Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation. Mol. Cell 2018, 69, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaac, R.S.; McShane, E.; Churchman, L.S. The multiple levels of mitonuclear coregulation. Annu. Rev. Genet. 2018, 52, 511–533. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.L.; Shiba, T.; Young, L.; Harada, S.; Kita, K.; Ito, K. Unraveling the heater: New insights into the structure of the alternative oxidase. Annu. Rev. Plant. Biol. 2013, 64, 637–663. [Google Scholar] [CrossRef] [Green Version]
- Shiba, T.; Kido, Y.; Sakamoto, K.; Inaoka, D.K.; Tsuge, C.; Tatsumi, R.; Takahashi, G.; Balogun, E.O.; Nara, T.; Aoki, T.; et al. Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 4580–4585. [Google Scholar] [CrossRef] [Green Version]
- May, B.; Young, L.; Moore, A.L. Structural insights into the alternative oxidases: Are all oxidases made equal? Biochem. Soc. Trans. 2017, 45, 731–740. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.E.; Vanlerberghe, G.C. Origins, evolutionary history, and taxonomic distribution of alternative oxidase and plastoquinol terminal oxidase. Comp. Biochem. Physiol. Part D 2006, 1, 357–364. [Google Scholar] [CrossRef]
- McDonald, A.E. Alternative oxidase: An inter-kingdom perspective on the function and regulation of this broadly distributed “cyanide-resistant” terminal oxidase. Funct. Plant. Biol. 2008, 35, 535–552. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.E. Alternative oxidase: What information can protein sequence comparisons give us? Physiol. Plant. 2009, 137, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Neimanis, K.; Staples, J.F.; Huner, N.P.; McDonald, A.E. Identification, expression, and taxonomic distribution of alternative oxidases in non-angiosperm plants. Gene 2013, 526, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Joseph-Horne, T.; Holloman, D.W.; Wood, P.M. Fungal respiration: A fusion of standard and alternative components. Biochim. Biophys. Acta 2001, 1504, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Vanlerberghe, G.C.; McIntosh, L. Alternative oxidase: From gene to function. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 1997, 48, 703–734. [Google Scholar] [CrossRef] [PubMed]
- Nargang, F.E.; Kennell, J.C. Mitochondria and respiration. In Cellular and Molecular Biology of Filamentous Fungi; Borkovich, K.A., Ebbole, D.J., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 155–178. [Google Scholar]
- Vanlerberghe, G.C. Alternative oxidase: A mitochondrial respiratory pathway to maintain metabolic and signaling homeostasis during abiotic and biotic stress in plants. Int. J. Mol. Sci. 2013, 14, 6805–6847. [Google Scholar] [CrossRef] [PubMed]
- Vanlerberghe, G.C.; Dahal, K.; Alber, N.A.; Chadee, A. Photosynthesis, respiration and growth: A carbon and energy balancing act for alternative oxidase. Mitochondrion 2020, 52, 197–211. [Google Scholar] [CrossRef]
- Li, Q.; Ritzel, R.G.; McLean, L.T.T.; McIntosh, L.; Ko, T.; Bertrand, H.; Nargang, F.E. Cloning and analysis of the alternative oxidase of Neurospora crassa. Genetics 1996, 142, 129–140. [Google Scholar]
- Tanton, L.L.; Nargang, C.E.; Kessler, K.E.; Li, Q.; Nargang, F.E. Alternative oxidase expression in Neurospora crassa. Fungal Genet. Biol. 2003, 39, 176–190. [Google Scholar] [CrossRef]
- Lambowitz, A.M.; Slayman, C.W. Cyanide-resistant respiration in Neurospora crassa. J. Bacteriol. 1971, 108, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Henry, M.F.; Nyns, E.J. Cyanide-insensitive respiration: An alternative mitochondrial pathway. Subcell. Biochem. 1975, 4, 1–65. [Google Scholar]
- Lambowitz, A.M.; Sabourin, J.R.; Bertand, H.; Nickels, R.; McIntosh, L. Immunological identification of the alternative oxidase of Neurospora crassa mitochondria. Mol. Cell. Biol. 1989, 9, 1362–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, H.; Argan, A.; Szakacs, N.A. Genetic control of the biogenesis of cyanide insensitive respiration in Neurospora crassa. In Mitochondria 1983; Schweyen, R.J., Wolf, K., Kaudewitz, F., Eds.; Walter de Gruyter Co.: Berlin, Germany, 1983; pp. 495–507. [Google Scholar]
- Tissieres, A.; Mitchell, H.K.; Haskins, F.A. Studies on the respiratory system of the poky strain of Neurospora. J. Biol. Chem. 1953, 205, 423–433. [Google Scholar] [PubMed]
- Lambowitz, A.M.; Smith, E.W.; Slayman, C.W. Electron transport in Neurospora mitochondria: Studies on wild type and poky. J. Biol. Chem. 1972, 247, 4850–4858. [Google Scholar] [PubMed]
- Kuntzel, H. Proteins of mitochondrial and cytoplasmic ribosomes from Neurospora crassa. Nature 1969, 222, 142–146. [Google Scholar] [CrossRef]
- McKee, E.E.; Ferguson, M.; Bentley, A.T.; Marks, T.A. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob Agents Chemother 2006, 50, 2042–2049. [Google Scholar] [CrossRef] [Green Version]
- Descheneau, A.T.; Cleary, I.A.; Nargang, F.E. Genetic evidence for a regulatory pathway controlling alternative oxidase production in Neurospora crassa. Genetics 2005, 169, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Nargang, F.E.; Adames, K.; Rub, C.; Cheung, S.; Easton, N.; Nargang, C.E.; Chae, M.S. Identification of genes required for alternative oxidase production in the Neurospora crassa gene knockout library. G3 2012, 2, 1345–1356. [Google Scholar] [CrossRef] [Green Version]
- Chae, M.S.; Nargang, C.E.; Cleary, I.A.; Lin, C.C.; Todd, A.T.; Nargang, F.E. Two zinc cluster transcription factors control induction of alternative oxidase in Neurospora crassa. Genetics 2007, 177, 1997–2006. [Google Scholar] [CrossRef] [Green Version]
- Chae, M.S.; Lin, C.C.; Kessler, K.E.; Nargang, C.E.; Tanton, L.L.; Hahn, L.B.; Nargang, F.E. Identification of an alternative oxidase induction motif in the promoter region of the aod-1 gene in Neurospora crassa. Genetics 2007, 175, 1597–1606. [Google Scholar] [CrossRef] [Green Version]
- Chae, M.S.; Nargang, F.E. Investigation of regulatory factors required for alternative oxidase production in Neurospora crassa. Physiol. Plant. 2009, 137, 407–418. [Google Scholar] [CrossRef]
- Qi, Z.; Smith, K.M.; Bredeweg, E.L.; Bosnjak, N.; Freitag, M.; Nargang, F.E. Alternative oxidase transcription factors AOD2 and AOD5 of Neurospora crassa control the expression of genes involved in energy production and metabolism. G3 2017, 7, 449–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellem, C.H.; Bovier, E.; Lorin, S.; Sainsard-Chanet, A. Mutations in two zinc cluster proteins activate alternative respiratory and gluconeogenic pathways and restore senescence in long-lived respiratory mutants of Podospora Anserina. Genetics 2009, 182, 69–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Murray, S.L.; Wong, K.H.; Davis, M.A.; Hynes, M.J. Reprogramming of carbon metabolism by the transcriptional activators AcuK and AcuM in Aspergillus nidulans. Mol. Microbiol. 2012, 84, 942–964. [Google Scholar] [CrossRef] [PubMed]
- Bovier, E.; Sellem, C.H.; Humbert, A.; Sainsard-Chanet, A. Genetic and functional investigation of Zn2Cys6 transcription factors RSE2 and RSE3 in Podospora anserina. Eukaryot Cell 2014, 13, 53–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosnjak, N.; Smith, K.M.; Asaria, I.; Lahola-Chomiak, A.; Kishore, N.; Todd, A.T.; Freitag, M.; Nargang, F.E. Involvement of a G protein regulatory circuit in alternative oxidase production in Neurospora crassa. G3 2019, 9, 3453–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colot, H.V.; Park, G.; Turner, G.E.; Ringelberg, C.; Crew, C.; Litvinkova, L.; Weiss, R.L.; Borkovich, K.A.; Dunlap, J.C. A high-throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc. Natl. Acad. Sci. USA 2006, 103, 10352–10357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chang, A. A mutant plasma membrane protein is stabilized upon loss of Yvh1, a novel ribosome assembly factor. Genetics 2009, 181, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Lo, K.Y.; Li, Z.; Wang, F.; Marcotte, E.M.; Johnson, A.W. Ribosome stalk assembly requires the dual-specificity phosphatase Yvh1 for the exchange of Mrt4 with P0. J. Cell Biol. 2009, 186, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Kemmler, S.; Occhipinti, L.; Veisu, M.; Panse, V.G. Yvh1 is required for a late maturation step in the 60S biogenesis pathway. J. Cell Biol. 2009, 186, 863–880. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, M.; Nugroho, S.; Iida, N.; Sakai, T.; Kaneko, Y.; Harashima, S. Genetic interactions of ribosome maturation factors Yvh1 and Mrt4 influence mRNA decay, glycogen accumulation, and the expression of early meiotic genes in Saccharomyces cerevisiae. J. Biochem. 2011, 150, 103–111. [Google Scholar] [CrossRef]
- Geng, Q.; Xhabija, B.; Knuckle, C.; Bonham, C.A.; Vacratsis, P.O. The atypical dual specificity phosphatase hYVH1 associates with multiple ribonucleoprotein particles. J. Biol. Chem. 2017, 292, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.H.; De Serres, F.J. Genetic and microbiological research techniques for Neurospora crassa. Method. Enzym. 1970, 17, 79–143. [Google Scholar]
- Metzenberg, R.L. Vogel’s medium N salts: Avoiding the need for ammonium nitrate. Fungal Genet. Newslett. 2003, 50, 14. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laoratory Press: New York, NY, USA, 2001. [Google Scholar]
- Pall, M.L.; Brunelli, J.P. A series of six compact fungal transformation vectors containing polylinkers with multiple unique restriction sites. Fungal Genet. Newslett. 1993, 40, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Hoppins, S.C.; Go, N.E.; Klein, A.; Schmitt, S.; Neupert, W.; Rapaport, D.; Nargang, F.E. Alternative splicing gives rise to different isoforms of the Neurospora crassa Tob55 protein that vary in their ability to insert β-barrel proteins into the outer mitochondrial membrane. Genetics 2007, 177, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Nargang, F.E.; Rapaport, D. Neurospora crassa as a model organism for mitochondrial biogenesis. In Mitochondria. Practical Protocols; Leister, D.L., Herrmann, J., Eds.; Humana Press: Totowa, NJ, USA, 2007; pp. 107–123. [Google Scholar]
- Lambowitz, A.M. Preparation and analysis of mitochondrial ribosomes. Methods Enzymol. 1979, 54, 421–433. [Google Scholar]
- Talbot, K.J.; Russell, P.J. Nuclear buoyant density determination and the purification and characterization of wild-type neurospora nuclei using percoll density gradients. Plant. Physiol. 1982, 70, 704–708. [Google Scholar] [CrossRef]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A common enzyme with uncommon functions. Clin. Exp. Pharm. Physiol. 2012, 39, 674–679. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bertrand, H.; Pittenger, T.H. Cytoplasmic mutants selected from continuously growing cultures of Neurospora crassa. Genetics 1969, 61, 643–659. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Towbin, H.; Staehelin, T.; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 79, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Iyer, P.; Herkal, A.; Abdullah, J.; Stout, A.; Free, S.J. Identification and characterization of genes required for cell-to-cell fusion in Neurospora crassa. Eukaryot Cell 2011, 10, 1100–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnici, J.L.; Fu, C.; Caccamise, L.M.; Arnold, J.W.; Free, S.J. Neurospora crassa female development requires the PACC and other signal transduction pathways, transcription factors, chromatin remodeling, cell-to-cell fusion, and autophagy. PLoS ONE 2014, 9, e110603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, K.; Hakes, D.J.; Wang, Y.; Park, H.D.; Cooper, T.G.; Dixon, J.E. A yeast protein phosphatase related to the vaccinia virus VH1 phosphatase is induced by nitrogen starvation. Proc. Natl. Acad. Sci. USA 1992, 89, 12175–12179. [Google Scholar] [CrossRef] [Green Version]
- Muda, M.; Manning, E.R.; Orth, K.; Dixon, J.E. Identification of the human YVH1 protein-tyrosine phosphatase orthologue reveals a novel zinc binding domain essential for in vivo function. J. Biol. Chem. 1999, 274, 23991–23995. [Google Scholar] [CrossRef] [Green Version]
- Beeser, A.E.; Cooper, T.G. The dual-specificity protein phosphatase Yvh1p regulates sporulation, growth, and glycogen accumulation independently of catalytic activity in Saccharomyces cerevisiae via the cyclic AMP-dependent protein kinase cascade. J. Bacteriol. 2000, 182, 3517–3528. [Google Scholar] [CrossRef] [Green Version]
- Grad, L.; Descheneau, A.; Neupert, W.; Lill, R.; Nargang, F. Inactivation of the Neurospora crassa mitochondrial outer membrane protein TOM70 by repeat-induced point mutation (RIP) causes defects in mitochondrial protein import and morphology. Curr. Genet. 1999, 36, 137–146. [Google Scholar] [CrossRef]
- Attardi, G.; Schatz, G. The biogenesis of mitochondria. Annu. Rev. Cell Biol. 1988, 4, 289–333. [Google Scholar] [CrossRef]
- Korb, H.; Neupert, W. Biogenesis of cytochrome c in mitochondria: Synthesis of apocytochrome c, transfer to mitochondria and conversion to holocytochrome c. Eur. J. Biochem. 1978, 91, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Pfanner, N.; Neupert, W. The mitochondrial protein import apparatus. Annu. Rev. Biochem. 1990, 59, 331–353. [Google Scholar] [CrossRef]
- Bottorff, D.A.; Parmaksizoglu, S.; Lemire, E.G.; Coffin, J.W.; Bertrand, H.; Nargang, F.E. Mutations in the structural gene for cytochrome c result in deficiency of both cytochromes aa3 and c in Neurospora crassa. Curr. Genet. 1994, 26, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Herrmann, J. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkovich, K.A.; Weiss, R.L. Purification and characterization of arginase from Neurospora crassa. J. Biol. Chem. 1987, 262, 7081–7086. [Google Scholar]
- Addison, R. A cell-free translation-translocation system reconstituted with subcellular fractions from the wall-less variant fz; sg; ox-1V of Neurospora crassa. Fungal Genet. Biol. 1998, 24, 345–353. [Google Scholar] [CrossRef]
- Sharda, P.R.; Bonham, C.A.; Mucaki, E.J.; Butt, Z.; Vacratsis, P.O. The dual-specificity phosphatase hYVH1 interacts with Hsp70 and prevents heat-shock-induced cell death. Biochem. J. 2009, 418, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, D.L.; Rosenberg, E.; Maroney, P.A. Induction of cyanide-insensitive respiration in Neurospora crassa. J. Biol. Chem. 1974, 249, 3551–3556. [Google Scholar] [PubMed]
- Kumar, R.; Musiyenko, A.; Cioffi, E.; Oldenburg, A.; Adams, B.; Bitko, V.; Krishna, S.S.; Barik, S. A zinc-binding dual-specificity YVH1 phosphatase in the malaria parasite, Plasmodium falciparum, and its interaction with the nuclear protein, pescadillo. Mol. Biochem. Parasitol. 2004, 133, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Park, H.D.; Beeser, A.E.; Clancy, M.J.; Cooper, T.G. The S. cerevisiae nitrogen starvation-induced Yvh1p and Ptp2p phosphatases play a role in control of sporulation. Yeast 1996, 12, 1135–1151. [Google Scholar] [CrossRef]
- Beeser, A.E.; Cooper, T.G. The dual-specificity protein phosphatase Yvh1p acts upstream of the protein kinase mck1p in promoting spore development in Saccharomyces cerevisiae. J. Bacteriol. 1999, 181, 5219–5224. [Google Scholar] [CrossRef] [Green Version]
- Sakumoto, N.; Mukai, Y.; Uchida, K.; Kouchi, T.; Kuwajima, J.; Nakagawa, Y.; Sugioka, S.; Yamamoto, E.; Furuyama, T.; Mizubuchi, H.; et al. A series of protein phosphatase gene disruptants in Saccharomyces cerevisiae. Yeast 1999, 15, 1669–1679. [Google Scholar] [CrossRef]
- Liu, X.; Qian, B.; Gao, C.; Huang, S.; Cai, Y.; Zhang, H.; Zheng, X.; Wang, P.; Zhang, Z. The putative protein phosphatase MoYvh1 functions upstream of MoPdeH to regulate the development and pathogenicity in Magnaporthe oryzae. Mol. Plant Microbe Interact. 2016, 29, 496–507. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Alonso, M.J.; Guillemain, G.; Kassis, N.; Girard, J.; Burnol, A.F.; Leturque, A. A novel cytosolic dual specificity phosphatase, interacting with glucokinase, increases glucose phosphorylation rate. J. Biol. Chem. 2000, 275, 32406–32412. [Google Scholar] [CrossRef] [Green Version]
- Hirai, M.; Yoshida, S.; Kashiwagi, H.; Kawamura, T.; Ishikawa, T.; Kaneko, M.; Ohkawa, H.; Nakagawara, A.; Miwa, M.; Uchida, K. 1q23 gain is associated with progressive neuroblastoma resistant to aggressive treatment. Genes Chromosomes Cancer 1999, 25, 261–269. [Google Scholar] [CrossRef]
- Gratias, S.; Schuler, A.; Hitpass, L.K.; Stephan, H.; Rieder, H.; Schneider, S.; Horsthemke, B.; Lohmann, D.R. Genomic gains on chromosome 1q in retinoblastoma: Consequences on gene expression and association with clinical manifestation. Int. J. Cancer 2005, 116, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Mendrzyk, F.; Korshunov, A.; Benner, A.; Toedt, G.; Pfister, S.; Radlwimmer, B.; Lichter, P. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin. Cancer Res. 2006, 12, 2070–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biernacki, M.A.; Marina, O.; Zhang, W.; Liu, F.; Bruns, I.; Cai, A.; Neuberg, D.; Canning, C.M.; Alyea, E.P.; Soiffer, R.J.; et al. Efficacious immune therapy in chronic myelogenous leukemia (CML) recognizes antigens that are expressed on CML progenitor cells. Cancer Res. 2010, 70, 906–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen le, B.; Diskin, S.J.; Capasso, M.; Wang, K.; Diamond, M.A.; Glessner, J.; Kim, C.; Attiyeh, E.F.; Mosse, Y.P.; Cole, K.; et al. Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility loci. PLoS Genet. 2011, 7, e1002026. [Google Scholar] [CrossRef] [PubMed]
- Cain, E.L.; Braun, S.E.; Beeser, A. Characterization of a human cell line stably over-expressing the candidate oncogene, dual specificity phosphatase 12. PLoS ONE 2011, 6, e18677. [Google Scholar] [CrossRef] [PubMed]
- Kozarova, A.; Hudson, J.W.; Vacratsis, P.O. The dual-specificity phosphatase hYVH1 (DUSP12) is a novel modulator of cellular DNA content. Cell Cycle 2011, 10, 1669–1678. [Google Scholar] [CrossRef] [Green Version]
- Bonham, C.A.; Vacratsis, P.O. Redox regulation of the human dual specificity phosphatase YVH1 through disulfide bond formation. J. Biol. Chem. 2009, 284, 22853–22864. [Google Scholar] [CrossRef] [Green Version]
- Sakumoto, N.; Yamashita, H.; Mukai, Y.; Kaneko, Y.; Harashima, S. Dual-specificity protein phosphatase Yvh1p, which is required for vegetative growth and sporulation, interacts with yeast pescadillo homolog in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2001, 289, 608–615. [Google Scholar] [CrossRef]
- Oeffinger, M.; Leung, A.; Lamond, A.; Tollervey, D. Yeast pescadillo is required for multiple activities during 60S ribosomal subunit synthesis. RNA 2002, 8, 626–636. [Google Scholar] [CrossRef] [Green Version]
- Tarassov, K.; Messier, V.; Landry, C.R.; Radinovic, S.; Serna Molina, M.M.; Shames, I.; Malitskaya, Y.; Vogel, J.; Bussey, H.; Michnick, S.W. An in vivo map of the yeast protein interactome. Science 2008, 320, 1465–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, A.; Pech, M.; Thoms, M.; Beckmann, R.; Hurt, E. Ribosome-stalk biogenesis is coupled with recruitment of nuclear-export factor to the nascent 60S subunit. Nat. Struct. Mol. Biol. 2016, 23, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Musalgaonkar, S.; Johnson, A.W.; Taylor, D.W. Tightly-orchestrated rearrangements govern catalytic center assembly of the ribosome. Nat. Commun. 2019, 10, 958. [Google Scholar] [CrossRef] [Green Version]
- Klingauf-Nerurkar, P.; Gillet, L.C.; Portugal-Calisto, D.; Oborska-Oplova, M.; Jager, M.; Schubert, O.T.; Pisano, A.; Pena, C.; Rao, S.; Altvater, M.; et al. The GTPase Nog1 co-ordinates the assembly, maturation and quality control of distant ribosomal functional centers. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Comments | Origin |
|---|---|---|
| NCN251 | Mating type A. Also called 74-OR23-1VA. | FGSC #2489 |
| 76-26 | Mating type a. his-3, mtr. | R.L. Metzenberg |
| Δaod-1 (also known as 96H9) | Mating type A. aod-1 gene replaced with a hygromycin resistance cassette. | N. crassa single gene deletion library. FGSC #18947 |
| Δyvh1 (also known as 100B5) | Mating type A. yvh1 gene replaced with a hygromycin resistance cassette. | N. crassa single gene deletion library. FGSC #19644 |
| N-HA-2 | Strain Δyvh1 with an ectopic copy of the genomic yvh1 gene that also encodes an N-terminal 3xHA tag. Basta and hygromycin resistant. | This study |
| C-HA-1 | Strain Δyvh1 with an ectopic copy of the genomic yvh1 gene that also encodes a C-terminal 3xHA tag. Basta and hygromycin resistant. | This study |
| P-mutants 8, 9, 10, 11, and 12. | YVH1 phosphatase domain mutants. Obtained via transformation of Δyvh1 with an N-terminal 3xHA tagged version of yvh1 that contained codons for amino acids H149, C150, and R156 in the HC(X)5R dual specificity phosphatase domain mutated to QS(X)5Q. Created by transformation of Δyvh1 with plasmid that also carried resistance to basta. | This study |
| Z mutants 4, 6, 9, 11, and 18. | YVH1 zinc binding domain mutants. Obtained via transformation of Δyvh1 with an N-terminal 3xHA tagged version of yvh1 that contained codons for amino acids cysteine 340 and 345 of the zinc binding domain mutated to serine. Created by transformation of Δyvh1 with plasmid that also carried resistance to basta. | This study |
| 23H2 | NCU08887 replaced by HygR cassette, mating type A. | N. crassa single gene deletion library. FGSC# 15957 |
| 40 E6 | NCU05600 replaced by HygR cassette, mating type a. | N. crassa single gene deletion library. FGSC# 13805 |
| 41G7 | NCU03589 replaced by HygR cassette, mating type A. | N. crassa single gene deletion library. FGSC# 13924 |
| 47H10 | NCU00778 replaced by HygR cassette, mating type a. | N. crassa single gene deletion library. FGSC# 16938 |
| 52D8 | NCU07281 replaced by HygR cassette, mating type A. | N. crassa single gene deletion library. FGSC# 14469 |
| 83H3 | NCU08365 replaced by HygR cassette, mating type a. | N. crassa single gene deletion library. FGSC# 18277 |
| 88H8 | NCU01542 replaced by HygR cassette, mating type a. | N. crassa single gene deletion library. FGSC# 19221 |
| 113G8 | NCU03071 replaced by HygR cassette, mating type a. | N. crassa single gene deletion library. FGSC# 18202 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desaulniers, A.B.; Kishore, N.; Adames, K.; Nargang, F.E. Characterization of Single Gene Deletion Mutants Affecting Alternative Oxidase Production in Neurospora crassa: Role of the yvh1 Gene. Microorganisms 2020, 8, 1186. https://doi.org/10.3390/microorganisms8081186
Desaulniers AB, Kishore N, Adames K, Nargang FE. Characterization of Single Gene Deletion Mutants Affecting Alternative Oxidase Production in Neurospora crassa: Role of the yvh1 Gene. Microorganisms. 2020; 8(8):1186. https://doi.org/10.3390/microorganisms8081186
Chicago/Turabian StyleDesaulniers, Adrien Beau, Nishka Kishore, Kelly Adames, and Frank E. Nargang. 2020. "Characterization of Single Gene Deletion Mutants Affecting Alternative Oxidase Production in Neurospora crassa: Role of the yvh1 Gene" Microorganisms 8, no. 8: 1186. https://doi.org/10.3390/microorganisms8081186