Post-Translational Regulation of ARF: Perspective in Cancer

1
Environmental Disease Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Daejeon 34141, Korea
2
Department of Biochemistry, College of Life science and Biotechnology, Yonsei University, Seoul 03722, Korea
3
Department of Biosystems and Bioengineering, KRIBB School of Biotechnology, University of Science and Technology (UST), Daejeon 34113, Korea
*
Author to whom correspondence should be addressed.
†
Contributed equally to this manuscript.
Biomolecules 2020, 10(8), 1143; https://doi.org/10.3390/biom10081143
Submission received: 25 June 2020
/
Revised: 25 July 2020
/
Accepted: 29 July 2020
/
Published: 4 August 2020
(This article belongs to the Special Issue Deciphering alternative functions of the INK4a/ARF locus)
Abstract
:Tumorigenesis can be induced by various stresses that cause aberrant DNA mutations and unhindered cell proliferation. Under such conditions, normal cells autonomously induce defense mechanisms, thereby stimulating tumor suppressor activation. ARF, encoded by the CDKN2a locus, is one of the most frequently mutated or deleted tumor suppressors in human cancer. The safeguard roles of ARF in tumorigenesis are mainly mediated via the MDM2-p53 axis, which plays a prominent role in tumor suppression. Under normal conditions, low p53 expression is stringently regulated by its target gene, MDM2 E3 ligase, which induces p53 degradation in a ubiquitin-proteasome-dependent manner. Oncogenic signals induced by MYC, RAS, and E2Fs trap MDM2 in the inhibited state by inducing ARF expression as a safeguard measure, thereby activating the tumor-suppressive function of p53. In addition to the MDM2-p53 axis, ARF can also interact with diverse proteins and regulate various cellular functions, such as cellular senescence, apoptosis, and anoikis, in a p53-independent manner. As the evidence indicating ARF as a key tumor suppressor has been accumulated, there is growing evidence that ARF is sophisticatedly fine-tuned by the diverse factors through transcriptional and post-translational regulatory mechanisms. In this review, we mainly focused on how cancer cells employ transcriptional and post-translational regulatory mechanisms to manipulate ARF activities to circumvent the tumor-suppressive function of ARF. We further discussed the clinical implications of ARF in human cancer.
1. Introduction
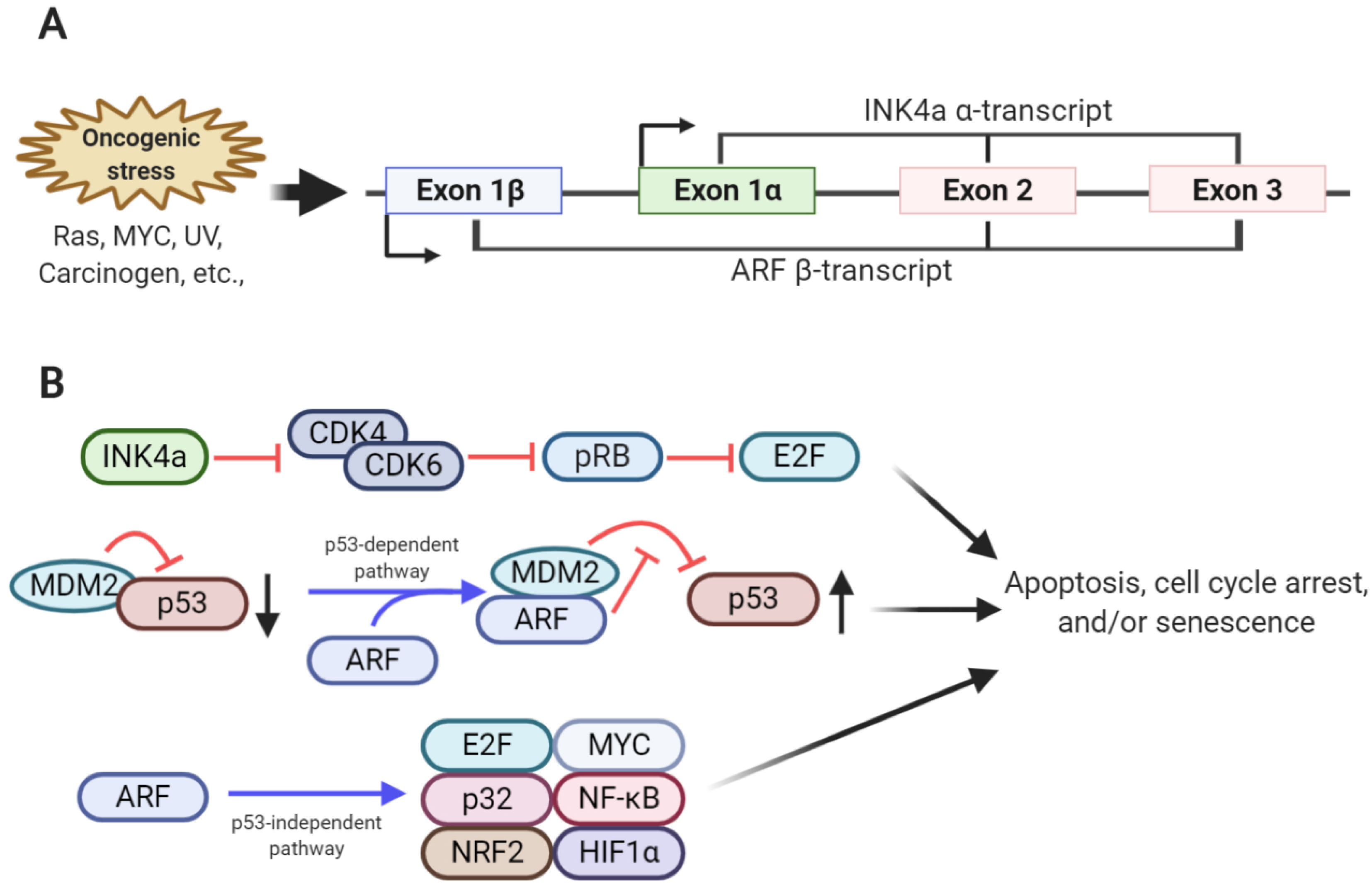
The cyclin-dependent kinase inhibitor 2A (CDKN2a) locus—frequently mutated or deleted in human cancer—encodes two different tumor suppressors, INK4a (referred to as p16INK4a) and ARF (referred to as p14ARF in humans and p19ARF in mice) [1,2]. These two proteins are translated from two different transcripts, α-transcript and β-transcript, respectively, which share sequences in exon 2 and 3. Although the INK4a and ARF transcripts share these sequences, both tumor suppressors display different amino acid sequences with distinctive functions due to the alternative reading frame and different transcription induction sites [3,4] (Figure 1).
Normal cells usually protect themselves from oncogenic signals, such as those induced by RAS and MYC signaling, by expressing tumor suppressors, such as INK4a and ARF, which prevent abnormal cellular growth that leads to tumor formation [5,6]. INK4a inhibits CDK4 and CDK6 activities required for G1 to S progression in the cell cycle by directly interacting with CDK4 and CDK6. This, in turn, interrupts the assembly of D-type cyclin and the CDK4/6 complex or inhibits the kinase activity of the cyclin D-CDK4/6 complex [7,8]. Due to the inhibition of CDK4/6, which phosphorylate RB, the hypo-phosphorylated form of RB predominates and interferes with the transcriptional activities of the E2F transcription factor 2 (E2F2) family members via direct binding, ultimately leading to cell cycle arrest [7] (Figure 1).
The major function of ARF is to stabilize and activate p53, resulting in cellular senescence or apoptosis. Mechanistically, ARF sequesters MDM2 (HDM2 in humans) via direct interaction, thereby blocking direct physical interaction of MDM2 with p53 in the cytosol and nucleus. As MDM2 is the E3 ligase for p53, ARF-induced MDM2 sequestration and inhibition prevents ubiquitination and proteasome-dependent degradation of p53, resulting in p53 stabilization [9,10]. p53 accumulation induces the formation of p53 tetrameric complexes in the nucleus, thereby activating the expression of genes related to cell cycle arrest or apoptosis (Figure 1). ARF involvement in MDM2 sequestration and the p53 axis has been considered a major part of the tumor-suppressive function of ARF; however, p53-independent roles of ARF are also emerging. The interactions of ARF with various proteins associated with cell proliferation and protection from oncogenic signals have been identified. For example, ARF inhibits E2F transcriptional activity by binding to E2F, thereby inducing cell cycle arrest [11,12,13,14]. The mitochondrial protein p32 enables ARF translocation to the mitochondria by interacting with the C-terminus of ARF, thereby promoting apoptosis [15]. Moreover, in response to oncogenic signaling, ARF interacts with various transcription factors, including MYC, nuclear factor-κB, and hypoxia-inducible factor 1-alpha, involved in various signaling pathways, thereby regulating cellular proliferation [16,17,18,19]. Recently, ARF has been shown to inhibit the transcriptional activity of nuclear factor E2-related factor 2, which protects cells from oxidative stress by activating the antioxidant program by binding to ARF, resulting in increased oxidative stress-induced ferroptosis [20]. Additionally, small mitochondrial ARF, p15smArf, has been identified as one of the ARF variants that is predominantly expressed in the mitochondria [21]. Although p15smArf, an N-terminal truncated form of full-length ARF, cannot activate p53, it could correct abnormal focal phenotype and spermatogenesis defects in ARF-null mice [22]. Structurally, ARF is composed of a high portion of basic and hydrophobic amino acids, allowing the aggregation of its recombinant form. This hampers the efforts made by many researchers to reveal its tertiary structure, further making its investigation difficult [23,24,25].
However, based on these observations, it was deduced that ARF formed a complex with numerous proteins to neutralize its charge and stabilize its spatial conformation. The importance of ARF in protection from aberrant tumor cell development has been further understood due to recent knowledge regarding the transcriptional and post-translational regulation of ARF by the newly revealed ARF regulators. This review focused on the newly extended regulatory network of ARF and its therapeutic implications in cancer.
2. Transcriptional Regulation of ARF
The paradoxical nature of oncogene-induced ARF expression has drawn attention to the search for ARF transcriptional factors (Table 1). While low ARF expression is maintained under normal conditions, significantly increased ARF expression is induced by oncogenic signals or DNA damage under stress conditions, subsequently prompting the activation of the fail-safe program. Fine-tuning of ARF expression by various transcription factors indicates that the tumor-suppressive activity of ARF can be manipulated under various stresses, depending on the nature of the stress signals (Figure 2). Here, we described several key transcription factors in the regulation of ARF mRNA expression.
2.1. Activators of ARF Transcription
MYC is an oncoprotein that regulates cell proliferation, differentiation, inflammation, and metabolism by regulating the transcription of various target genes [43]. To avoid unwanted effects resulting from its deregulation, MYC also induces ARF transcription, in turn initiating oncogene-induced senescence (OIS) [6]. OIS mechanisms can be either p53-dependent or -independent. As previously explained, ARF stabilizes p53 by trapping MDM2, inducing senescence or apoptosis [9,10]. Further, MYC expression also increased ARF mRNA in p53-null and wild type mouse embryonic fibroblasts (MEFs), suggesting that ARF might have p53-independent roles. Indeed, Arf-null cells showed resistance to MYC-induced apoptosis, indicating that ARF induction by MYC expression could be an important checkpoint in preventing aberrant cell proliferation through the activation of the fail-safe program [6]. As E2F is regulated by MYC, it has been identified as a regulator of MYC-mediated ARF expression [44,45]. The E2F family consists of five distinct E2F members. In particular, E2F transcription factor 1 (E2F1) and E2F2 activate ARF transcription. The conserved sequences of E2F1 and E2F2 are located in the ARF promoter, and activate ARF transcription in an RB-independent manner [44]. The E2F1-binding site on the ARF promoter (-231 to -205) is known as the E2F-responsive element [46]. On the contrary, another E2F member, E2F3b, functions as a negative regulator of ARF expression [47]. E2F3b deletion blocks mitogen-induced cell cycle entry and suppresses E2F-responsive gene expression, thereby inhibiting cell proliferation. The ARF promoter contains the E2F3b-specific binding site, which in response to E2F3b binding represses (rather than activating) ARF transcription. ARF stabilization, along with suppression of E2F-responsive genes, occurs in E2F3b-deficient cells, leading to p53 activation and triggering of G0/G1 cell cycle arrest [45].
DMP1, also referred to as cyclin D-binding Myb-like protein, can also induce cell growth arrest by inducing ARF expression [31,48]. Three DMP1 isoforms, DMP1α, β, and γ, are produced via alternative splicing, and they show differential functions in cell cycle progression [49]. DMP1α can bind to the consensus sequence of the ARF promoter (−189 to −181 in the transcription start site), thereby increasing ARF transcription [31]. DMP1α arrests the cell cycle in G0/G1 by increasing ARF expression. Dmp1-null MEFs show a delay in ARF accumulation when cells are sub-cultured and grow faster than wild type MEFs. H-RasV12 expression results in tumorigenic phenotypes, and the delay in replicative senescence of Dmp1-null MEFs have been observed, suggesting that DMP1 functions as a tumor suppressor by increasing ARF expression [49]. In contrast to DMP1α, DMP1β inhibits ARF transcription [32]. DMP1β, which does not contain the DNA binding site of DMP1α, sequesters DMP1α, thereby inhibiting its binding to ARF promoter. DMP1β dose-dependently inhibits DMP1α-induced ARF promoter activity, implying that DMP1β antagonizes DMP1α transcriptional function and disturbs the interaction between DMP1α and ARF promoter [48]. In breast cancer patients, DMP1β is more expressed than DMP1α, suggesting the distinct role of DMP1β as an oncogenic factor rather than as a tumor suppressor [32].
FoxO is a subtype of the forkhead-box transcription factor, and has been identified as an ARF transcription factor [28]. Lymphomas, which express a dominant-negative FoxO mutant (dnFoxO), exhibit low ARF mRNA levels, regardless of the p53 status, implying that FoxO is involved in ARF transcriptional regulation. FoxO specifically binds to the FoxO-binding site in the first intron of ARF, thereby activating ARF transcription. FoxO inhibition in dnFoxO-expressing MYC-driven lymphomas promotes cell proliferation, while that in ARF-deficient MYC-driven lymphomas does not affect tumorigenicity, suggesting that FoxO inhibits MYC-derived lymphomagenesis by binding to the ARF promoter and inducing ARF expression.
Although ARF transcriptional regulation under conditions of oncogenic stresses has drawn significant attention, ARF regulation is an important process in embryogenesis [50,51,52]. Similar to Arf-deficient embryos, Tgfβ2-deficient embryos showed eyes with a hyperplasia phenotype and low ARF expression and acidic β-galactosidase on embryonic day 13.5, implying that TGFβ2 might function as a positive regulator of ARF transcription during embryogenesis [29]. Smad 2 and 3 appear to bind to the proximal region of the ARF locus in a TGFβ2-dependent manner, and inducing histone acetylation and RNA pol II recruitment. This in turn promotes ARF promoter remodeling and activation [30]. Unlike TGFβ2, TGFβ1 participates in a signaling pathway that negatively regulates ARF transcription in B-cell lymphoma [41]. In mutant p53-expressing B-cell lymphoma cells, ARF showed tumorigenic effects, different from its original tumor-suppressive function, by stabilizing mutant p53 through MDM2 inhibition. In this case, TGFβ1 activation in the B-cell lymphoma resulted in decreased E2F1 expression, leading to reduced ARF transcription. Low ARF expression leads to destabilization of mutant p53, thereby inducing cell cycle arrest.
2.2. Suppressors of ARF Transcription
The polycomb group transcription factor, BMI-1, was identified as an oncogene that cooperates with MYC in mouse lymphomas [53,54,55,56]. Bmi-1-deficient cells showed cellular senescence phenotypes, including decreased cell proliferation, defective S-phase cell cycle, cytoplasmic enlargement, unresponsiveness to growth factors, and increased acidic β-galactosidase, indicating that Bmi-1 deficiency was closely related to cellular senescence [57]. While there was no change in p21, p27, and p53 expression in Bmi-1-deficient cells, ARF and INK4a were upregulated. In the Bmi-1-deficient mouse model, a significantly small size and an ataxia phenotype were observed, and these phenotypes could be restored by intercrossing the BMI-1-deficient mice with Ink4a/Arf locus-deficient mice [57].
Another polycomb group protein, CBX7, has been identified as a transcriptional repressor of ARF [58]. CBX7 expression prevents ARF and INK4a mRNA accumulation, thereby delaying replicative senescence. When cells were sub-cultured, CBX7 levels gradually decreased, and thus, CBX7 expression was completely lost in the senescent cells. Furthermore, Cbx7-deficient cells showed severe growth arrest and high ARF and INK4a levels, thereby indicating that CBX7 and senescence suppressed each other. CBX7-induced suppression of ARF and INK4a expression occurs in a BMI-1-independent manner, suggesting that CBX7 may repress ARF and INK4a transcription by interacting with another polycomb-repressive complex-1 (PRC1) subset, but not with BMI-1.
Polycomb-repressive complex-2 (PRC2) represses ARF transcription by associating with Twist-1 [42]. Twist-1 expression in bone marrow-derived mesenchymal stem/stromal cells (BMSCs) results in increased proliferation and decreased β-galactosidase-positive cell counts. ARF expression is reduced by Twist-1, implying that Twist-1 may function as a negative regulator of ARF and prevent cellular senescence. Twist-1 recognizes H3K27me3—which interacts with Ezh2, a PRC2 component—on the ARF locus, subsequently recruiting PRC2, which suppresses ARF transcription. Additionally, Twist-1 suppresses the expression of E47, which is an INK4a transcriptional activator. Collectively, Twist-1 suppresses ARF and INK4a expression by recruiting PRC2 and decreasing E47 expression, thereby functioning as a negative regulator of cellular senescence.
Through senescence bypass library screening, TBX2 has been identified as a transcriptional suppressor of ARF [37]. TBX2 prevents BMI-1 deletion-induced premature senescence. The accumulation of ARF in Bmi-1-deficient cells was reduced upon TBX2 expression without any change in INK4a, suggesting that TBX2 specifically inhibits ARF expression.
EGFR, a membrane-bound receptor tyrosine kinase, serves as a mediator of proliferative signaling by activating diverse downstream components [59]. Suppression of ARF expression has recently been identified as being among the EGFR oncogenic mechanisms [34]. Upon binding to the epidermal growth factor, EGFR activates and interacts with the catalytic subunit type 3 (VPS34) of phosphatidylinositol 3-kinase, while translocating from the plasma membrane to the nucleus. The EGFR-VPS34 complex inhibits ARF transcription by binding to the AT-rich sequence in the ARF promoter, thereby suppressing the ARF-mediated fail-safe program [35].
3. Post-Translational Regulation of ARF Regulates Its Functional Roles in Cellular Physiology
The control of ARF expression at the transcriptional level has been sufficiently described; accumulating evidence regarding the post-translational regulation of ARF suggests that oncogenic stress-mediated ARF induction may be associated with molecular mechanisms that regulate protein levels and functions. In particular, the involvement of phosphorylation, ubiquitination, and proteasome- and lysosome-dependent pathways in the post-translational regulation of ARF has recently been observed.
3.1. ARF Phosphorylation
Phosphorylation is one of the well-known post-translational modifications involved in numerous cellular signaling pathways. Serine, threonine, or tyrosine residues of proteins are commonly phosphorylated by kinases, which are enzymes that add the phosphate group onto the target amino acids via esterification reactions [60]. Phosphorylation at these amino acids leads to conformational changes in proteins as a result of the change in charge of proteins, thereby inducing protein activation/deactivation or promoting protein degradation in a proteasome-dependent manner. Additionally, phosphorylation can change the affinity in protein–protein interactions (PPIs); thus, in diverse signaling pathways, phosphorylation cascades function as key events in regulating cellular signaling.
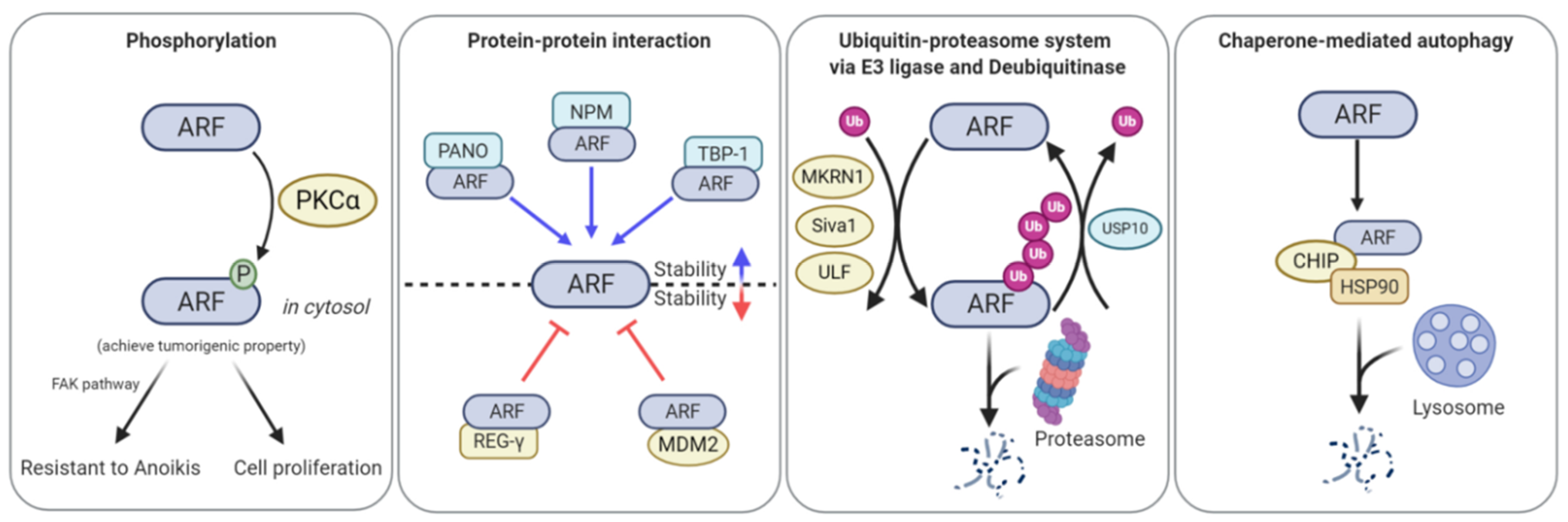
Despite the important roles of phosphorylation in protein activity regulation, only one phosphorylation site has been identified in ARF (Figure 3). Inoue et al. (2005) found that 12-o-tetradecanoyl-phorbol 13-acetate (TPA)-mediated protein kinase C alpha (PKCα) activation stabilized the ARF protein [61]. TPA treatment stabilizes ARF by activating PKCα, but not members of other PKC families. Here, binding of PKCα to ARF in the cytoplasmic compartment induces ARF phosphorylation at threonine 8, thereby resulting in the stabilization of ARF [62]. The phospho-mimetic mutant of ARF (T8D) showed a longer half-life and lesser nucleolar localization than wild type ARF. Additionally, T8D was no longer involved in cell growth retardation and could not affect the MDM2/p53 axis, suggesting that ARF phosphorylation inhibited ARF tumor-suppressive function. Recently, Fontana et al. (2018) reported that ARF phosphorylation at threonine 8 by PKCα was involved in the regulation of the focal adhesion kinase (FAK) pathway. ARF phosphorylation in the cytoplasm promotes cell spreading and alleviates anoikis [63]. Collectively, ARF phosphorylation appears to function as a cell survival factor rather than a tumor suppressor. Further molecular studies on the roles of ARF phosphorylation induced by PKCα or other kinases are required.
3.2. ARF Regulation via Degradation
3.2.1. The Ubiquitin-Proteasome System (UPS)
Ubiquitination is an important cellular process in which small molecules called ubiquitins are added to the target proteins; it occurs in three steps, requiring three enzymes, i.e., E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and E3 ubiquitin ligases. Ubiquitin forms a thioester bond with E1 in an ATP-dependent manner, resulting in the initial activation of ubiquitin. Then, ubiquitin is transferred to E2. E2-conjugated ubiquitins are transferred to the target proteins by E3 ligases; this is the rate-determining step of ubiquitination [64]. More than 1000 E3 ligases have been identified in humans, implying that ubiquitination regulates numerous target proteins and cell signaling pathways, and by extension, cellular physiology. The carboxyl-terminal residue of ubiquitins (G76) is linked to the lysine residue of target proteins via covalent bonds, leading to target protein activation or a change in localization. Additionally, ubiquitins can form polymers, known as polyubiquitin chains; this process is known as polyubiquitination [65]. Polyubiquitin chains are generated by adding ubiquitins to one of seven lysines (K6, 11, 27, 29, 33, 48, or 63) or methionine 1 (M1). Each polyubiquitin chain specifically regulates protein functions. In particular, the K48-linked polyubiquitin chain promotes target protein degradation in a proteasome-dependent manner, and the K63- or M1-linked polyubiquitin chains provide a platform for signaling transduction.
As human ARF does not contain any lysine residue, studies on ARF ubiquitination mainly focused on exploring the possibility of N-terminal ubiquitination of ARF [66]. In humans and mice, ARF turnover is inhibited upon treatment with MG132, a proteasome inhibitor, suggesting that ARF protein is continuously degraded in a proteasome-dependent manner. Overexpression of ubiquitin and human ARF or lysine-deficient mouse ARF (K26R) induces ARF ubiquitination, resulting in the accumulation of ubiquitinated ARF upon MG132 treatment. The N-terminal mutant ARF, which is recalcitrant to processing by methionine aminopeptidase, has fewer ubiquitinated forms and a longer turnover time than wild type ARF, thereby implying that N-terminal ubiquitination of ARF by an unknown E3 ligase destabilizes ARF in a proteasome-dependent manner.
ULF, also referred to as thyroid hormone receptor interactor 12, has been identified as the first E3 ligase for ARF [67]. ULF depletion stabilizes p53 by increasing ARF protein levels without altering those of ARF mRNA, leading to cell cycle arrest. ARF ubiquitination by ULF increases when NPM, which binds to ARF and arrests it in the nucleolus, is inhibited, implying that ULF-mediated ARF ubiquitination occurs in the nucleolus. Therefore, NPM appears to protect ARF by sequestering it away from the E3 ULF in the nucleus. Recently, several proteins have been reported as regulators of ULF-mediated ARF degradation. MYC binds to ULF and then inhibits ARF-ULF interaction [67,68]. The inhibitory function of MYC in ULF-mediated ARF ubiquitination does not depend on MYC transcriptional activity, suggesting that MYC can control ARF levels via transcriptional as well as post-translational regulation. Tumor necrosis factor receptor (TNFR)-associated death domain (TRADD) independently inhibits ULF-mediated ARF ubiquitination and stabilizes ARF proteins via the TNFR signaling pathway [69]. TRADD actively shuttles between the cytoplasm and nucleus. Nuclear TRADD interacts with ULF and abrogates ARF-ULF interaction. Upon expression of H-RasV12 in cells, increased TRADD expression, followed by ARF stabilization is observed, resulting in the promotion of OIS. Nucleostemin (NS) is a GTPase that is localized in the nucleolus and is involved in overall protein synthesis [70]. NS stabilizes ARF in two ways. The first involves the solidification of the NPM-ARF complex in the nucleolus. NS depletion leads to reduced NPM-ARF interaction, subsequently resulting in ARF destabilization. The second involves the inhibition of ULF-ARF interaction. NS binds to ULF and abolishes ULF-mediated ARF ubiquitination. Glioma tumor-suppressor candidate region gene 2 (GLTSCR2), which is a nucleolar protein, has also been identified as a modulator of ARF nucleoplasmic localization and stability [71]. GLTSCR2 can interact with ARF, leading to ARF nucleoplasmic localization. This increases ULF-ARF interaction, thereby inducing ARF degradation in a proteasome-dependent manner.
MKRN1 is an E3 ligase that regulates diverse cellular signaling pathways by leading to the ubiquitin-mediated proteasome-dependent degradation of target proteins. Ko et al. (2012) reported that MKRN1 ubiquitinates ARF, thereby leading to the proteasome-dependent degradation of ARF [72]. MKRN1 depletion promotes cellular senescence in gastric cancer cell lines, which can be rescued by ARF co-depletion. MKRN1 overexpression decreases ARF protein levels and half-life, and this can be blocked by MG132 treatment. A change in ARF localization from the nucleolus to the nucleus and cytoplasm can be observed upon the induction of MKRN1 expression, indicating that MKRN1-mediated ARF ubiquitination—followed by proteasomal degradation—occurs in the nucleus and cytoplasm. Another E3 ligase, Siva1, has been identified as a direct E3 ligase for ARF [73]. Siva1 interacts with ARF and induces the export of ARF from the nucleolus. Siva1-ARF interaction increases K48-linked polyubiquitination of ARF, resulting in ARF destabilization via the proteasome pathway. Siva1 depletion leads to p53 stabilization through the ARF-MDM2 axis, resulting in cell cycle arrest at the G1 stage. Beta-transducin repeat-containing protein 2 (β-TrCP2), a phosphorylation-dependent E3 ligase, ubiquitinates and degrades mouse ARF (p19ARF) in response to serum stimulation, thereby promoting cell proliferation via growth factor signaling [74]. β-TrCP2 deletion in MEFs increased p19ARF levels and suppressed cell proliferation. β-TrCP2-induced p19ARF ubiquitination and degradation require phosphorylation at serine 75. This is performed by ribosomal protein S6 kinase beta-1 (S6K1), which is activated in response to growth factor- or nutrient-dependent mammalian target of rapamycin (mTOR) signaling. As only p19ARF—but not p14ARF or other species—contains the β-TrCP2 and S6K1 degron, the regulation of p19ARF via the mTOR-S6K1-β-TrCP2 axis was restricted in the mouse model.
Recently, deubiquitinating enzymes (DUBs), E3 ligase counter enzymes that remove ubiquitin molecules from the target proteins by cleaving the thioester bonds of ubiquitins, have been reported to regulate ARF. USP10 has been identified as the first direct DUB for ARF [75]. USP10 is transcriptionally activated during MYC expression-induced OIS. MYC activates USP10 transcription by binding to the second E-box sequence located upstream of the USP10 transcription start site. USP10 deubiquitinates ARF by interacting with the N-terminus of ARF, thereby stabilizing the ARF protein by blocking the proteasome-mediated ARF degradation. USP10 depletion or knockout alleviates MYC expression-induced OIS, suggesting that USP10 functions as a key factor in the oncogenic stress-induced fail-safe program. Additionally, ubiquitin-specific-processing protease 7 (USP7) regulates ARF levels [76]. USP7 depletion in hepatocellular carcinoma (HCC) cell lines decreases proliferation and increases the number of cells arrested at the G1 stage. USP7 forms a complex with ULF, and protects it from proteasome-mediated degradation by deubiquitination. ULF stabilization promotes ARF degradation, thereby promoting HCC cell proliferation.
3.2.2. Chaperone-Mediated Autophagy (CMA)
Substrates containing the HSC70 canonical binding motif are recognized by a chaperone-co-chaperone complex, and are then bound by lysosome associated membrane glycoprotein 2 (LAMP2A) [77]. Interaction of the substrate-chaperone complex with LAMP2A results in LAMP2A multimerization and substrate internalization into the lysosome followed by substrate degradation in a process known as CMA [77].
Recently, Han et al. (2017) reported that HSP90, a molecular chaperone, regulated ARF turnover via CMA [78]. Treatment with geldanamycin, an inhibitor of HSP90, or HSP90 depletion by siRNA promotes cellular senescence, while ARF co-depletion rescues the increase in senescence. Interestingly, HSP90 binds to ARF in a CHIP-dependent manner. CHIP is a co-chaperone protein and an E3 ligase [79]. The formation of the HSP90, ARF, and CHIP complex allows ARF to interact with LAMP2A, which is subsequently degraded via the lysosomal degradation pathway. It must be noted that the E3 ligase activity of CHIP is not essential for HSP90-ARF-CHIP ternary complex formation and subsequent ARF degradation, suggesting that ubiquitination is not required.
3.3. Protein–Protein Interaction (PPI)
NPM (also called B23) is a multifunctional protein that controls a variety of cellular phenomena, including proliferation, genomic stability maintenance, cell death, rRNA processing, and ribosome biogenesis [80]. It is abundantly expressed in the nucleolus, and actively shuttles between the nucleus and cytoplasm. Over the past few years, the NPM-ARF interplays have been extensively studied. NPM enhances the nucleolar translocation of ARF by forming an NPM-ARF complex, thereby blocking ARF-MDM2 interaction—in the nucleus—and ARF-mediated p53 stabilization [81,82]. NPM depletion promotes ARF translocation from the nucleolus to the nucleus and cytoplasm, and subsequently leads to ARF destabilization. Furthermore, ARF was found to be localized in the cytoplasm of NPM-deficient cells or acute myeloid leukemia cells containing mutant NPM, suggesting that NPM played a central role in ARF nucleolar localization and stability [82,83]. Under genotoxic stress, the NPM-ARF complex translocates to the nucleus in a c-Jun-NH2-kinase (JNK) pathway-dependent manner [84]. c-Jun interacts with NPM in the nucleolus under normal conditions. When cells are exposed in ultraviolet radiation, JNK-induced phosphorylation of c-Jun at threonine 91 and 93 leads to the nuclear translocation of the c-Jun-NPM-ARF complex. Recently, another DNA damage response pathway—the ataxia telangiectasia mutated (ATM)-mediated pathway—has been identified as a novel signaling pathway that regulates ARF localization by modulating serine/threonine-protein kinase Nek 2 (Nek2)-dependent NPM phosphorylation [85]. Under normal conditions, Nek2 phosphorylates NPM at serine 70 and 88, thereby enhancing NPM-ARF interaction. When cells are treated with doxorubicin, ATM activates protein phosphatase-1 (PP1)—a counter phosphatase of Nek2—thereby, inducing NPM dephosphorylation, which causes nuclear localization of ARF by abrogating NPM-ARF interaction, and destabilizing ARF via ULF-mediated ubiquitination. Another kinase, protein kinase B (AKT), also regulates ARF stability and localization via NPM phosphorylation [86]. AKT interacts with NPM, resulting in NPM phosphorylation at serine 48 (located in the oligomerization domain). Structure analysis revealed that AKT-induced NPM phosphorylation at serine 48 is incompatible with NPM oligomerization due to steric clashes, thereby resulting in the nuclear and cytoplasmic localization of the NPM-ARF complex. The nuclear localization of the NPM-ARF complex inhibits MDM2 function and stabilizes p53. As AKT directly phosphorylates and activates MDM2, MDM2 inhibition by AKT-mediated NPM phosphorylation appears to occur in an ARF expression-dependent manner, but the detailed molecular mechanisms remain unclear.
In addition to NPM, several other ARF binding partners have been reported as regulators of ARF localization and stability. TBP-1 has been identified as an ARF stabilizer [87]. TBP-1, a proteasome component, forms a complex with ARF by binding to the 1-39 amino acids of ARF, which blocks proteasome-dependent ARF degradation, thereby resulting in ARF stabilization [88]. As the first two-three amino acids of ARF cannot influence TBP-1-mediated stabilization, TBP-1-mediated ARF stabilization appears to occur in an N-terminal ubiquitination-independent manner. Another component of the proteasome, REGγ, promotes ARF degradation in a ubiquitination-independent manner [89]. REGγ binds to ARF and directly induces its destabilization in a proteasome-dependent manner. Recently, MDM2 has been identified as a negative regulator of ARF [90]. MDM2 overexpression results in ARF destabilization via the ubiquitination-independent proteasome pathway. PKCα-induced ARF phosphorylation appears to block MDM2-mediated ARF degradation, indicating that MDM2 interacts with dephosphorylated ARF and transports it to the proteasome. Additionally, PANO, a nucleolar protein, interacts with ARF [91]. PANO overexpression increases p53 levels via ARF stabilization, thereby leading to increased apoptosis.
4. Post-Translational Regulation of ARF in Human Cancer
Over the last three decades, many mechanistic and clinical observations have indicated a close relationship between ARF and tumor progression in animal and human cancer models. ARF-null mice generated by specifically targeting exon 1β in the CDKN2a locus spontaneously develop numerous tumors, including sarcomas, lymphomas, and lung carcinomas, resulting in death within one year [92,93,94]. Treatment of ARF-knockout mice using carcinogens, such as dimethylbenz(a)anthracene (DMBA) or X-ray, promotes tumor progression, leading to a short life span of six months with drastic tumor development. Epigenetic modifications on ARF promoters have been identified in patients with a wide spectrum of tumors. As the ARF promoter contains a CpG island, ARF silencing by hypermethylation of these promoter sequences has frequently been reported in various human cancers, such as breast, bladder, colon, liver, gastric, lung, oral, prostate, and brain cancer [95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110]. Additionally, homologous deletion or loss of heterozygosity on the CDKN2a locus has also been frequently reported in numerous cancers, including breast, bladder, liver, lung, oral, prostate, and kidney cancer [97,102,106,107,108,109,110,111,112]. Furthermore, point mutations, including short deletion, insertion, and missense mutations, on the ARF exon 1β or exon 2 have been found in familial melanoma [113,114,115,116].
Genetic and epigenetic modifications in the CDKN2a locus have been well characterized in diverse cancers; the uncoupling of ARF mRNA and protein expression has also been observed in lung cancer [117,118]. Low ARF expression without deleterious mutations in the CDKN2a locus is found in non-small cell lung cancer (NSCLC), implying that post-translational regulation of ARF may be involved in cancer development (Table 2). In malignant gastric cancer patients, increased MKRN1 and low ARF expression is observed in well-differentiated adenocarcinoma, while low MKRN1 and high ARF expression is detected in poorly differentiated adenocarcinoma [72]. MKRN1 and ARF expression in gastric cancer patients shows an inverse correlation, suggesting that MKRN1-mediated ARF degradation may have an important clinical implication in gastric cancer. Additionally, a significant correlation between TRADD and ARF has been observed in invasive breast cancer [69]. TRADD expression does not correlate with estrogen, progesterone, and receptor tyrosine-protein kinase erbB-2 (HER2) receptor expression; however, it shows a positive correlation with ARF expression and relapse-free survival rates. In HCC patients, high USP7 and ULF expression, and low ARF expression, is observed [76]. Low USP7, ULF expression indicates high overall survival rates in HCC patients, suggesting that USP7-ULF-mediated ARF regulation is a useful parameter for predicting HCC prognosis. Various studies have been performed on the correlation between post-translational regulators and ARF in NSCLC. A panel of lung carcinomas with reduced ATM levels showed high ARF expression and low PP1 phosphorylation [85]. Furthermore, the inverse correlation between ATM and ARF was strengthened in a high INK4a expression panel, indicating that the association between ATM and ARF in NSCLC was mediated by post-translational regulation, not CDKN2a locus aberration. High HSP90 and CHIP expression, with reduced ARF expression, has also been identified in NSCLC [78]; high HSP90 or combined HSP90/CHIP expression, with low ARF expression, indicates worse overall survival rates. The inverse correlation between combined HSP90/CHIP expression and ARF is more substantial in advanced NSCLC than in the early stage, suggesting that combined HSP90/CHIP expression may be an independent prognostic marker for early detection of NSCLC. Recently, MYC, USP10, and ARF expression in NSCLC have been determined [75]. USP10 and ARF expression show a positive correlation, whereas MYC expression does not correlate with USP10 and ARF expression. MYC appears to be closely related to the disruption of the fail-safe program in NSCLC. Additionally, the dual loss of USP10 and ARF expression is frequently observed in small intestinal adenocarcinoma and ovarian cancer patients, thus implying that low combined USP10/ARF expression is a prognostic marker of small intestinal adenocarcinoma and ovarian cancer [119,120].
5. Conclusions and Perspectives
Since its discovery, ARF regulation has been extensively studied because of its importance in the determination of cell fate in response to oncogenic signals. ARF induces cellular senescence, cell cycle arrest, and apoptosis via p53-dependent or -independent pathways in response to oncogenic stress, which activates the fail-safe program [2,4]. When cells are abrogated in the ARF-induced fail-safe program due to deregulated ARF expression, cells fail to activate defense mechanisms in response to oncogenic stresses, such as DNA damage, oncogene activation, and oxidative stresses, leading to uncontrolled proliferation [2,4].
Numerous transcription factors have been identified as transcriptional regulators of ARF, and their clinical implications in cancer have been reported. However, the uncoupling of ARF mRNA and protein expression in human cancer remains unclear. For a decade, the molecular mechanisms underlying post-translational regulation of ARF have been suggested as key events in maintaining the balance of ARF expression in response to various signaling pathways. Most post-translational regulatory mechanisms control the nucleolar, nuclear, and cytoplasmic localizations of ARF, leading to stabilization/destabilization. In particular, PKCα-induced ARF phosphorylation leads to cytoplasmic localization and a change in the functional role of ARF (Figure 3). Phosphorylated ARF regulates cell spreading, rather than cell cycle arrest, via the FAK signaling pathway, suggesting that post-translational modification of ARF alters its function [63,121]. Recently, the oncogenic functions of ARF have been reported. ARF promotes tumorigenesis in prostate cancer by stabilizing Slug, interacts with metallopeptidase-7, and shows high protein expression in aggressive lymphoma, invasive bladder cancer, and thyroid cancer [122,123,124,125,126,127,128]. Furthermore, the fail-safe program-independent functions of ARF such as autophagic and oxidative stress-sensing functions have been continually accumulated. However, the mechanisms by which ARF switches from tumor-suppressive to oncogenic (or other) functions remain unclear [20,127,129,130,131]. Further studies on the post-translational regulation of ARF will provide a comprehensive understanding of these contradictory functions, thereby presenting a broad understanding of the ARF network in human cancer.
Many cancer patients show genetic and epigenetic modifications in the CDKN2a locus. However, these modifications do not explain why several cancer patients show low ARF expression without CDKN2a locus aberration. As described above, diverse factors are involved in the post-translational regulation of ARF, demonstrating meaningful clinical implications in cancer patients. Targeting the fine-tuned post-translational regulation of ARF would enable the development of independent prognostic markers and therapeutic strategies for various cancers.
Author Contributions
J.S. (Jinho Seo), D.S. and J.S. (Jaewhan Song) wrote and conceived the manuscript. S.R.L. helped in designing the figures. D.-B.O. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning, grant numbers 2015R1A3A2066581 and 2020R1C1C1006833; Brain Korea 21 (BK21) PLUS program; Next-Generation BioGreen 21 Program of the Rural Development Administration, grant number PJ013320; and Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program.
Acknowledgments
All figures were created with biorender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hall, M.; Peters, G. Genetic alterations of cyclins, cyclin-dependent kinases, and cdk inhibitors in human cancer. Adv. Cancer Res. 1996, 68, 67–108. [Google Scholar] [PubMed]
- Fontana, R.; Ranieri, M.; La Mantia, G.; Vivo, M. Dual role of the alternative reading frame arf protein in cancer. Biomolecules 2019, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Han, S.Y.; Song, J. Dynamics of arf regulation that control senescence and cancer. BMB Rep. 2016, 49, 598–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.; Han, S.Y.; Song, J. Regulatory network of arf in cancer development. Mol. Cells 2018, 41, 381–389. [Google Scholar] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16ink4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Zindy, F.; Eischen, C.M.; Randle, D.H.; Kamijo, T.; Cleveland, J.L.; Sherr, C.J.; Roussel, M.F. Myc signaling via the arf tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev. 1998, 12, 2424–2433. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin d/cdk4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Russo, A.A.; Tong, L.; Lee, J.O.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the cyclin-dependent kinase cdk6 by the tumour suppressor p16ink4a. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef]
- Kamijo, T.; Weber, J.D.; Zambetti, G.; Zindy, F.; Roussel, M.F.; Sherr, C.J. Functional and physical interactions of the arf tumor suppressor with p53 and mdm2. Proc. Natl. Acad. Sci. USA 1998, 95, 8292–8297. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The ink4a tumor suppressor gene product, p19arf, interacts with mdm2 and neutralizes mdm2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Eymin, B.; Karayan, L.; Seite, P.; Brambilla, C.; Brambilla, E.; Larsen, C.J.; Gazzeri, S. Human arf binds e2f1 and inhibits its transcriptional activity. Oncogene 2001, 20, 1033–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martelli, F.; Hamilton, T.; Silver, D.P.; Sharpless, N.E.; Bardeesy, N.; Rokas, M.; DePinho, R.A.; Livingston, D.M.; Grossman, S.R. P19arf targets certain e2f species for degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 4455–4460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Nag, A.; Raychaudhuri, P. Differential regulation of e2f1, dp1, and the e2f1/dp1 complex by arf. Mol. Cell. Biol. 2002, 22, 8398–8408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, A.; Sen, J.; Hagen, J.; Korgaonkar, C.K.; Caffrey, M.; Quelle, D.E.; Hughes, D.E.; Ackerson, T.J.; Costa, R.H.; Raychaudhuri, P. Arf directly binds dp1: Interaction with dp1 coincides with the g1 arrest function of arf. Mol. Cell. Biol. 2005, 25, 8024–8036. [Google Scholar] [CrossRef] [Green Version]
- Itahana, K.; Zhang, Y. Mitochondrial p32 is a critical mediator of arf-induced apoptosis. Cancer Cell 2008, 13, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Nag, A.; Pan, W.; Hay, N.; Gartel, A.L.; Colamonici, O.; Mori, Y.; Raychaudhuri, P. Myc-arf (alternate reading frame) interaction inhibits the functions of myc. J. Biol. Chem. 2004, 279, 36698–36707. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Gregory, M.A.; Li, Z.; Brousal, J.P.; West, K.; Hann, S.R. P19arf directly and differentially controls the functions of c-myc independently of p53. Nature 2004, 431, 712–717. [Google Scholar] [CrossRef]
- Rocha, S.; Campbell, K.J.; Perkins, N.D. P53- and mdm2-independent repression of nf-kappa b transactivation by the arf tumor suppressor. Mol. Cell 2003, 12, 15–25. [Google Scholar] [CrossRef]
- Fatyol, K.; Szalay, A.A. The p14arf tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1alpha (hif-1alpha ) and inhibits hif-1-mediated transcription. J. Biol. Chem. 2001, 276, 28421–28429. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. Nrf2 is a major target of arf in p53-independent tumor suppression. Mol. Cell 2017, 68, 224–232. [Google Scholar] [CrossRef]
- Reef, S.; Zalckvar, E.; Shifman, O.; Bialik, S.; Sabanay, H.; Oren, M.; Kimchi, A. A short mitochondrial form of p19arf induces autophagy and caspase-independent cell death. Mol. Cell 2006, 22, 463–475. [Google Scholar] [CrossRef] [PubMed]
- van Oosterwijk, J.G.; Li, C.; Yang, X.; Opferman, J.T.; Sherr, C.J. Small mitochondrial arf (smarf) protein corrects p53-independent developmental defects of arf tumor suppressor-deficient mice. Proc. Natl. Acad. Sci. USA 2017, 114, 7420–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiGiammarino, E.L.; Filippov, I.; Weber, J.D.; Bothner, B.; Kriwacki, R.W. Solution structure of the p53 regulatory domain of the p19arf tumor suppressor protein. Biochemistry 2001, 40, 2379–2386. [Google Scholar] [PubMed]
- Bothner, B.; Lewis, W.S.; DiGiammarino, E.L.; Weber, J.D.; Bothner, S.J.; Kriwacki, R.W. Defining the molecular basis of arf and hdm2 interactions. J. Mol. Biol. 2001, 314, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The arf tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef]
- Müller-Tidow, C.; Metzelder, S.; Buerger, H.; Packeisen, J.; Ganser, A.; Heil, G.; Kügler, K.; Adigüzel, G.; Schwäble, J.; Steffen, B. Expression of the p14 arf tumor suppressor predicts survival in acute myeloid leukemia. Leukemia 2004, 18, 720–726. [Google Scholar] [CrossRef]
- Elliott, M.J.; Dong, Y.B.; Yang, H.; McMasters, K.M. E2f-1 up-regulates c-myc and p14arf and induces apoptosis in colon cancer cells. Clin. Cancer Res. 2001, 7, 3590–3597. [Google Scholar]
- Bouchard, C.; Lee, S.; Paulus-Hock, V.; Loddenkemper, C.; Eilers, M.; Schmitt, C.A. Foxo transcription factors suppress myc-driven lymphomagenesis via direct activation of arf. Genes Dev. 2007, 21, 2775–2787. [Google Scholar] [CrossRef] [Green Version]
- Freeman-Anderson, N.E.; Zheng, Y.; McCalla-Martin, A.C.; Treanor, L.M.; Zhao, Y.D.; Garfin, P.M.; He, T.C.; Mary, M.N.; Thornton, J.D.; Anderson, C.; et al. Expression of the arf tumor suppressor gene is controlled by tgfbeta2 during development. Development 2009, 136, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Zhao, Y.D.; Gibbons, M.; Abramova, T.; Chu, P.Y.; Ash, J.D.; Cunningham, J.M.; Skapek, S.X. Tgfbeta signaling directly induces arf promoter remodeling by a mechanism involving smads 2/3 and p38 mapk. J. Biol. Chem. 2010, 285, 35654–35664. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Roussel, M.F.; Sherr, C.J. Induction of arf tumor suppressor gene expression and cell cycle arrest by transcription factor dmp1. Proc. Natl. Acad. Sci. USA 1999, 96, 3993–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglic, D.; Stovall, D.B.; Cline, J.M.; Fry, E.A.; Mallakin, A.; Taneja, P.; Caudell, D.L.; Willingham, M.C.; Sui, G.; Inoue, K. Dmp1beta, a splice isoform of the tumour suppressor dmp1 locus, induces proliferation and progression of breast cancer. J. Pathol. 2015, 236, 90–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Fry, E.A. Aberrant splicing of the dmp1-arf-mdm2-p53 pathway in cancer. Int. J. Cancer 2016, 139, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Ozenne, P.; Dayde, D.; Brambilla, E.; Eymin, B.; Gazzeri, S. P14(arf) inhibits the growth of lung adenocarcinoma cells harbouring an egfr l858r mutation by activating a stat3-dependent pro-apoptotic signalling pathway. Oncogene 2013, 32, 1050–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayde, D.; Guerard, M.; Perron, P.; Hatat, A.S.; Barrial, C.; Eymin, B.; Gazzeri, S. Nuclear trafficking of egfr by vps34 represses arf expression to promote lung tumor cell survival. Oncogene 2016, 35, 3986–3994. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, W. Functional characterization of e2f3b in human hepg2 liver cancer cell line. J. Cell. Biochem. 2018, 119, 3429–3439. [Google Scholar] [CrossRef]
- Jacobs, J.J.; Keblusek, P.; Robanus-Maandag, E.; Kristel, P.; Lingbeek, M.; Nederlof, P.M.; van Welsem, T.; van de Vijver, M.J.; Koh, E.Y.; Daley, G.Q.; et al. Senescence bypass screen identifies tbx2, which represses cdkn2a (p19(arf)) and is amplified in a subset of human breast cancers. Nat. Genet. 2000, 26, 291–299. [Google Scholar] [CrossRef]
- Pietersen, A.M.; Horlings, H.M.; Hauptmann, M.; Langerød, A.; Ajouaou, A.; Cornelissen-Steijger, P.; Wessels, L.F.; Jonkers, J.; Van De Vijver, M.J.; van Lohuizen, M. Ezh2 and bmi1 inversely correlate with prognosis and tp53 mutation in breast cancer. Breast Cancer Res. 2008, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; He, L.; Kapoor, A.; Gillis, A.; Rybak, A.P.; Cutz, J.-C.; Tang, D. Bmi1 promotes prostate tumorigenesis via inhibiting p16ink4a and p14arf expression. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2008, 1782, 642–648. [Google Scholar] [CrossRef] [Green Version]
- Bernard, D.; Martinez-Leal, J.F.; Rizzo, S.; Martinez, D.; Hudson, D.; Visakorpi, T.; Peters, G.; Carnero, A.; Beach, D.; Gil, J. Cbx7 controls the growth of normal and tumor-derived prostate cells by repressing the ink4a/arf locus. Oncogene 2005, 24, 5543–5551. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Ghosh, P.; O’Farrell, T.; Munk, R.; Rezanka, L.J.; Sasaki, C.Y.; Longo, D.L. Transforming growth factor beta1 (tgf-beta1) suppresses growth of b-cell lymphoma cells by p14(arf)-dependent regulation of mutant p53. J. Biol. Chem. 2012, 287, 23184–23195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakouros, D.; Isenmann, S.; Cooper, L.; Zannettino, A.; Anderson, P.; Glackin, C.; Gronthos, S. Twist-1 induces ezh2 recruitment regulating histone methylation along the ink4a/arf locus in mesenchymal stem cells. Mol. Cell. Biol. 2012, 32, 1433–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of myc and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, 1–24. [Google Scholar] [CrossRef] [PubMed]
- DeGregori, J.; Leone, G.; Miron, A.; Jakoi, L.; Nevins, J.R. Distinct roles for e2f proteins in cell growth control and apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 7245–7250. [Google Scholar] [CrossRef] [Green Version]
- Aslanian, A.; Iaquinta, P.J.; Verona, R.; Lees, J.A. Repression of the arf tumor suppressor by e2f3 is required for normal cell cycle kinetics. Genes Dev. 2004, 18, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Komori, H.; Enomoto, M.; Nakamura, M.; Iwanaga, R.; Ohtani, K. Distinct e2f-mediated transcriptional program regulates p14arf gene expression. EMBO J. 2005, 24, 3724–3736. [Google Scholar] [CrossRef] [Green Version]
- Humbert, P.O.; Verona, R.; Trimarchi, J.M.; Rogers, C.; Dandapani, S.; Lees, J.A. E2f3 is critical for normal cellular proliferation. Genes Dev. 2000, 14, 690–703. [Google Scholar]
- Tschan, M.P.; Federzoni, E.A.; Haimovici, A.; Britschgi, C.; Moser, B.A.; Jin, J.; Reddy, V.A.; Sheeter, D.A.; Fischer, K.M.; Sun, P.; et al. Human dmtf1beta antagonizes dmtf1alpha regulation of the p14(arf) tumor suppressor and promotes cellular proliferation. Biochim. Biophys. Acta 2015, 1849, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Wen, R.; Rehg, J.E.; Adachi, M.; Cleveland, J.L.; Roussel, M.F.; Sherr, C.J. Disruption of the arf transcriptional activator dmp1 facilitates cell immortalization, ras transformation, and tumorigenesis. Genes Dev. 2000, 14, 1797–1809. [Google Scholar]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.L.; Thornton, J.D.; Martin, A.C.; Rehg, J.E.; Bertwistle, D.; Zindy, F.; Skapek, S.X. Arf-dependent regulation of pdgf signaling in perivascular cells in the developing mouse eye. EMBO J. 2005, 24, 2803–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromley, A.; Churchman, M.L.; Zindy, F.; Sherr, C.J. Transient expression of the arf tumor suppressor during male germ cell and eye development in arf-cre reporter mice. Proc. Natl. Acad. Sci. USA 2009, 106, 6285–6290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Lohuizen, M.; Verbeek, S.; Scheijen, B.; Wientjens, E.; van der Gulden, H.; Berns, A. Identification of cooperating oncogenes in e mu-myc transgenic mice by provirus tagging. Cell 1991, 65, 737–752. [Google Scholar] [CrossRef]
- Haupt, Y.; Alexander, W.S.; Barri, G.; Klinken, S.P.; Adams, J.M. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in e mu-myc transgenic mice. Cell 1991, 65, 753–763. [Google Scholar] [CrossRef]
- van Lohuizen, M.; Frasch, M.; Wientjens, E.; Berns, A. Sequence similarity between the mammalian bmi-1 proto-oncogene and the drosophila regulatory genes psc and su(z)2. Nature 1991, 353, 353–355. [Google Scholar] [CrossRef]
- Alkema, M.J.; Jacobs, J.; Voncken, J.W.; Jenkins, N.A.; Copeland, N.G.; Satijn, D.P.; Otte, A.P.; Berns, A.; van Lohuizen, M. Mpc2, a new murine homolog of the drosophila polycomb protein is a member of the mouse polycomb transcriptional repressor complex. J. Mol. Biol. 1997, 273, 993–1003. [Google Scholar] [CrossRef]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; DePinho, R.A.; van Lohuizen, M. The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef]
- Gil, J.; Bernard, D.; Martinez, D.; Beach, D. Polycomb cbx7 has a unifying role in cellular lifespan. Nat. Cell Biol. 2004, 6, 67–72. [Google Scholar] [CrossRef]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The egfr odyssey—From activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Inoue, R.; Shiraishi, T. Pkcalpha is involved in phorbol ester tpa-mediated stabilization of p14arf. Biochem. Biophys. Res. Commun. 2005, 330, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Vivo, M.; Ranieri, M.; Sansone, F.; Santoriello, C.; Calogero, R.A.; Calabro, V.; Pollice, A.; La Mantia, G. Mimicking p14arf phosphorylation influences its ability to restrain cell proliferation. PLoS ONE 2013, 8, 1–12. [Google Scholar] [CrossRef]
- Fontana, R.; Guidone, D.; Sangermano, F.; Calabro, V.; Pollice, A.; La Mantia, G.; Vivo, M. Pkc dependent p14arf phosphorylation on threonine 8 drives cell proliferation. Sci. Rep. 2018, 8, 7056–7065. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, M.W.; Bae, K.H.; Lee, S.C.; Song, J.; Lee, E.W. The roles of ubiquitination in extrinsic cell death pathways and its implications for therapeutics. Biochem. Pharmacol. 2019, 162, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of e3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.L.; den Besten, W.; Bertwistle, D.; Roussel, M.F.; Sherr, C.J. N-terminal polyubiquitination and degradation of the arf tumor suppressor. Genes Dev. 2004, 18, 1862–1874. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Shan, J.; Zhu, W.G.; Qin, J.; Gu, W. Transcription-independent arf regulation in oncogenic stress-mediated p53 responses. Nature 2010, 464, 624–627. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Kon, N.; Zhong, J.; Zhang, P.; Yu, L.; Gu, W. Differential effects on arf stability by normal versus oncogenic levels of c-myc expression. Mol. Cell 2013, 51, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Chio, I.I.C.; Sasaki, M.; Ghazarian, D.; Moreno, J.; Done, S.; Ueda, T.; Inoue, S.; Chang, Y.L.; Chen, N.J.; Mak, T.W. Tradd contributes to tumour suppression by regulating ulf-dependent p19arf ubiquitylation. Nat. Cell Biol 2012, 14, 625–633. [Google Scholar] [CrossRef]
- Lo, D.; Zhang, Y.; Dai, M.S.; Sun, X.X.; Zeng, S.X.; Lu, H. Nucleostemin stabilizes arf by inhibiting the ubiquitin ligase ulf. Oncogene 2015, 34, 1688–1697. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Cho, Y.E.; Kim, S.H.; Kim, Y.J.; Park, J.H. Gltscr2 promotes the nucleoplasmic translocation and subsequent degradation of nucleolar arf. Oncotarget 2017, 8, 16293–16302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.; Shin, J.Y.; Seo, J.; Lee, K.D.; Lee, E.W.; Lee, M.S.; Lee, H.W.; Choi, I.J.; Jeong, J.S.; Chun, K.H.; et al. Acceleration of gastric tumorigenesis through mkrn1-mediated posttranslational regulation of p14arf. J. Natl. Cancer Inst. 2012, 104, 1660–1672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zha, M.; Zhao, X.; Jiang, P.; Du, W.; Tam, A.Y.; Mei, Y.; Wu, M. Siva1 inhibits p53 function by acting as an arf e3 ubiquitin ligase. Nat. Commun. 2013, 4, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Araki, T.; Nakagawa, M.; Hirao, A.; Unno, M.; Nakayama, K. S6 kinase- and beta-trcp2-dependent degradation of p19arf is required for cell proliferation. Mol. Cell Biol. 2015, 35, 3517–3527. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Han, S.Y.; Choi, C.H.; Cho, H.; Lee, M.S.; Kim, S.Y.; Song, J.S.; Hong, K.M.; Lee, H.W.; Hewitt, S.M.; et al. Oncogene-induced senescence mediated by c-myc requires usp10 dependent deubiquitination and stabilization of p14arf. Cell Death Differ. 2018, 25, 1050–1062. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.B.; Shi, G.M.; Dong, Z.R.; Ke, A.W.; Ma, H.H.; Gao, Q.; Shen, Z.Z.; Huang, X.Y.; Chen, H.; Yu, D.D.; et al. Ubiquitin-specific protease 7 accelerates p14(arf) degradation by deubiquitinating thyroid hormone receptor-interacting protein 12 and promotes hepatocellular carcinoma progression. Hepatology 2015, 61, 1603–1614. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Han, S.Y.; Ko, A.; Kitano, H.; Choi, C.H.; Lee, M.S.; Seo, J.; Fukuoka, J.; Kim, S.Y.; Hewitt, S.M.; Chung, J.Y.; et al. Molecular chaperone hsp90 is necessary to prevent cellular senescence via lysosomal degradation of p14arf. Cancer Res. 2017, 77, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Han, S.Y.; Seong, D.; Han, H.J.; Song, J. Multifaceted c-terminus of hsp70-interacting protein regulates tumorigenesis via protein quality control. Arch. Pharm. Res. 2019, 42, 63–75. [Google Scholar] [CrossRef]
- Grisendi, S.; Mecucci, C.; Falini, B.; Pandolfi, P.P. Nucleophosmin and cancer. Nat. Rev. Cancer 2006, 6, 493–505. [Google Scholar] [CrossRef]
- Korgaonkar, C.; Hagen, J.; Tompkins, V.; Frazier, A.A.; Allamargot, C.; Quelle, F.W.; Quelle, D.E. Nucleophosmin (b23) targets arf to nucleoli and inhibits its function. Mol. Cell. Biol. 2005, 25, 1258–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, E.; Bonetti, P.; Lazzerini Denchi, E.; Martinelli, P.; Zamponi, R.; Marine, J.C.; Helin, K.; Falini, B.; Pelicci, P.G. Nucleophosmin is required for DNA integrity and p19arf protein stability. Mol. Cell. Biol. 2005, 25, 8874–8886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Besten, W.; Kuo, M.L.; Williams, R.T.; Sherr, C.J. Myeloid leukemia-associated nucleophosmin mutants perturb p53-dependent and independent activities of the arf tumor suppressor protein. Cell Cycle 2005, 4, 1593–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogev, O.; Saadon, K.; Anzi, S.; Inoue, K.; Shaulian, E. DNA damage-dependent translocation of b23 and p19 arf is regulated by the jun n-terminal kinase pathway. Cancer Res. 2008, 68, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase atm and arf tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef]
- Hamilton, G.; Abraham, A.G.; Morton, J.; Sampson, O.; Pefani, D.E.; Khoronenkova, S.; Grawenda, A.; Papaspyropoulos, A.; Jamieson, N.; McKay, C.; et al. Akt regulates npm dependent arf localization and p53mut stability in tumors. Oncotarget 2014, 5, 6142–6167. [Google Scholar] [CrossRef]
- Pollice, A.; Nasti, V.; Ronca, R.; Vivo, M.; Lo Iacono, M.; Calogero, R.; Calabro, V.; La Mantia, G. Functional and physical interaction of the human arf tumor suppressor with tat-binding protein-1. J. Biol Chem 2004, 279, 6345–6353. [Google Scholar] [CrossRef] [Green Version]
- Pollice, A.; Sepe, M.; Villella, V.R.; Tolino, F.; Vivo, M.; Calabro, V.; La Mantia, G. Tbp-1 protects the human oncosuppressor p14arf from proteasomal degradation. Oncogene 2007, 26, 5154–5162. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Barton, L.F.; Chi, Y.; Clurman, B.E.; Roberts, J.M. Ubiquitin-independent degradation of cell-cycle inhibitors by the reggamma proteasome. Mol. Cell 2007, 26, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Vivo, M.; Matarese, M.; Sepe, M.; Di Martino, R.; Festa, L.; Calabro, V.; La Mantia, G.; Pollice, A. Mdm2-mediated degradation of p14arf: A novel mechanism to control arf levels in cancer cells. PLoS ONE 2015, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Watari, A.; Li, Y.; Higashiyama, S.; Yutsudo, M. A novel proapoptotic gene pano encodes a post-translational modulator of the tumor suppressor p14arf. Exp. Cell Res. 2012, 318, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, T.; Zindy, F.; Roussel, M.F.; Quelle, D.E.; Downing, J.R.; Ashmun, R.A.; Grosveld, G.; Sherr, C.J. Tumor suppression at the mouse ink4a locus mediated by the alternative reading frame product p19arf. Cell 1997, 91, 649–659. [Google Scholar] [CrossRef] [Green Version]
- Kamijo, T.; Bodner, S.; van de Kamp, E.; Randle, D.H.; Sherr, C.J. Tumor spectrum in arf-deficient mice. Cancer Res. 1999, 59, 2217–2222. [Google Scholar] [PubMed]
- Sharpless, N.E.; Ramsey, M.R.; Balasubramanian, P.; Castrillon, D.H.; DePinho, R.A. The differential impact of p16(ink4a) or p19(arf) deficiency on cell growth and tumorigenesis. Oncogene 2004, 23, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.; Dominguez, G.; Silva, J.M.; Garcia, J.M.; Gallego, I.; Corbacho, C.; Provencio, M.; Espana, P.; Bonilla, F. Analysis of genetic and epigenetic processes that influence p14arf expression in breast cancer. Oncogene 2001, 20, 4586–4590. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, G.; Carballido, J.; Silva, J.; Silva, J.M.; Garcia, J.M.; Menendez, J.; Provencio, M.; Espana, P.; Bonilla, F. P14arf promoter hypermethylation in plasma DNA as an indicator of disease recurrence in bladder cancer patients. Clin. Cancer Res. 2002, 8, 980–985. [Google Scholar] [PubMed]
- Silva, J.; Silva, J.M.; Dominguez, G.; Garcia, J.M.; Cantos, B.; Rodriguez, R.; Larrondo, F.J.; Provencio, M.; Espana, P.; Bonilla, F. Concomitant expression of p16ink4a and p14arf in primary breast cancer and analysis of inactivation mechanisms. J. Pathol. 2003, 199, 289–297. [Google Scholar] [CrossRef]
- Dominguez, G.; Silva, J.; Garcia, J.M.; Silva, J.M.; Rodriguez, R.; Munoz, C.; Chacon, I.; Sanchez, R.; Carballido, J.; Colas, A.; et al. Prevalence of aberrant methylation of p14arf over p16ink4a in some human primary tumors. Mutat. Res. 2003, 530, 9–17. [Google Scholar] [CrossRef]
- Lee, M.; Sup Han, W.; Kyoung Kim, O.; Hee Sung, S.; Sun Cho, M.; Lee, S.N.; Koo, H. Prognostic value of p16ink4a and p14arf gene hypermethylation in human colon cancer. Pathol. Res. Pract. 2006, 202, 415–424. [Google Scholar] [CrossRef]
- Esteller, M.; Tortola, S.; Toyota, M.; Capella, G.; Peinado, M.A.; Baylin, S.B.; Herman, J.G. Hypermethylation-associated inactivation of p14(arf) is independent of p16(ink4a) methylation and p53 mutational status. Cancer Res. 2000, 60, 129–133. [Google Scholar]
- Tannapfel, A.; Busse, C.; Geissler, F.; Witzigmann, H.; Hauss, J.; Wittekind, C. Ink4a-arf alterations in liver cell adenoma. Gut 2002, 51, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannapfel, A.; Sommerer, F.; Benicke, M.; Weinans, L.; Katalinic, A.; Geissler, F.; Uhlmann, D.; Hauss, J.; Wittekind, C. Genetic and epigenetic alterations of the ink4a-arf pathway in cholangiocarcinoma. J. Pathol. 2002, 197, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Iida, S.; Akiyama, Y.; Nakajima, T.; Ichikawa, W.; Nihei, Z.; Sugihara, K.; Yuasa, Y. Alterations and hypermethylation of the p14(arf) gene in gastric cancer. Int. J. Cancer 2000, 87, 654–658. [Google Scholar] [CrossRef]
- Zochbauer-Muller, S.; Fong, K.M.; Virmani, A.K.; Geradts, J.; Gazdar, A.F.; Minna, J.D. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61, 249–255. [Google Scholar]
- Chaar, I.; Amara, S.; Elamine, O.E.; Khiari, M.; Ounissi, D.; Khalfallah, T.; Ben Hmida, A.; Mzabi, S.; Bouraoui, S. Biological significance of promoter hypermethylation of p14/arf gene: Relationships to p53 mutational status in tunisian population with colorectal carcinoma. Tumour Biol. 2014, 35, 1439–1449. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.S.; Wang, Y.C.; Tseng, R.C.; Chang, J.W.; Chen, J.T.; Shih, C.M.; Chen, C.Y.; Wang, Y.C. 5’ cytosine-phospho-guanine island methylation is responsible for p14arf inactivation and inversely correlates with p53 overexpression in resected non-small cell lung cancer. Clin. Cancer Res. 2004, 10, 4734–4741. [Google Scholar] [CrossRef] [Green Version]
- Berggren, P.; Kumar, R.; Sakano, S.; Hemminki, L.; Wada, T.; Steineck, G.; Adolfsson, J.; Larsson, P.; Norming, U.; Wijkstrom, H.; et al. Detecting homozygous deletions in the cdkn2a(p16(ink4a))/arf(p14(arf)) gene in urinary bladder cancer using real-time quantitative pcr. Clin. Cancer Res. 2003, 9, 235–242. [Google Scholar]
- Shintani, S.; Nakahara, Y.; Mihara, M.; Ueyama, Y.; Matsumura, T. Inactivation of the p14(arf), p15(ink4b) and p16(ink4a) genes is a frequent event in human oral squamous cell carcinomas. Oral Oncol. 2001, 37, 498–504. [Google Scholar] [CrossRef]
- Konishi, N.; Nakamura, M.; Kishi, M.; Nishimine, M.; Ishida, E.; Shimada, K. Heterogeneous methylation and deletion patterns of the ink4a/arf locus within prostate carcinomas. Am. J. Pathol. 2002, 160, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Sailasree, R.; Abhilash, A.; Sathyan, K.M.; Nalinakumari, K.R.; Thomas, S.; Kannan, S. Differential roles of p16ink4a and p14arf genes in prognosis of oral carcinoma. Cancer Epidemiol. Biomark. Prev. 2008, 17, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Nishida, N.; Fukuda, Y.; Nishimura, T.; Komeda, T.; Nakao, K. Alteration of the p14(arf) gene and p53 status in human hepatocellular carcinomas. J. Gastroenterol. 2004, 39, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Bilim, V.; Hara, N.; Takahashi, K.; Tomita, Y. Homozygous deletions of the ink4a/arf locus in renal cell cancer. Anticancer Res. 2006, 26, 4299–4305. [Google Scholar] [PubMed]
- Rizos, H.; Puig, S.; Badenas, C.; Malvehy, J.; Darmanian, A.P.; Jimenez, L.; Mila, M.; Kefford, R.F. A melanoma-associated germline mutation in exon 1beta inactivates p14arf. Oncogene 2001, 20, 5543–5547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randerson-Moor, J.A.; Harland, M.; Williams, S.; Cuthbert-Heavens, D.; Sheridan, E.; Aveyard, J.; Sibley, K.; Whitaker, L.; Knowles, M.; Bishop, J.N.; et al. A germline deletion of p14(arf) but not cdkn2a in a melanoma-neural system tumour syndrome family. Hum. Mol. Genet. 2001, 10, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizos, H.; Darmanian, A.P.; Holland, E.A.; Mann, G.J.; Kefford, R.F. Mutations in the ink4a/arf melanoma susceptibility locus functionally impair p14arf. J. Biol. Chem. 2001, 276, 41424–41434. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, C.; Lee Wu, C.; Evans, G.; Howell, A.; Elles, R.G.; Jordan, R.; Sloan, P.; Read, A.P.; Thakker, N. Germline mutation of arf in a melanoma kindred. Hum. Mol. Genet. 2002, 11, 1273–1279. [Google Scholar] [CrossRef] [Green Version]
- Vonlanthen, S.; Heighway, J.; Tschan, M.P.; Borner, M.M.; Altermatt, H.J.; Kappeler, A.; Tobler, A.; Fey, M.F.; Thatcher, N.; Yarbrough, W.G.; et al. Expression of p16ink4a/p16alpha and p19arf/p16beta is frequently altered in non-small cell lung cancer and correlates with p53 overexpression. Oncogene 1998, 17, 2779–2785. [Google Scholar] [CrossRef] [Green Version]
- Gazzeri, S.; Della Valle, V.; Chaussade, L.; Brambilla, C.; Larsen, C.J.; Brambilla, E. The human p19arf protein encoded by the beta transcript of the p16ink4a gene is frequently lost in small cell lung cancer. Cancer Res. 1998, 58, 3926–3931. [Google Scholar]
- Song, J.S.; Yi, J.M.; Cho, H.; Choi, C.H.; Park, Y.; Chung, E.J.; Song, J.; Chung, J.Y.; Hong, S.M. Dual loss of usp10 and p14arf protein expression is associated with poor prognosis in patients with small intestinal adenocarcinoma. Tumour Biol. 2018, 40, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Han, G.H.; Chay, D.B.; Yi, J.M.; Cho, H.; Chung, J.Y.; Kim, J.H. Loss of both usp10 and p14arf protein expression is an independent prognostic biomarker for poor prognosis in patients with epithelial ovarian cancer. Cancer Genom. Proteom. 2019, 16, 553–562. [Google Scholar] [CrossRef]
- Vivo, M.; Fontana, R.; Ranieri, M.; Capasso, G.; Angrisano, T.; Pollice, A.; Calabro, V.; La Mantia, G. P14arf interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 2017, 36, 4913–4928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Liu, S.; Lu, W.; Yang, Q.; Williams, K.D.; Binhazim, A.A.; Carver, B.S.; Matusik, R.J.; Chen, Z. Slug regulates e-cadherin repression via p19arf in prostate tumorigenesis. Mol. Oncol. 2014, 8, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Lu, W.; Liu, S.; Yang, Q.; Goodwin, J.S.; Sathyanarayana, S.A.; Pratap, S.; Chen, Z. Mmp7 interacts with arf in nucleus to potentiate tumor microenvironments for prostate cancer progression in vivo. Oncotarget 2016, 7, 47609–47619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Aguilera, A.; Sanchez-Beato, M.; Garcia, J.F.; Prieto, I.; Pollan, M.; Piris, M.A. P14(arf) nuclear overexpression in aggressive b-cell lymphomas is a sensor of malfunction of the common tumor suppressor pathways. Blood 2002, 99, 1411–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.K.; Park, J.Y.; Kang, H.J.; Cho, H.C. Overexpression of p16ink4a and p14arf in haematological malignancies. Clin. Lab. Haematol. 2003, 25, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Ferru, A.; Fromont, G.; Gibelin, H.; Guilhot, J.; Savagner, F.; Tourani, J.M.; Kraimps, J.L.; Larsen, C.J.; Karayan-Tapon, L. The status of cdkn2a alpha (p16ink4a) and beta (p14arf) transcripts in thyroid tumour progression. Br. J. Cancer 2006, 95, 1670–1677. [Google Scholar] [CrossRef] [Green Version]
- Humbey, O.; Pimkina, J.; Zilfou, J.T.; Jarnik, M.; Dominguez-Brauer, C.; Burgess, D.J.; Eischen, C.M.; Murphy, M.E. The arf tumor suppressor can promote the progression of some tumors. Cancer Res. 2008, 68, 9608–9613. [Google Scholar] [CrossRef] [Green Version]
- Owczarek, T.B.; Kobayashi, T.; Ramirez, R.; Rong, L.; Puzio-Kuter, A.M.; Iyer, G.; Teo, M.Y.; Sanchez-Vega, F.; Wang, J.; Schultz, N.; et al. Arf confers a context-dependent response to chemotherapy in muscle-invasive bladder cancer. Cancer Res. 2017, 77, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Pimkina, J.; Humbey, O.; Zilfou, J.T.; Jarnik, M.; Murphy, M.E. Arf induces autophagy by virtue of interaction with bcl-xl. J. Biol. Chem. 2009, 284, 2803–2810. [Google Scholar] [CrossRef] [Green Version]
- Budina-Kolomets, A.; Hontz, R.D.; Pimkina, J.; Murphy, M.E. A conserved domain in exon 2 coding for the human and murine arf tumor suppressor protein is required for autophagy induction. Autophagy 2013, 9, 1553–1565. [Google Scholar] [CrossRef] [Green Version]
- Horvat, A.; Noto, J.M.; Ramatchandirin, B.; Zaika, E.; Palrasu, M.; Wei, J.; Schneider, B.G.; El-Rifai, W.; Peek, R.M., Jr.; Zaika, A.I. Helicobacter pylori pathogen regulates p14arf tumor suppressor and autophagy in gastric epithelial cells. Oncogene 2018, 37, 5054–5065. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Overview of the genomic structure and function of the CDKN2a locus. (A) Two transcripts, α-transcript (encoding INK4a) and β-transcript (encoding ARF), are transcribed from the CDKN2a locus in response to oncogenic stresses. Although these two transcripts share exon 2 and 3 sequences, they have alternative reading frames, and thus are translated into two different proteins. (B) INK4a inhibits cyclin-dependent kinase 4/6 (CDK4/CDK6) activity, leading to an increase in hypo-phosphorylated retinoblastoma (RB) levels. Hypo-phosphorylated RB blocks E2F function, subsequently inducing cell cycle arrest. ARF binds to mouse double minute 2 homolog (MDM2), which accumulates in the nucleolus and inhibits E3 ligase activity. This leads to p53 stabilization, inducing cell cycle arrest and apoptosis. ARF induces apoptosis, cell cycle arrest, and senescence in a p53-independent manner.
Figure 1.
Overview of the genomic structure and function of the CDKN2a locus. (A) Two transcripts, α-transcript (encoding INK4a) and β-transcript (encoding ARF), are transcribed from the CDKN2a locus in response to oncogenic stresses. Although these two transcripts share exon 2 and 3 sequences, they have alternative reading frames, and thus are translated into two different proteins. (B) INK4a inhibits cyclin-dependent kinase 4/6 (CDK4/CDK6) activity, leading to an increase in hypo-phosphorylated retinoblastoma (RB) levels. Hypo-phosphorylated RB blocks E2F function, subsequently inducing cell cycle arrest. ARF binds to mouse double minute 2 homolog (MDM2), which accumulates in the nucleolus and inhibits E3 ligase activity. This leads to p53 stabilization, inducing cell cycle arrest and apoptosis. ARF induces apoptosis, cell cycle arrest, and senescence in a p53-independent manner.

Figure 2.
A number of transcriptional factors positively or negatively regulate ARF transcription. Smads, DMP1α, E2Fs, MYC, and FoxO activate ARF transcription. E2F3b, enhancer of zeste homolog 2 (Ezh2)/Twist-1, chromobox protein homolog 7 (CBX7), T-box transcription factor 2 (TBX2), B-cell-specific Moloney murine leukemia virus integration site 1 (BMI-1), and epidermal growth factor receptor (EGFR) directly binds to the ARF promoter and suppress ARF transcription. DMP1β interacts with DMP1α, subsequently blocking the binding of DMP1α to the ARF promoter. Furthermore, the transforming growth factor beta 1 (TGFβ1) signaling pathway negatively regulates ARF transcription by inhibiting E2F1 expression.
Figure 2.
A number of transcriptional factors positively or negatively regulate ARF transcription. Smads, DMP1α, E2Fs, MYC, and FoxO activate ARF transcription. E2F3b, enhancer of zeste homolog 2 (Ezh2)/Twist-1, chromobox protein homolog 7 (CBX7), T-box transcription factor 2 (TBX2), B-cell-specific Moloney murine leukemia virus integration site 1 (BMI-1), and epidermal growth factor receptor (EGFR) directly binds to the ARF promoter and suppress ARF transcription. DMP1β interacts with DMP1α, subsequently blocking the binding of DMP1α to the ARF promoter. Furthermore, the transforming growth factor beta 1 (TGFβ1) signaling pathway negatively regulates ARF transcription by inhibiting E2F1 expression.

Figure 3.
ARF function, stability, and localization are regulated by phosphorylation, ubiquitination, PPIs, and chaperone-mediated autophagy (CMA). PKCα-induced ARF phosphorylation leads to the cytoplasmic localization of ARF, thereby promoting cell spreading and alleviating anoikis. Makorin ring finger protein 1 (MKRN1), Siva1, and ubiquitin ligase for ARF 1 (ULF1) ubiquitinate ARF, leading to its proteasome-mediated degradation. Ubiquitin-specific peptidase 10 (USP10) directly binds to ARF and then detaches ubiquitin from ARF, thus stabilizing ARF. Proapoptotic nuclear protein 1 (PANO), nucleophosmin (NPM), and tat-binding protein-1 (TBP-1) can bind to ARF, thereby preventing its degradation. Proteasome activator complex subunit 3 (REG-γ) and MDM2 interact with ARF and then transport ARF to the proteasome, thereby resulting in ARF degradation. C-terminus heat shock cognate 71 kDa protein (HSC70)-interacting protein (CHIP)-heat shock protein 90 (HSP90) forms a complex with ARF, leading to degradation of ARF in a lysosome-dependent manner.
Figure 3.
ARF function, stability, and localization are regulated by phosphorylation, ubiquitination, PPIs, and chaperone-mediated autophagy (CMA). PKCα-induced ARF phosphorylation leads to the cytoplasmic localization of ARF, thereby promoting cell spreading and alleviating anoikis. Makorin ring finger protein 1 (MKRN1), Siva1, and ubiquitin ligase for ARF 1 (ULF1) ubiquitinate ARF, leading to its proteasome-mediated degradation. Ubiquitin-specific peptidase 10 (USP10) directly binds to ARF and then detaches ubiquitin from ARF, thus stabilizing ARF. Proapoptotic nuclear protein 1 (PANO), nucleophosmin (NPM), and tat-binding protein-1 (TBP-1) can bind to ARF, thereby preventing its degradation. Proteasome activator complex subunit 3 (REG-γ) and MDM2 interact with ARF and then transport ARF to the proteasome, thereby resulting in ARF degradation. C-terminus heat shock cognate 71 kDa protein (HSC70)-interacting protein (CHIP)-heat shock protein 90 (HSP90) forms a complex with ARF, leading to degradation of ARF in a lysosome-dependent manner.


{kind=link}
{kind=link}
{kind=link}
Table 1.
The relationship between the transcription factors and ARF expression in human cancer.
| Transcription Factor | Cancer Type | Correlation with ARF Expression | Molecular Mechanism | Ref. |
|---|---|---|---|---|
| MYC | Acute myeloid leukemia | Positive correlation with ARF The combined expression of high MYC and ARF in AML Patients with low ARF expression worsen overall survival rates | MYC overexpression increases ARF mRNA transcription. ARF null mice exhibit resistance to MYC-driven apoptosis. | [26] |
| E2F1/E2F2 | Colon cancer | Positive correlation with ARF The combined expression of high E2Fs and ARF in colon cancer | E2Fs bind to the conserved sequence of ARF promoter, increasing ARF transcription. Overexpression of E2F1 leads to G2/M arrest with increase in ARF protein levels. | [27] |
| FoxO | Primary lymphoma | Positive correlation with ARF FoxO proteins have an instructive role in regulating ARF expression during MYC-induced lymphomagenesis | FoxO increases ARF transcription via interacting with FoxO-binding site region in the first intron of ARF. Lymphomas expressing a dominant-negative mutant of FoxO (dnFoxO) have low levels of ARF mRNA regardless of the p53 status. | [28] |
| TGF-β2/ SMAD2/3 | Unknown | Positive correlation with ARF TGFβ2-deficient embryos show hyperplasia phenotype in the eyes at embryonic day 13.5 with low ARF expression | SMAD2/3 bind to a proximal region of the ARF locus in a TGFβ2-dependent manner. | [29,30] |
| DMP1α | Unknown | Positive correlation with ARF | DMP1α binds to the consensus sequence of the ARF promoter, leading to an increase in ARF transcription. | [31] |
| DMP1β | Breast cancer | Inverse correlation with ARF The correlation between high DMP1β expression and shorter survival of breast cancer patients | DMP1β binds to DMP1α, which inhibit its transcriptional activity, thereby leading to a decrease in ARF transcription. High DMP1β and low DMP1α expression due to alternative splicing is frequently observed in breast cancer patients. | [32,33] |
| EGFR/VPS34 | Lung cancer | Inverse correlation with ARF The expression of low ARF in lung tumors harbouring constitutive active mutant EGFR | Active EGFR interacts with VPS34, which moves to the nucleus, thus inhibiting ARF expression via binding to the AT-rich sequence of the ARF promoter. | [34,35] |
| E2F3b | Hepatocarcinoma | Inverse correlation with ARF The expression of high E2F3 in hepatocellular carcinoma (HCC) | E2F3b represses ARF mRNA expression via binding to ARF promoter. E2F3b induces G1/S phase transition and markedly increases cell proliferation, but has a minor effect on apoptosis. | [36] |
| TBX2 | Breast cancer | Inverse correlation with ARF TBX2 amplification in human breast cancer | ARF expression in BMI-1 deficient cells is suppressed by TBX2 without any change in INK4a level. | [37] |
| BMI-1 | Breast cancer | Inverse correlation with ARF | Overexpression of BMI-1 results in the elevation of expression of polycomb group (PcG)-target genes followed by the inhibition of ARF expression. | [38] |
| Prostate cancer | Inverse correlation with ARF The combined expression of high BMI-1 and low ARF in prostate cancer | BMI-1-expressing DU145 cells form drastic large tumors in NOD/SCID mice. | [39] | |
| CBX7 | Prostate cancer | Inverse correlation with ARF | CBX7 ablation retards cell proliferation via the ARF/p53 and INK4a/Rb pathways. | [40] |
| TGF-β1 | B-cell lymphoma | Inverse correlation with ARF | In B-cell lymphoma expressing mutant p53, activation of TGFβ1 leads to a decrease in E2F1 expression, leading to the reduction in ARF transcription. The low expression of ARF induces the destabilization of mutant p53. | [41] |
| Twist/Ezh2 | Unknown | Inverse correlation with ARF | Twist-1 recognizes H3K27me3 on the ARF locus followed by interaction with Ezh2, which leads to suppression of ARF transcription via PRC2 complex. | [42] |
(Related to ‘2. Transcriptional regulation of ARF’).
Table 2.
The relationship between the ARF post-translational regulator and ARF expression in human cancer.
Table 2.
The relationship between the ARF post-translational regulator and ARF expression in human cancer.
| Post-Translational Regulator | Cancer Type | Correlation with ARF Expression | Molecular Mechanism | Ref. |
|---|---|---|---|---|
| MKRN1 | Gastric adenocarcinoma | Inverse correlation with ARF The combined expression of high MKRN1 and low ARF in well-differentiated adenocarcinoma | MKRN1 promotes ARF ubiquitination, which leads to the proteasome-dependent degradation of ARF | [72] |
| TRADD | Invasive breast cancer | Positive correlation with ARF Low TRADD expression correlates with poor prognosis. | TRADD competes with ULF for interaction with ARF, protecting ARF from ULF-mediated ubiquitination. | [69] |
| ATM | Lung carcinoma | Inverse correlation with ARF | ATM-PP1 axis inhibits Nek2 kinase activity, which induces the de-phosphorylation of NPM, thus leading to the nucleoplasm localization and degradation of ARF. | [85] |
| USP7/ULF | Hepatocarcinoma | Inverse correlation with ARF The combined expression of low USP7 and ULF worsen overall survival rates. | USP7 forms a complex with ULF that protects ULF protein from proteasome-mediated degradation via removal of ubiquitin. | [76] |
| HSP90/CHIP | NSCLC | Inverse correlation with ARF The combined expression of high HSP90, CHIP, and low ARF worsen overall survival rates. | HSP90 and CHIP complex form an interaction with ARF, which induces lysosomal degradation of ARF through binding to LAMP2A. The E3 ligase activity of CHIP is not required for formation of a tertiary complex and lysosomal degradation of ARF. | [78,79] |
| USP10 | NSCLC | Positive correlation with ARF The combined expression of low USP10 and ARF worsen overall survival rates. | MYC increases the stability of ARF protein via induction of USP10, which is a deubiquitinase of ARF. | [75] |
| Small intestine cancer | Positive correlation with ARF The combined expression of high USP10 and ARF are negatively correlated with vascular and lymphatic invasion. The combined expression of low USP10 and ARF worsen overall survival rates. | Several patients with intestinal adenocarcinoma contain aberrant hyper-methylations in the USP10 and ARF promoter regions with low expression of both proteins. | [119] | |
| Ovarian Cancer | Positive correlation with ARF The combined expression of low USP10 and ARF is displayed in cancer. The combined expression of low USP10 and ARF worsen overall survival rates. | High degree of methylation in USP10 and ARF CpG islands detected by methylation specific PCR analysis in ovarian cancer patients | [120] |
(Related to ‘4. Post-translational regulation of ARF in human cancer’).
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seo, J.; Seong, D.; Lee, S.R.; Oh, D.-B.; Song, J. Post-Translational Regulation of ARF: Perspective in Cancer. Biomolecules 2020, 10, 1143. https://doi.org/10.3390/biom10081143
AMA Style
Seo J, Seong D, Lee SR, Oh D-B, Song J. Post-Translational Regulation of ARF: Perspective in Cancer. Biomolecules. 2020; 10(8):1143. https://doi.org/10.3390/biom10081143
Chicago/Turabian StyleSeo, Jinho, Daehyeon Seong, Seung Ri Lee, Doo-Byoung Oh, and Jaewhan Song. 2020. "Post-Translational Regulation of ARF: Perspective in Cancer" Biomolecules 10, no. 8: 1143. https://doi.org/10.3390/biom10081143
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.