Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
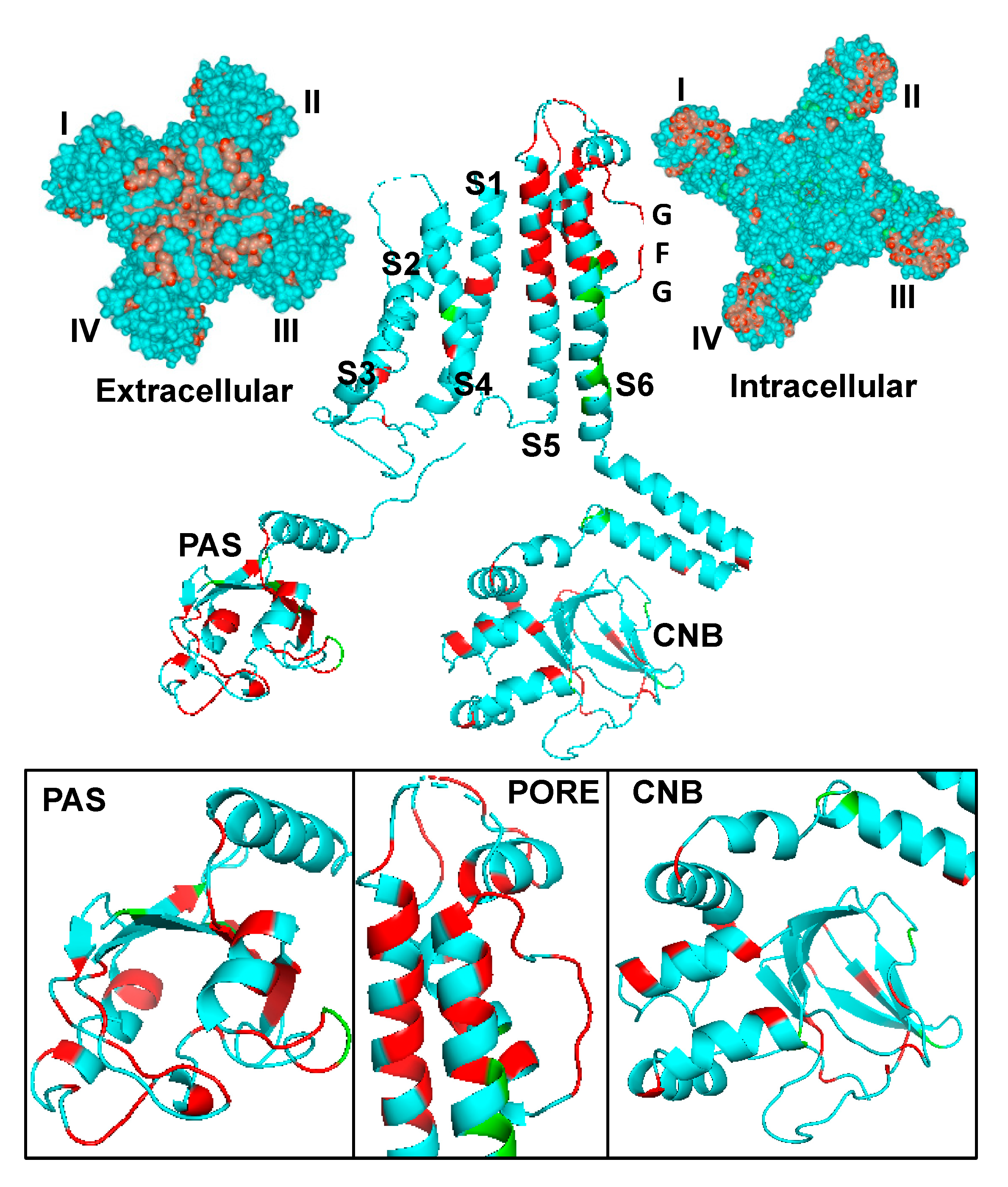
1.1. KCNH2, Kv11.1 and IKr
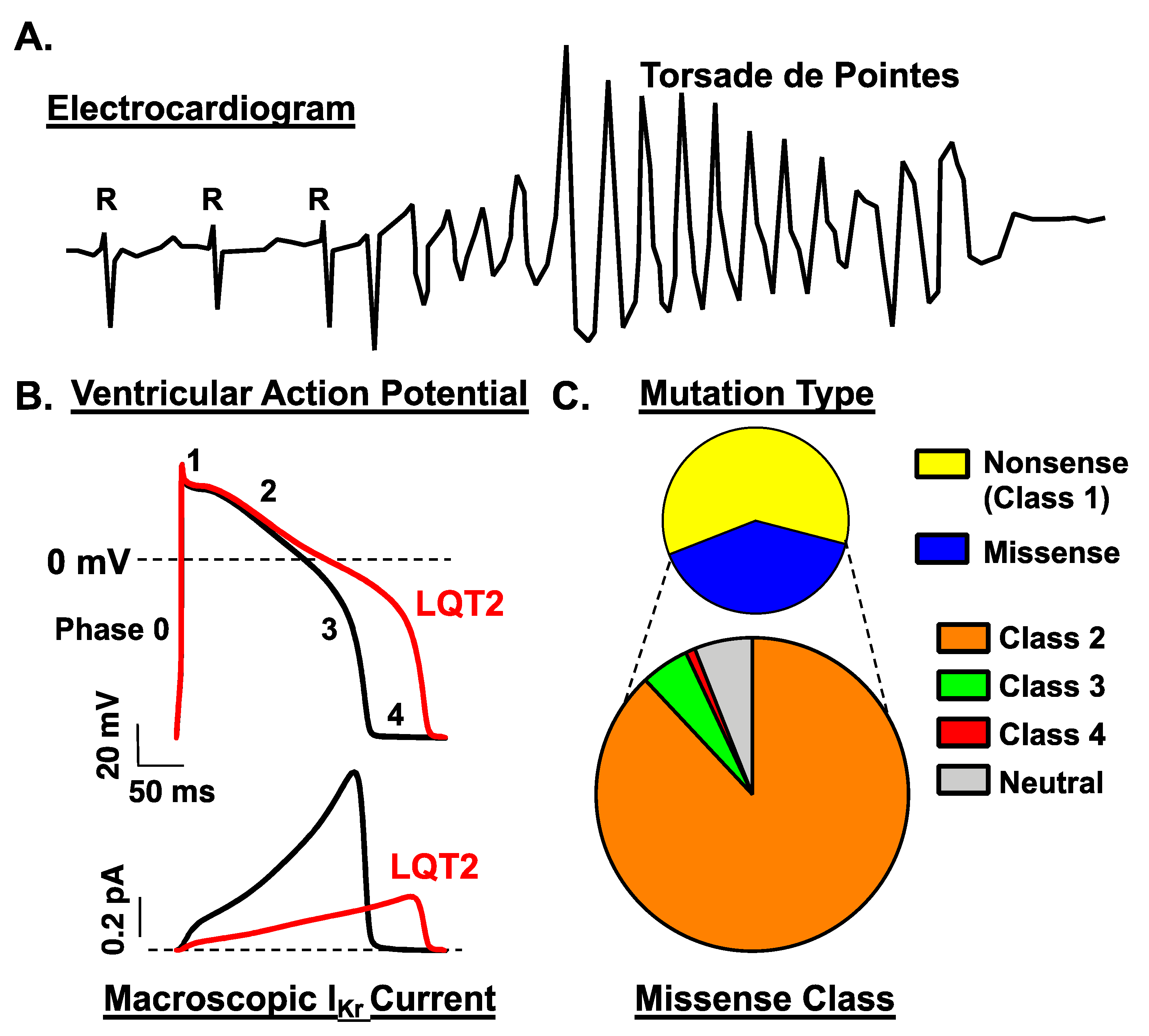
1.2. Long QT Syndrome Type 2
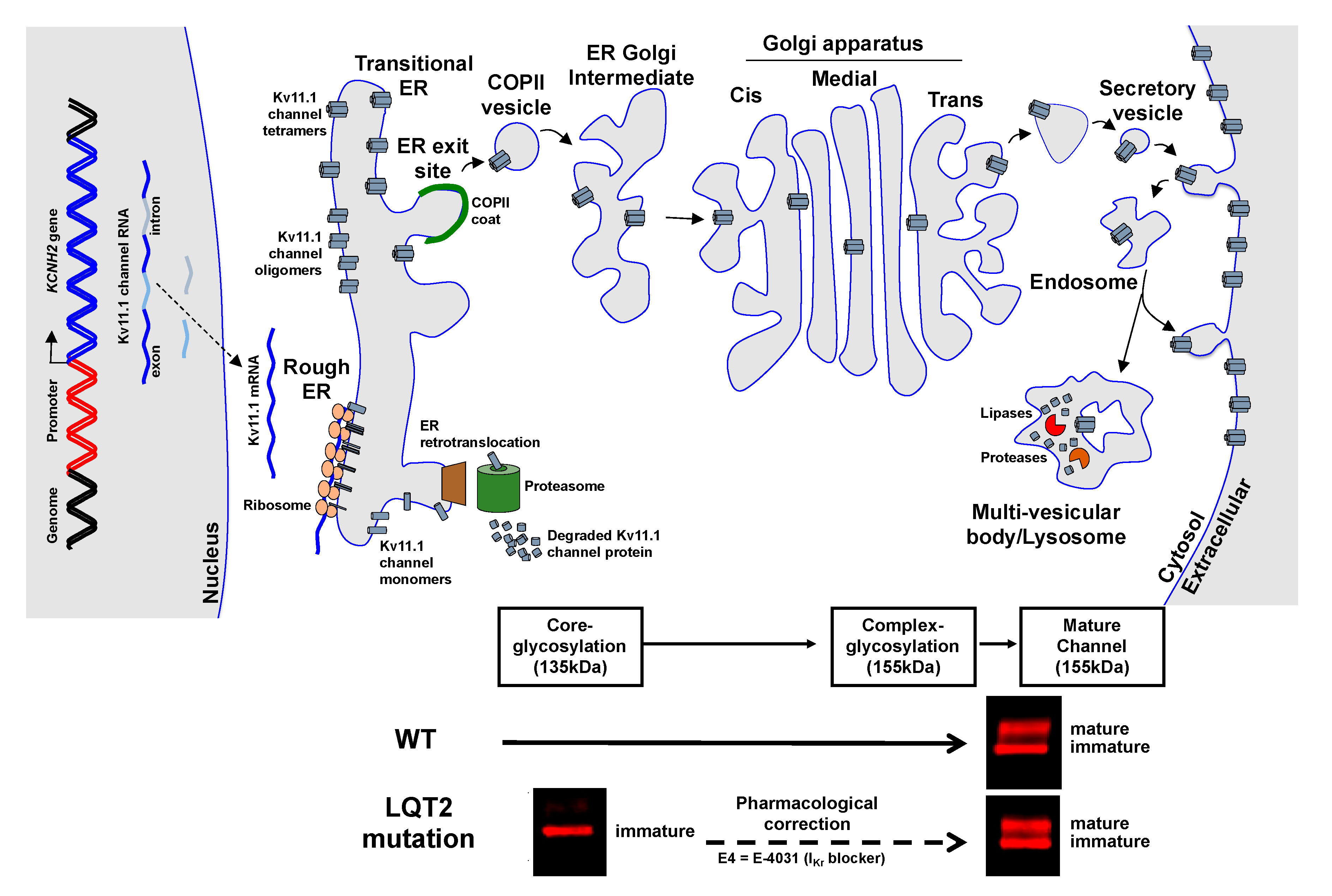
1.3. Novel Therapeutic Approaches to Treat LQT2
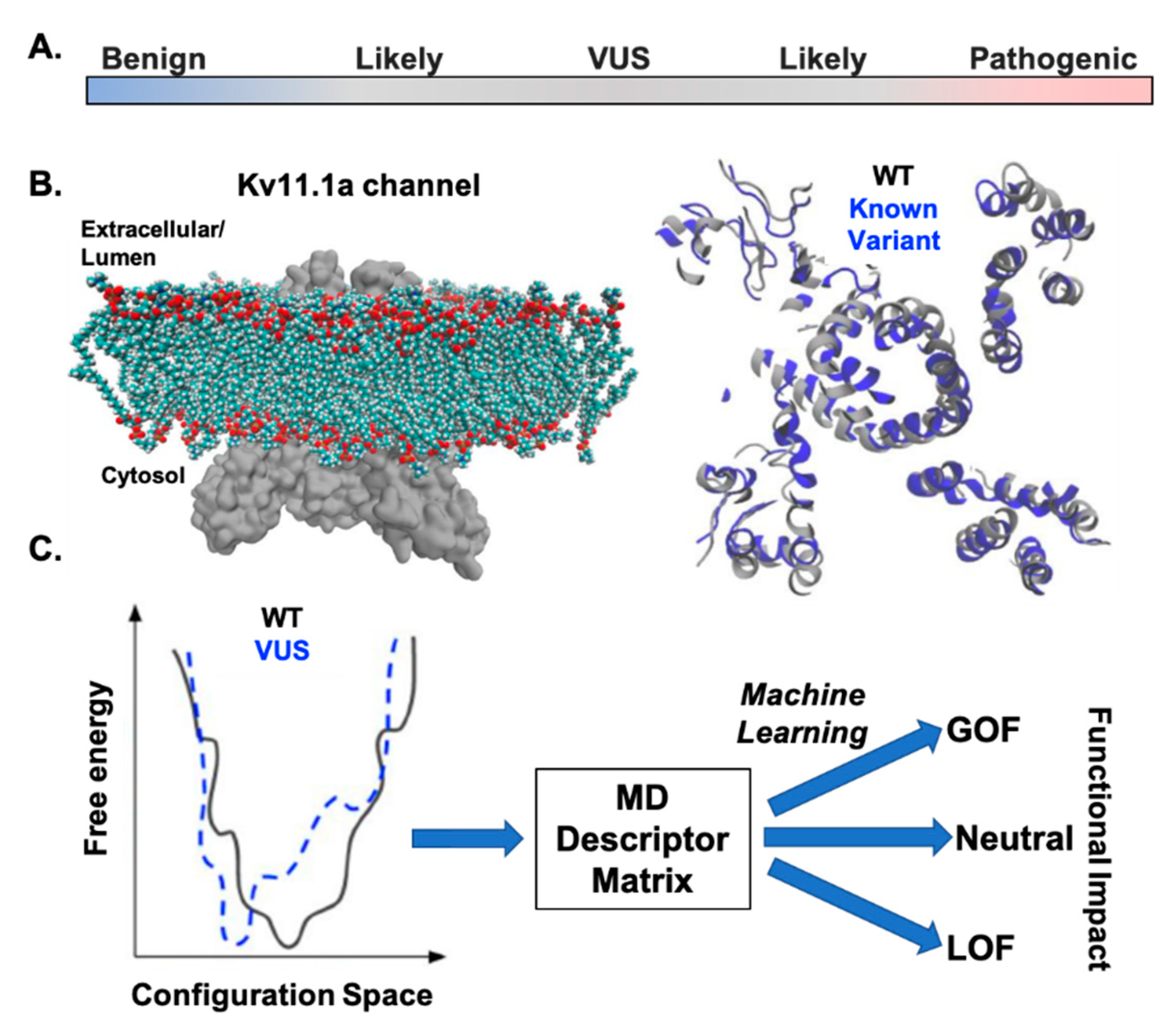
1.4. KCNH2 Variants of Uncertain Significance (VUS)
2. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Moss, A.J. Long QT syndrome. JAMA 2003, 289, 2041–2044. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Diagnostic criteria for the long Qt syndrome. An update. Circulation 1993, 88, 782–784. [Google Scholar] [CrossRef] [Green Version]
- El-Sherif, N.; Turitto, G.; Boutjdir, M. Congenital Long QT syndrome and torsade de pointes. Ann. Noninvasive Electrocardiol. 2017, 22, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, M.; Warmke, J.; Ganetzky, B.; Robertson, G. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 1995, 269, 92–95. [Google Scholar] [CrossRef]
- Wang, Q.; Curran, M.; Splawski, I.; Burn, T.C.; Millholland, J.; VanRaay, T.; Shen, J.; Timothy, K.; Vincent, G.; De Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Donger, C.; Denjoy, I.; Berthet, M.; Neyroud, N.; Cruaud, C.; Bennaceur, M.; Chivoret, G.; Schwartz, K.; Coumel, P.; Guicheney, P. KVLQT1 C-terminal missense mutation causes a forme fruste long-QT syndrome. Circulation 1997, 96, 2778–2781. [Google Scholar] [CrossRef]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincen, G.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef] [Green Version]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive Summary: Hrs/Ehra/Aphrs expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013, 10, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Crotti, L.; Insolia, R. Long-Qt syndrome: From genetics to management. Circ. Arrhythmia Electrophysiol. 2012, 5, 868–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Chen, Q.; Towbin, J.A. Genetics, molecular mechanisms and management of long QT syndrome. Ann. Med. 1998, 30, 58–65. [Google Scholar] [CrossRef]
- Delisle, B.P.; Anson, B.D.; Rajamani, S.; January, C.T. Biology of cardiac Arrhythmias. Circ. Res. 2004, 94, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.L.; Anderson, C.L.; Burgess, D.E.; Elayi, C.S.; January, C.T.; Delisle, B.P. Molecular pathogenesis of long QT syndrome type. J. Arrhythmia 2016, 32, 373–380. [Google Scholar] [CrossRef] [Green Version]
- London, B.; Trudeau, M.C.; Newton, K.P.; Beyer, A.K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Satler, C.A.; Robertson, G.A. Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ. Res. 1997, 81, 870–878. [Google Scholar] [CrossRef]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 forms I Kr potassium channels with HERG and is associated with cardiac Arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, M.; Van Der Heyden, M.A.; Van Veen, T.A. Deciphering hERG channels: Molecular basis of the rapid component of the delayed rectifier potassium current. J. Mol. Cell. Cardiol. 2012, 53, 369–374. [Google Scholar] [CrossRef]
- Gong, Q.; Stump, M.R.; Dunn, A.R.; Deng, V.; Zhou, Z. Alternative splicing and Polyadenylation contribute to the generation of hERG1 C-terminal Isoforms. J. Boil. Chem. 2010, 285, 32233–32241. [Google Scholar] [CrossRef] [Green Version]
- Kupershmidt, S.; Snyders, D.J.; Raes, A.; Roden, D.M. A K+ channel splice variant common in human heart lacks a C-terminal domain required for expression of rapidly activating delayed rectifier current. J. Boil. Chem. 1998, 273, 27231–27235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, G.W. KCNE2 and the K+ channel. Channels 2012, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.D.; Krahn, A.D.; Ackerman, M.J.; Rohatgi, R.K.; Moss, A.J.; Nazer, B.; Tadros, R.; Gerull, B.; Sanatani, S.; Wijeyeratne, Y.D.; et al. Loss-of-Function KCNE2 Variants. Circ. Arrhythmia Electrophysiol. 2017, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Delisle, B.P.; Anson, B.D.; Kilby, J.A.; Will, M.L.; Tester, D.J.; Gong, Q.; Zhou, Z.; Ackerman, M.J.; January, C.T. Most Lqt2 mutations reduce Kv11.1 (Herg) current by a class 2 (Trafficking-Deficient) mechanism. Circulation 2006, 113, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.L.; Kuzmicki, C.E.; Childs, R.R.; Hintz, C.J.; Delisle, B.P.; January, C.T. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 2014, 5, 5535–5548. [Google Scholar] [CrossRef]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001. [Google Scholar]
- Wang, W.; MacKinnon, R. Cryo-EM structure of the open human ether-à-go-go related K+ channel hERG. Cell 2017, 169, 422–430. [Google Scholar] [CrossRef] [Green Version]
- Schulteis, C.T.; Nagaya, N.; Papazian, D.M. Subunit folding and assembly steps are interspersed during shaker potassium channel biogenesis. J. Boil. Chem. 1998, 273, 26210–26217. [Google Scholar] [CrossRef] [Green Version]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Boil. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Zhou, Z.; Gong, Q.; Ye, B.; Fan, Z.; Makielski, J.C.; Robertson, G.A.; January, C.T. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys. J. 1998, 74, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Foo, B.; Williamson, B.; Young, J.C.; Lukacs, G.L.; Shrier, A. hERG quality control and the long QT syndrome. J. Physiol. 2016, 594, 2469–2481. [Google Scholar] [CrossRef] [Green Version]
- Ficker, E.; Dennis, A.T.; Wang, L.; Brown, A.M. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel hERG. Circ. Res. 2003, 92, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delisle, B.P.; Underkofler, H.A.S.; Moungey, B.M.; Slind, J.K.; Kilby, J.A.; Best, J.M.; Foell, J.D.; Balijepalli, R.C.; Kamp, T.J.; January, C.T. Small GTPase determinants for the Golgi processing and plasmalemmal expression of human ether-a-go-go Related (hERG) K+Channels. J. Boil. Chem. 2008, 284, 2844–2853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Q.; Anderson, C.L.; January, C.T.; Zhou, Z. Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am. J. Physiol. Circ. Physiol. 2002, 283, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, B.; Barbier, C.; Guo, K.; Vasantharuban, J.; Lukacs, G.L.; Shrier, A. Mutation-specific peripheral and ER quality control of hERG channel cell-surface expression. Sci. Rep. 2019, 9, 10113–10121. [Google Scholar] [CrossRef] [Green Version]
- Kanner, S.A.; Jain, A.; Colecraft, H.M. Development of a high-throughput flow cytometry assay to monitor defective trafficking and rescue of Long QT2 Mutant hERG channels. Front. Physiol. 2018, 9, 397–407. [Google Scholar] [CrossRef]
- Perry, M.D.; Ng, C.A.; Mann, S.A.; Sadrieh, A.; Imtiaz, M.; Hill, A.P.; Vandenberg, J.I. Getting to the heart of Herg K(+) channel Gating. J. Physiol. 2015, 593, 2575–2585. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Zhang, L.; Vincent, G.M.; Horne, B.D.; Zhou, Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation 2007, 116, 17–24. [Google Scholar] [CrossRef] [Green Version]
- Stump, M.R.; Gong, Q.; Zhou, Z. LQT2 nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation. Am. J. Physiol. Circ. Physiol. 2013, 305, H1397–H1404. [Google Scholar] [CrossRef] [Green Version]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Gong, Q.; Epstein, M.L.; January, C.T. HERG channel dysfunction in human Long QT syndrome. J. Boil. Chem. 1998, 273, 21061–21066. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zou, A.; Splawski, I.; Keating, M.T.; Sanguinetti, M.C. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J. Boil. Chem. 1999, 274, 10113–10118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakana, Y.; Takai, S.; Nakajima, K.-I.; Tani, K.; Yamamoto, A.; Watson, P.; Stephens, D.; Hauri, H.-P.; Tagaya, M. Bap31 is an itinerant protein that moves between the Peripheral Endoplasmic Reticulum (ER) and a Juxtanuclear compartment Related to ER-associated degradation. Mol. Boil. Cell 2008, 19, 1825–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.L.; Reloj, A.R.; Nataraj, P.S.; Bartos, D.C.; Schroder, E.A.; Moss, A.J.; Ohno, S.; Horie, M.; Anderson, C.L.; January, C.T.; et al. Pharmacological correction of long QT-linked mutations in KCNH2 (hERG) increases the trafficking of Kv11.1 channels stored in the transitional endoplasmic reticulum. Am. J. Physiol. Physiol. 2013, 305, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ficker, E.; Obejero-Paz, C.A.; Zhao, S.; Brown, A.M. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2ether-a-gogo-related gene (HERG) Mutations. J. Boil. Chem. 2001, 277, 4989–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furutani, M.; Trudeau, M.C.; Hagiwara, N.; Seki, A.; Gong, Q.; Zhou, Z.; Imamura, S.; Nagashima, H.; Kasanuki, H.; Takao, A.; et al. Novel mechanism associated with an inherited cardiac arrhythmia: Defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation 1999, 99, 2290–2294. [Google Scholar] [CrossRef]
- Hall, A.R.; Anderson, C.L.; Smith, J.L.; Mirshahi, T.; Elayi, C.S.; January, C.T.; Delisle, B.P. Visualizing mutation-specific differences in the trafficking-deficient Phenotype of Kv11.1 proteins linked to long QT syndrome type. Front. Physiol. 2018, 9, 584–595. [Google Scholar] [CrossRef] [PubMed]
- Ficker, E.; Thomas, D.; Viswanathan, P.C.; Dennis, A.T.; Priori, S.G.; Napolitano, C.; Memmi, M.; Wible, B.A.; Kaufman, E.S.; Iyengar, S.K.; et al. Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am. J. Physiol. Circ. Physiol. 2000, 279, 1748–1756. [Google Scholar] [CrossRef]
- Hirsch, C.; Ploegh, H.L. Intracellular targeting of the proteasome. Trends Cell Boil. 2000, 10, 268–272. [Google Scholar] [CrossRef]
- Gong, Q.; Keeney, D.R.; Molinari, M.; Zhou, Z. Degradation of trafficking-defective long QT syndrome type II mutant channels by the ubiquitin-proteasome Pathway. J. Boil. Chem. 2005, 280, 19419–19425. [Google Scholar] [CrossRef] [Green Version]
- Ficker, E.; Dennis, A.T.; Obejero-Paz, C.A.; Castaldo, P.; Taglialatela, M.; Brown, A.M. Retention in the endoplasmic reticulum as a mechanism of dominant-negative current suppression in human long QT syndrome. J. Mol. Cell. Cardiol. 2000, 32, 2327–2337. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Spazzolini, C.; Silvia, P.G.; Crotti, L.; Vicentini, A.; Landolina, M.; Gasparini, M.; Wilde, A.A.M.; Knops, R.E.; Denjoy, I.; et al. Who are the long-QT syndrome patients who receive an implantable cardioverter-defibrillator and what happens to them? Circulation 2010, 122, 1272–1282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Gong, Q.; January, C.T. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J. Boil. Chem. 1999, 274, 31123–31126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lees-Miller, J.P.; Duan, Y.; Teng, G.Q.; Duff, H.J. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: Involvement of S6 sites. Mol. Pharmacol. 2000, 57, 367–374. [Google Scholar] [PubMed]
- Mitcheson, J.S.; Chen, J.; Lin, M.; Culberson, C.; Sanguinetti, M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 2000, 97, 12329–12333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delisle, B.P.; Slind, J.K.; Kilby, J.A.; Anderson, C.L.; Anson, B.D.; Balijepalli, R.C.; Tester, D.J.; Ackerman, M.J.; Kamp, T.J.; January, C.T. Intragenic suppression of trafficking-defective KCNH2 channels associated with long QT syndrome. Mol. Pharmacol. 2005, 68, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C. HERG1 channel agonists and cardiac arrhythmia. Curr. Opin. Pharmacol. 2013, 15, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Qile, M.; Beekman, H.D.; Sprenkeler, D.J.; Houtman, M.J.; Van Ham, W.B.; Stary-Weinzinger, A.; Beyl, S.; Hering, S.; Berg, D.V.D.; De Lange, E.C.M.; et al. LUF7244, an allosteric modulator/activator of K v 11.1 channels, counteracts dofetilide-induced torsades de pointes arrhythmia in the chronic atrioventricular block dog model. Br. J. Pharmacol. 2019, 176, 3871–3885. [Google Scholar] [CrossRef] [Green Version]
- Qile, M.; Ji, Y.; Golden, T.D.; Houtman, M.J.; Romunde, F.; Fransen, D.; Van Ham, W.B.; Ijzerman, A.P.; January, C.T.; Heitman, L.H.; et al. LUF7244 plus Dofetilide rescues aberrant Kv11.1 trafficking and produces functional IKv11. Mol. Pharmacol. 2020, 97, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Aridor, M.; Hannan, L.A. Traffic Jam: A compendium of human diseases that affect intracellular transport processes. Traffic 2000, 1, 836–851. [Google Scholar] [CrossRef]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef]
- Welsh, M.J.; Smith, A.E. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993, 73, 1251–1254. [Google Scholar] [CrossRef]
- McClure, M.L.; Barnes, S.; Brodsky, J.L.; Sorscher, E.J. Trafficking and function of the cystic fibrosis transmembrane conductance regulator: A complex network of posttranslational modifications. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, 719–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serohijos, A.W.R.; Hegedűs, T.; Aleksandrov, A.A.; He, L.; Cui, L.; Dokholyan, N.V.; Riordan, J.R. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. USA 2008, 105, 3256–3261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukacs, G.L.; Chang, X.B.; Bear, C.; Kartner, N.; Mohamed, A.; Riordan, J.R.; Grinstein, S. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J. Boil. Chem. 1993, 268, 21592–21598. [Google Scholar]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [Green Version]
- Connett, G. Lumacaftor-ivacaftor in the treatment of cystic fibrosis: Design, development and place in therapy. Drug Des. Dev. Ther. 2019, 13, 2405–2412. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Ramachandra, C.J.A.; Singh, P.; Chitre, A.; Lua, C.H.; Mura, M.; Crotti, L.; Wong, P.; Schwartz, P.J.; Gnecchi, M.; et al. Identification of a targeted and testable antiarrhythmic therapy for long-QT syndrome type 2 using a patient-specific cellular model. Eur. Hear. J. 2017, 39, 1446–1455. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Gnecchi, M.; Dagradi, F.; Castelletti, S.; Parati, G.; Spazzolini, C.; Sala, L.; Crotti, L. From patient-specific induced pluripotent stem cells to clinical translation in long QT syndrome type. Eur. Hear. J. 2019, 40, 1832–1836. [Google Scholar] [CrossRef]
- Braam, S.; Tertoolen, L.; Casini, S.; Matsa, E.; Lu, H.; Teisman, A.; Passier, R.; Denning, C.; Gallacher, D.; Towart, R.; et al. Repolarization reserve determines drug responses in human pluripotent stem cell derived cardiomyocytes. Stem Cell Res. 2013, 10, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, E.S.; Ficker, E. Is restoration of intracellular trafficking clinically feasible in the long QT syndrome?: The example of HERG mutations. J. Cardiovasc. Electrophysiol. 2003, 14, 320–322. [Google Scholar] [CrossRef]
- Shah, S.H.; Arnett, D.; Houser, S.R.; Ginsburg, G.S.; Macrae, C.; Mital, S.; Loscalzo, J.; Hall, J.L. Mhs opportunities for the cardiovascular community in the precision medicine initiative. Circulation 2016, 133, 226–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council Committee on, A. Framework for Developing a New Taxonomy of Disease. The National Academies Collection: Reports Funded by National Institutes of Health. In Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Kapa, S.; Tester, D.J.; Salisbury, B.A.; Harris-Kerr, C.; Pungliya, M.S.; Alders, M.; Wilde, A.A.M.; Ackerman, M.J. Genetic testing for long-QT syndrome: Distinguishing pathogenic mutations from benign variants. Circulation 2009, 120, 1752–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taggart, N.W.; Haglund, C.M.; Tester, D.J.; Ackerman, M.J. Diagnostic miscues in congenital long-QT syndrome. Circulation 2007, 115, 2613–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, P.J.; Ackerman, M.J.; George, A.L.; Wilde, A.A. Impact of genetics on the clinical management of Channelopathies. J. Am. Coll. Cardiol. 2013, 62, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tester, D.J.; Schwartz, P.J.; Ackerman, M.J. Congenital long QT syndrome. Electr. Dis. Heart 2013, 3, 439–468. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Wells, Q.S.; Stallings, S.; Bush, W.S.; Gordon, A.; Nickerson, D.A.; Kim, J.H.; Crosslin, D.R.; Jarvik, G.P.; Carrell, D.S.; et al. Association of Arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA 2016, 315, 47–57. [Google Scholar] [CrossRef]
- Gaba, P.; Bos, J.M.; Cannon, B.C.; Cha, Y.-M.; Friedman, P.A.; Asirvatham, S.J.; Ackerman, M.J. Implantable cardioverter-defibrillator explantation for overdiagnosed or overtreated congenital long QT syndrome. Hear. Rhythm. 2016, 13, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. Hrs/Ehra expert consensus statement on the state of genetic testing for the Channelopathies and Cardiomyopathies. This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011, 8, 1308–1339. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Boil. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Vanoye, C.G.; George, A.L. Decoding KCNH2 variants of unknown significance. Hear. Rhythm. 2020, 17, 501–502. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.-A.; Perry, M.D.; Liang, W.; Smith, N.J.; Foo, B.; Shrier, A.; Lukacs, G.L.; Hill, A.P.; Vandenberg, J. High-throughput phenotyping of heteromeric human ether-à-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants. Hear. Rhythm. 2020, 17, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Routes, T.C.; Eckhardt, L.L.; Delisle, B.P.; January, C.T.; Kamp, T.J. A rapid solubility assay of protein domain misfolding for pathogenicity assessment of rare DNA sequence variants. Genet. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Alexov, E. Investigating the linkage between disease-causing amino acid variants and their effect on protein stability and binding. Proteins Struct. Funct. Bioinform. 2016, 84, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyulkhandanyan, A.; Rezaie, A.R.; Roumenina, L.; Lagarde, N.; Fremeaux-Bacchi, V.; Miteva, M.A.; Villoutreix, B.O. Analysis of protein missense alterations by combining sequence- and structure-based methods. Mol. Genet. Genom. Med. 2020, 8, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Changeux, J.-P.; Christopoulos, A. Allosteric modulation as a unifying mechanism for receptor function and regulation. Diabetes Obes. Metab. 2017, 19, 4–21. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Eisenhaber, F.; Persson, B.; Argos, P. Protein structure prediction: Recognition of primary, secondary, and tertiary structural features from amino acid sequence. Crit. Rev. Biochem. Mol. Boil. 1995, 30, 1–94. [Google Scholar] [CrossRef]
- Sowmya, G.; Ranganathan, S. Protein-protein interactions and prediction: A comprehensive overview. Protein Pept. Lett. 2014, 21, 779–789. [Google Scholar] [CrossRef]
- Ben-Bassat, A.; Giladi, M.; Haitin, Y. Structure of KCNH2 cyclic nucleotide-binding homology domain reveals a functionally vital salt-bridge. J. Gen. Physiol. 2020, 152, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Muskett, F.W.; Thouta, S.; Thomson, S.J.; Bowen, A.; Stansfeld, P.J.; Mitcheson, J.S. Mechanistic insight into Humanether-à-go-go-related Gene (hERG) K+Channel deactivation Gating from the solution structure of the EAG domain. J. Boil. Chem. 2010, 286, 6184–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaixo, R.; Harley, C.A.; Castro-Rodrigues, A.F.; Morais-Cabral, J.H. Structural properties of PAS Domains from the KCNH Potassium Channels. PloS ONE 2013, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohning, A.E.; Levonis, S.M.; Williams-Noonan, B.; Schweiker, S.S. A Practical guide to molecular docking and homology modelling for medicinal chemists. Curr. Top. Med. Chem. 2017, 17, 2023–2040. [Google Scholar] [CrossRef] [Green Version]
- Nurisso, A.; Daina, A.; Walker, R.C. A Practical introduction to molecular dynamics simulations: Applications to homology modeling. Adv. Struct. Saf. Stud. 2011, 857, 137–173. [Google Scholar] [CrossRef]
- Miranda, W.E.; Demarco, K.R.; Guo, J.; Duff, H.J.; Vorobyov, I.; Clancy, C.E.; Noskov, S.Y. Selectivity filter modalities and rapid inactivation of the hERG1 channel. Proc. Natl. Acad. Sci. USA 2020, 117, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Man Yip, K.; Fischer, N.; Paknia, E.; Chari, A.; Stark, H. Breaking the next Cryo-Em resolution barrier—atomic resolution determination of proteins! bioRxiv 2020, 2020, 1–21. [Google Scholar] [CrossRef]
- Sula, A.; Booker, J.; Ng, L.C.T.; Naylor, C.E.; DeCaen, P.G.; Wallace, B. The complete structure of an activated open sodium channel. Nat. Commun. 2017, 8, 14205–14224. [Google Scholar] [CrossRef] [Green Version]
- Jing, Z.; Liu, C.; Cheng, S.Y.; Qi, R.; Walker, B.D.; Piquemal, J.-P.; Ren, P. Polarizable force fields for biomolecular simulations: Recent advances and applications. Annu. Rev. Biophys. 2019, 48, 371–394. [Google Scholar] [CrossRef]
- Demarco, K.R.; Bekker, S.; Vorobyov, I. Challenges and advances in atomistic simulations of potassium and sodium ion channel gating and permeation. J. Physiol. 2018, 597, 679–698. [Google Scholar] [CrossRef] [Green Version]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. Generalized born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the SC ’14 International Conference for High Performance Computing, Networking, Storage and Analysis, New Orleans, LA, USA, 16–21 November 2014. [Google Scholar]
- Kavakiotis, I.; Tsave, O.; Salifoglou, A.; Maglaveras, N.; Vlahavas, I.; Chouvarda, I. Machine learning and data mining methods in diabetes research. Comput. Struct. Biotechnol. J. 2017, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ono, M.; Burgess, D.E.; Schroder, E.A.; Elayi, C.S.; Anderson, C.L.; January, C.T.; Sun, B.; Immadisetty, K.; Kekenes-Huskey, P.M.; Delisle, B.P. Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients. Biomolecules 2020, 10, 1144. https://doi.org/10.3390/biom10081144
Ono M, Burgess DE, Schroder EA, Elayi CS, Anderson CL, January CT, Sun B, Immadisetty K, Kekenes-Huskey PM, Delisle BP. Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients. Biomolecules. 2020; 10(8):1144. https://doi.org/10.3390/biom10081144
Chicago/Turabian StyleOno, Makoto, Don E. Burgess, Elizabeth A. Schroder, Claude S. Elayi, Corey L. Anderson, Craig T. January, Bin Sun, Kalyan Immadisetty, Peter M. Kekenes-Huskey, and Brian P. Delisle. 2020. "Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients" Biomolecules 10, no. 8: 1144. https://doi.org/10.3390/biom10081144