 Qiao Yang1†
Qiao Yang1† Quanjun Hu
Quanjun Hu- 1Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, State Key Laboratory of Hydraulics and Mountain River Engineering, College of Life Sciences, Sichuan University, Chengdu, China
- 2State Key Laboratory of Grassland Agro-Ecosystem, Institute of Innovation Ecology, Lanzhou University, Lanzhou, China
Megacarpaea delavayi (Brassicaceae), a plant found the high mountains of southwest China at high altitudes (3000–4800 m), is used as a vegetable or medicine. Here, we report a draft genome for this species. The assembly genome of M. delavayi is 883 Mb, and 61.59% of the genome is composed of repeat sequences. Annotation of the genome identified a total of 41,114 protein-coding genes. We found that M. delavayi experienced an independent whole-genome duplication (WGD), paralleling those independent WGDs in Iberis, Biscutella, and Anastatica in the early Miocene. Phylogenetic analyses based on the single-copy genes confirmed the position of the genus Megacarpaea within the expanded lineage II of the family and resolved its basal divergence to a subclade consisting of Anastatica, Iberis, and Biscutella. Species-specific and fast-evolving genes in M. delavayi are mainly involved in “DNA repair” and “response to UV-B radiation.” These genetic changes may together help this species survive in high-altitude environments. The reference genome reported here provides a valuable resource for studying adaptation of this and other alpine plants to the high-altitude habitats.
Introduction
Polyploidy (whole-genome duplication, WGD), which occurs frequently through evolutionary histories of plants (Wu et al., 2020), contributes greatly to both species diversification and colonization of the new niches (Soltis and Soltis, 2016). Numerous independent WGDs were identified for angiosperm families, such as Asteraceae (Huang et al., 2016), Poaceae (McKain et al., 2016), and Brassicaceae (Edger et al., 2015). The Brassicaceae-specific WGD was named At-α WGDs (Bowers et al., 2003), which occurred 23–43 million years ago (Mya) (Mandáková et al., 2010). In addition, more independent WGDs were revealed to be specific to lineages or species within the family (Mandáková et al., 2017). It is increasingly clear that subsequent lineage-specific or species-specific WGD events laid the foundation for species diversification, environmental adaptability, and stress tolerance of the Brassicaceae species (Kagale et al., 2014).
In this study, we aimed to examine whether WGD occurred in one alpine herb, Megacarpaea delavayi (2n = 18) of Brassicaceae through sequencing its draft genome. This species grows on swampy meadows, steep grassy slopes, and open thickets of the high mountains (Hengduan Mountains) in southwest China at elevations of 3000–4800 m (Cheo et al., 2001). It has been collected as a wild vegetable and medicine for years by the local inhabitants in the high-altitude regions (Zhong et al., 2015). The dried plants of M. delavayi are used to treat dysentery, lung cough, and disordered indigestion by Bai and Tibetan people (Lei et al., 2009; Shen et al., 2009). Other species of the genus are distributed in the high-elevation regions from the Hengduan Mountains to Himalaya and central Asia (Cheo et al., 2001). However, phylogenetic relationships of the genus Megacarpae in the family Brassicaceae remain unclear although both recent studies suggested its likely position in the expanded lineage II of the family Brassicaceae (Guo et al., 2017; Nikolov et al., 2019). Maximum likelihood analyses based on targeted enrichment sequence data suggested the close relationship between Megacarpaea, Iberis, Cochlearia and others although this received little support according to coalescent analyses of these data (Nikolov et al., 2019). In addition, in these analyses, it remained unsolved whether Biscutella should be placed in this clade (Nikolov et al., 2019) although another study based on genome-scale single-copy genes suggested the well-supported close relationship between Biscutella and Iberis (Kiefer et al., 2019). Independent WGDs, which might have led to incorrect gene orthology alignments (Walker et al., 2017), seem to account for these conflicting phylogenies (Nikolov et al., 2019).
Here, we report the assembly and comparative genomic analysis of the M. delavayi genome. We revealed that one independent WGD occurred in this species in the early Miocene, paralleling those WGDs in other genera. We determined the phylogenetic relationship of the genus Megacarpaea based on genome-scale single-copy genes. We further found that numerous species-specific and fast-evolving genes existed in this species, which may be beneficial for its survival in the alpine habitat.
Materials and Methods
Plant Materials and Genomic DNA Extraction
One wild M. delavayi (2n = 18) individual (Figure 1A) was collected from Cangshan Mountain (3181 m, N25.659, E100.117) in Yunnan Province, China. Fresh and healthy leaves were immediately frozen at −80°C for DNA extraction. High-quality genomic DNA from leaf tissue was extracted with the CTAB method (Liu et al., 2009). We used 1% agarose gel electrophoresis to check the quality of the high-molecular-weight DNA. High-quality genomic DNA with an effective concentration of more than 2 nM was used to construct the library.
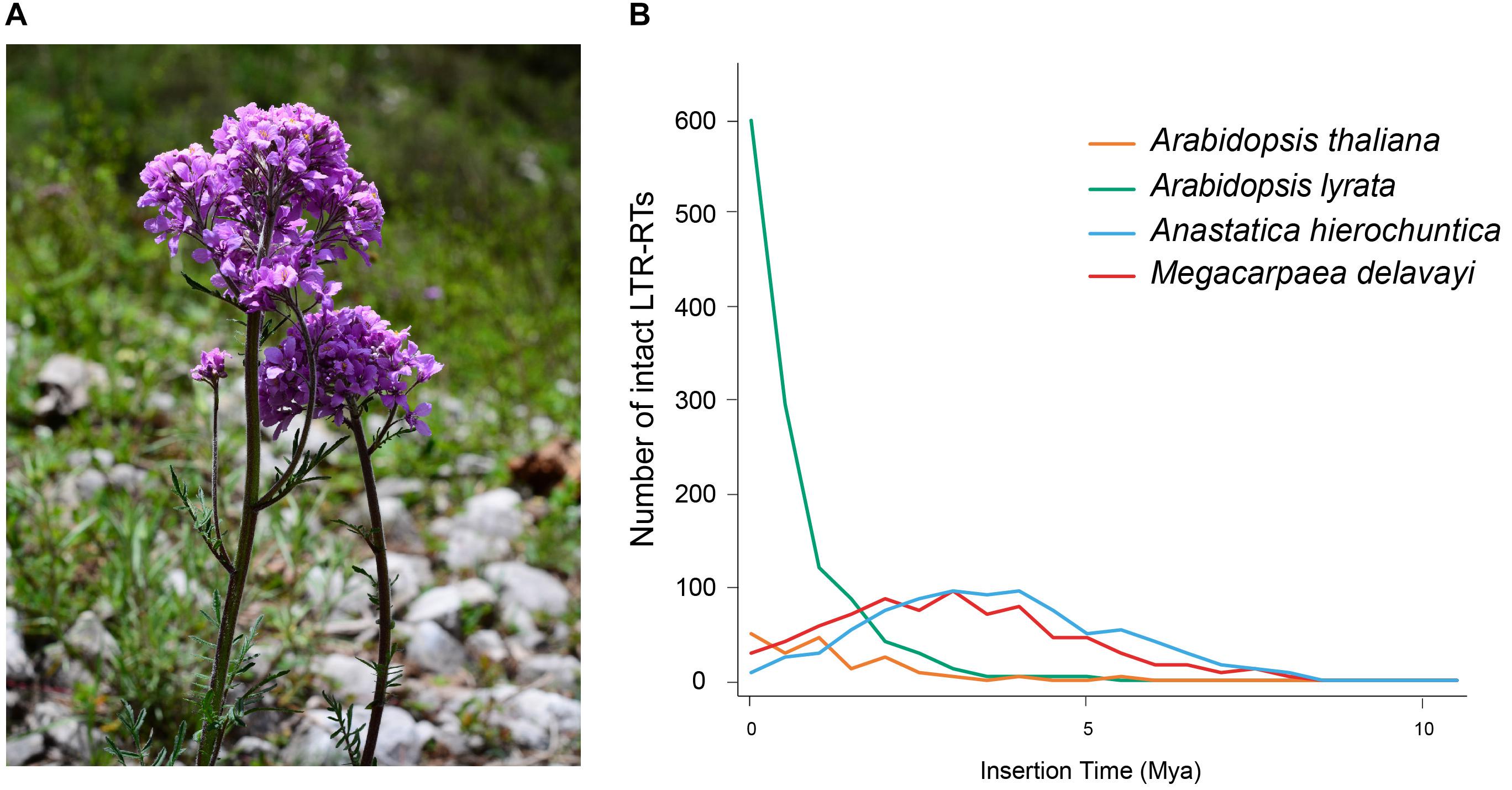
Figure 1. (A) M. delavayi in flowering stage of growth. Photo taken by Lei Zhang in Pingwu County (3,077 m a.s.l.), Sichuan Province, China. (B) Insertion time distribution of intact LTR-RTs of M. delavayi and other Brassicaceae species.
Genome Sequencing and Assembly
We constructed Illumina paired-end libraries with small (230, 500, and 800 bp) and large (2, 5, 10, and 20 kb) insert sizes and read lengths of 150 bp (Supplementary Table S1). We sequenced them using the Illumina HiSeq 2500 platform (Illumina, San Diego, CA, United States) at Novogene (Tianjin, China) following the manufacturer protocols. Short reads were first subjected to quality filtering with Trimmomatic v0.36 (Bolger et al., 2014), error correction with BFC v1.8 (Li, 2015), and mate-pair data deduplication with FastUniq v1.1 (Xu et al., 2012). The genome was initially assembled into scaffolds with Platanus v1.2.4 (Kajitani et al., 2014) with the first round of gap closing. An additional gap closing procedure was performed with GapCloser v1.12 (Luo et al., 2012). Finally, we evaluated genome assemblies for completeness using BUSCO v3.0.2 (Waterhouse et al., 2018) with “embryophyta_odb9” (Supplementary Table S2).
Genome Size Estimation
The sequencing reads were used to estimate genome size of M. delavayi based on k-mer frequencies. We used quality-filtered Illumina short reads and 17-mer frequency distribution to estimate the genome size with Jellyfish v2.2.9 (Marçais and Kingsford, 2011). The highest peak value of k-mer distribution was used to estimate the sequencing depth. We plotted the distribution of k-mer depth against the frequency with the main peak occurring at a depth of 19 (Supplementary Figure S1). A Perl script1 was used to calculate the size of the M. delavayi genome.
Repetitive Identification
We used a combination of homology-based and de novo search methods to identify repeat sequences in the M. delavayi genome. In terms of homology-based prediction, RepeatMasker v4.0.7 (Tarailo-Graovac and Chen, 2009) was used to find repeat elements at the DNA level with the Repbase library. De novo repeat annotation of the M. delavayi genome was performed with RepeatModeler v1.0.11 (Smit and Hubley, 2008).
We identified intact long-terminal repeat retrotransposons (LTR-RTs) by searching the M. delavayi genome with LTRharvest v1.5.10 (Ellinghaus et al., 2008; -motif tgca -motifmis 1) and LTR_Finder v1.06 (Xu and Wang, 2007; -D 20000 -d 1000 -L 5000 -l 100). Then, LTR_retriever v1.9 (Ou and Jiang, 2018) was used to integrate the results of LTR_Finder and LTRharvest (Supplementary Table S3). Using a substitution rate (r) of 7 × 10–9 substitutions per site per year (Ossowski et al., 2010), we calculated the insertion time (T) for each LTR retrotransposon as T = K/(2r), where K is genetic distance and r is the rate of nucleotide substitution per site per year (r = 7 × 10–9).
Gene Annotation
A combination of de novo, homology-based, and transcript-based approaches were used to predict protein-coding genes in the M. delavayi genome. Before the transcriptome could be aligned, RNA-Seq reads needed to be assembled into transcripts. We first filtered RNA-Seq reads with potential low-quality regions using Trimmomatic v0.36 (Bolger et al., 2014). After quality control was performed, all clean reads were assembled into de novo transcripts with Trinity v2.8.4 (Haas et al., 2013). Then, we used PASA v2.1.0 (Haas et al., 2003) to obtain information on the gene structure annotation by aligning the assembled transcripts with the genomes. Protein sequences of Aethionema arabicum, Arabidopsis lyrata, Arabidopsis thaliana, Brassica rapa, and Capsella rubella, were obtained for homology-based gene annotation. GlimmerHMM v3.0.4 (Majoros et al., 2004) was used to predict the gene structure in each protein-coding region. We performed de novo prediction with AUGUSTUS v3.2.3 (Stanke et al., 2006) to annotate protein-coding genes. The gene model parameters were trained from A. thaliana and our transcriptome data set. The above three gene prediction results were merged with EVidenceModeler v1.1.1 (Haas et al., 2008) to form a comprehensive and non-redundant reference gene list. Weights of evidence for gene models were defined as follows: de novo prediction weight (Augustus) = 1, homology-based prediction weight (GlimmerHMM) = 5, transcript-based prediction weight (PASA) = 10. The EVM merged result was updated with an additional round of PASA annotation to add UTRs and provide information on alternative splicing variants to gene models.
To obtain functional annotation of protein-coding genes, we used Blast2GO v2.5 (Conesa and Götz, 2008) for gene ontology (GO) annotation based on the NCBI-NR database. The predicted genes were mapped to KEGG pathways using KAAS (Moriya et al., 2007) to obtain the KEGG annotation. For Swiss-Prot annotations, we employed BLAST + v2.2.31 (Camacho et al., 2009; Blastp with the E-value cutoff 1 × 10−5) to align proteins to the Swiss-Prot databases. InterProScan v5.31-70 was used to determine the domains/motifs (Jones et al., 2014; Supplementary Table S4).
Gene Family Identification
We downloaded the protein-coding genes of Arabis alpina, Boechera stricta, Crucihimalaya himalaica, Eutrema heterophyllum, and Lepidium meyenii together with Megacarpaea delavayi to identify orthologous groups (Supplementary Table S5). To remove redundancy caused by alternative splicing variations, we retained only the gene models at each gene locus that encoded the longest protein sequence, and putative fragmented genes that encoded protein sequences shorter than 50 aa and stop codon ratios greater than 20% were filtered out. Then, we used Diamond v0.9.22 (Buchfink et al., 2014; E-value cutoff 1 × 10−5) to compare all filtered protein sequences and used OrthoMCL v2.0.9 (Li et al., 2003) to cluster genes into orthologous groups. Genes that could not be clustered into any gene family and for which only one species existed were considered species specific. Finally, we summarized the gene family cluster results for six species in Venn diagram format.
Phylogenetic Analyses
We downed the reported genomes of all related species from two main lineages of Brassiaceae, including Aethionema arabicum, Anastatica hierochuntica, Arabidopsis lyrate, Arabidopsis thaliana, Biscutella auriculate, Biscutella laevigata, Brassica rapa, Boechera stricta, Crucihimalaya himalaica, Eutrema heterophyllum, Iberis amara, Kernera saxatilis, Lepidium meyenii, Macropodium nivale, and Noccaea caerulescens (Supplementary Table S5). We used the single-copy orthologous genes identified in the gene family cluster analyses from these species and M. delavayi to construct phylogenetic tree. Multiple sequence alignments were performed for the protein sequence of each single-copy orthologous gene with MAFFT v7.313 (Katoh and Standley, 2013). Then, the alignments were concatenated to generate a super alignment matrix, which was used to generate a maximum likelihood tree with the PROTGAMMAILGX model in RAxML v8.2.11 (Stamatakis, 2014).
The divergence time between these species was estimated with the MCMCtree program in PAML v4.9 (Yang, 2007). The F84 model (model = 4) and independent rates molecular clock (clock = 2) were used for calculations in MCMCtree. The MCMC process was run for 1,500,000 iterations, with a sample frequency of 150, after a burn-in of 500,000 iterations. We ran the program twice for each data type to confirm that the results were convergent between runs. We looked up three calibration points in the TimeTree database (Kumar et al., 2017) to estimate the Brassicaceae divergence time: divergence time for Ae. arabicum and other Brassicaceae plants was 32–43 Mya, divergence time for Lineages II+ expanded lineage II and lineage I was 23.4–33.5 Mya, divergence time for L. meyenii and other lineage I plants was 11.9–20.6 Mya. The phylogenetic analyses for Brassicaceae was visualized with FigTree v1.4.32.
Gene Family Expansion and Contraction
CAFÉ (De Bie et al., 2006) is a tool for analyzing evolutionary changes of the gene families. This software uses the stochastic birth and death process to model gene gain and loss over a phylogeny. Based on the results of phylogeny and divergence time, we applied CAFE v4.2 to identify gene families that had undergone expansion and contraction on the phylogeny tree (p-value cutoff 0.05). For each significantly expanded and contracted gene family in M. delavayi, we inferred functional information based on its functional annotations.
WGD and Positively Selected Genes
We used MCScanX (Wang et al., 2012) to detect syntenic blocks (regions with at least five collinear genes) and duplication levels (depth) for four species: A. hierochuntica, A. thaliana, B. rapa, and M. delavayi. To recover the WGD event, we calculated synonymous substitution rates (Ks) for syntenic genes using codeml in PAML. To further examine whether the recent WGDs were shared by M. delavayi and the closely related species, we extracted homologous gene groups to construct phylogenetic trees of the orthologous genes. We used phylogenetic relationships of the homologous genes identified the likely WGD nodes.
To identify fast-evolving genes, we used MCScanX to search for syntenic blocks. Similar to previous research (Wu et al., 2019), we calculated non-synonymous substitution (Ka) and synonymous substitution (Ks) for the collinear orthologous gene pairs using the Perl script “add_ka_and_ks_to_collinearity.pl” in MCScanX. The ratio of Ka to Ks is a commonly used indicator of selective pressure acting on protein-coding genes with a ratio >1 representing positive selection.
Results
Genome Size Estimation
The distribution of short subsequence (k-mer) frequency, also known as the k-mer spectrum, is widely used to estimate genome size (Li et al., 2013; Zhang et al., 2015). A k-mer depth distribution was obtained from Jellyfish (Marçais and Kingsford, 2011) analyses, and the peak depth was clearly visible from the distribution data (Supplementary Figure S1). The genome size of M. delavayi calculated by the aforementioned Perl script was estimated to be approximately 899 Mb (Table 1).

Table 1. Statistics for the M. delavayi genome assembly and annotation.
Genome Assembly and Annotation
To sequence the whole genome of M. delavayi, we generated 133 Gb of paired-end and mate-pair clean reads (150 × assembled sequence coverage) with different insert sizes using an Illumina HiSeq 2500 platform (Supplementary Table S1). The final assembly genome of M. delavayi (883.81 Mb) consisted of 763,815 contigs (contig N50, 65.48 kb; longest contig, 794.13 kb; Table 1). Completeness of the genome regions was further assessed with BUSCO. Of a core set of 1440 single-copy ortholog genes of the Embryophyta lineage, 94.5% were complete in the genome, which contained 85.6% of the plant single-copy orthologs and 8.9% of the plant duplicate orthologs (Supplementary Table S2), which suggests that the genome of M. delavayi was well-assembled with high completeness and accuracy.
De novo, homology-based, and transcript-based approaches were combined to annotate protein-coding sequences (Chen et al., 2020; Kang et al., 2020; Zhang et al., 2020). In total, 41,114 genes were predicted with an average gene length and number of exons of 1853 bp and 6.57, respectively (Table 1). Moreover, we annotated functions of the predicted genes with Swiss-Prot, InterProScan, GO, and KEGG databases. The results showed that 85.78% of all the protein-coding genes were successfully annotated by at least one database (Supplementary Table S4).
Repetitive Elements Analysis
Through combination of de novo searches and homology-based methods, we identified nearly 536 Mb repetitive elements, representing 61.59% of the M. delavayi genome (Table 2). Retrotransposons (LTR, SINEs, and LINEs) were the most abundant, accounting for 44.06% of the genome. LTR-RTs represented 39.25% of the genome, and Ty3/Gypsy (24.71%) made up major elements of LTR-RTs (Table 2).
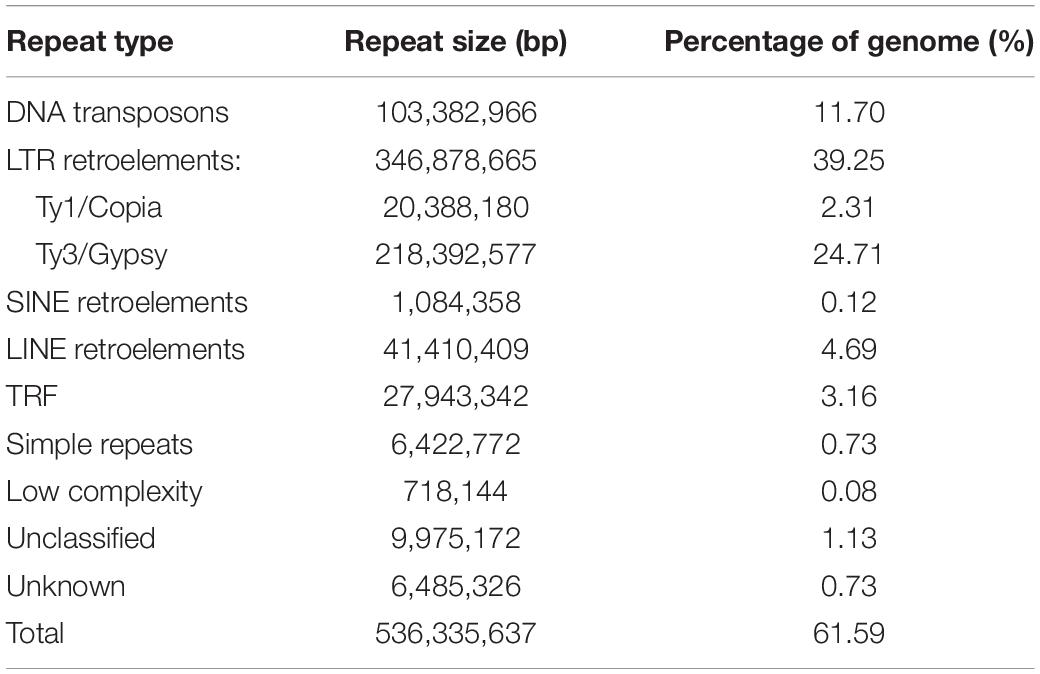
Table 2. Repeat content (subtypes) of M. delavayi genome.
A high proportion of repetitive elements in the M. delavayi genome were LTR-RTs, and the proliferation of retrotransposons might have been responsible for genome expansion (Zhang et al., 2019). To estimate insertion time of the LTR-RTs, we identified complete LTR-RTs in four Brassicaceae species (A. hierochuntica, A. thaliana, A. lyrate, and M. delavayi). We identified 814 complete LTR-RTs in the M. delavayi genome and 176 in A. thaliana and 1227 in A. lyrate (Supplementary Table S3). A. thaliana had more microdeletions in transposons than M. delavayi (Hu et al., 2011), and A. lyrata had a comparatively higher proportion of recent insertions (Slotte et al., 2013), consistent with our study. The insertion time distribution showed that M. delavayi LTR-RTs expanded within the past 5 million years (based on r of 7 × 10–9 substitutions per site per year; Figure 1B). In general, recent expansion of repeat sequences may have played a key role in increasing the genome size of M. delavayi.
Phylogenetic Analyses
Sixteen species were selected to identify orthologous groups, and they were clustered into 51,589 orthologous groups. A total of 361 single-copy gene families were identified and used to construct the maximum likelihood phylogenetic tree. Three major lineages were identified: lineages I, and traditionally recognized lineages II and those added to the expanded lineage II. Phylogenetic analysis confirmed the phylogenetic position of M. delavayi in the expanded lineage II of Brassicaceae (Figure 2A), consistent with previous published research (Guo et al., 2017; Kiefer et al., 2019; Nikolov et al., 2019). Within this lineage, M. delavayi diverged early as one subclade, and another comprised Anastatica, Iberis and Biscutella as suggested before based on the genome-scale single-copy genes. The close relationships between the latter three genera agree with phylogenetic analyses similarly based on the genome-scale, single-copy genes (Kiefer et al., 2019). Based the fossil-calibrated phylogeny, M. delavayi diverged from other three genera of the expanded lineage II around 20.53 Mya (Supplementary Figure S2).
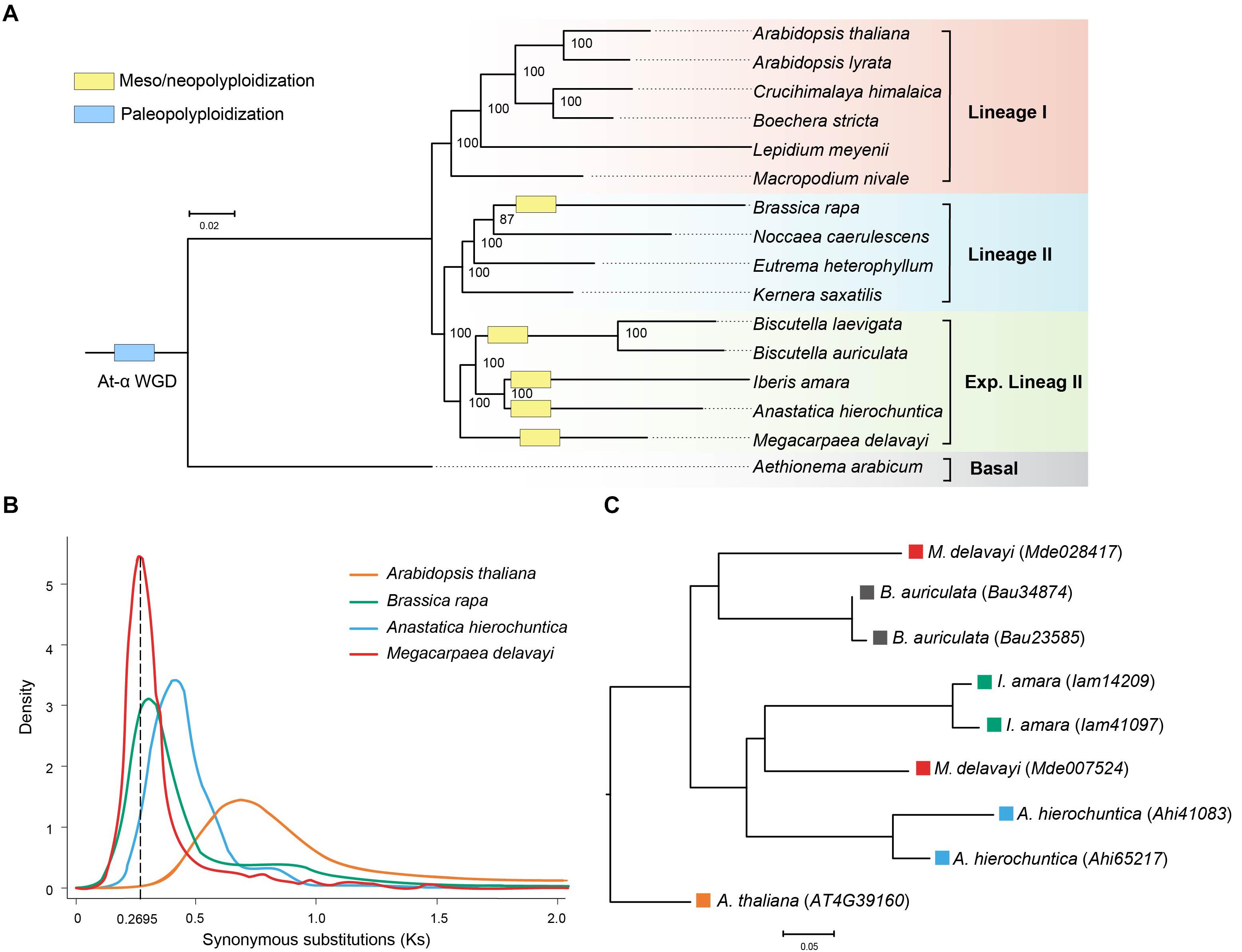
Figure 2. (A) Phylogenetic relationship of M. delavayi and other Brassicaceae species. Numbers on nodes represent bootstrap values. (B) Synonymous substitutions (Ks) estimated from M. delavayi-specific WGD. (C) A phylogenetic tree of the homologous genes from the expanded lineage II.
Whole-Genome Duplication
The distribution of Ks was analyzed to uncover and assess the frequency of WGD events in the Brassicaceae (Kagale et al., 2014). Using syntenic orthologs within each genome to construct the distribution of Ks, we found M. delavayi had undergone a further more species-specific WGD (Figure 2B) after the well-known ancient At-α (23–47 Mya) paleopolyploid WGD. Although the Brassicaceae diverged from other closely related eudicots at the beginning of the Cenozoic era, the rapid species diversification of the family occurred only within the Miocene (<23 Mya; Kagale et al., 2014). Importantly, the lineage- or species-specific polyploid or WGD events seemed to have promoted species diversification during this recent stage (Kagale et al., 2014; Kiefer et al., 2019). Four genera, Brassica, Anastatica, Iberis, and Biscutella, of the expanded lineage II were suggested to experience independent WGDs (Wang et al., 2011; Kiefer et al., 2019). We examined Ks distributions of these species. We confirmed these WGDs and found that they occurred between 16 and 23 Mya almost at the same stage after their divergences (Figure 2B). These WGDs seem to occur independently based on the Ks distributions despite the slight differences between them. It remains interesting to further examine whether Megacarpaea shared a WGD with the closely related Anastatica, Iberis, and Biscutella based the homologous genes. After the WGD, the duplicated gene may have been randomly lost in the derived lineages, and therefore, it is difficult to identify all paralogous genes between different lineages. We, therefore, used Arabidopsis without further WGD as an out-group. We extracted a set of homologous gene groups: Arabidopsis (1): Megacarpaea (2): Anastatica (2): Iberis (2): Biscutella (2). We recovered 24 groups of homologous genes, but phylogenetic analyses suggested that only one group could be used to construct a gene tree with most subclades statistically supported (Figure 2C). On this tree, two genes from Megacarpaea did not cluster together, and their clustering with other subclades failed to receive statistical support. Because two genes from each of the other three genera comprised a monophyletic clade, respectively, it is highly likely that WGDs occurred in these genera independently.
Gene Family Expansion and Contraction
Gene loss and gain are the primary reasons for functional changes (Xing et al., 2019). To better understand the relationships between the gene families of M. delavayi and other crucifer, we performed a systematic comparison of genes among different species. Phylogenetic analyses indicated that M. delavayi was phylogenetically categorized into expanded lineage II. Further comparisons of these species revealed 2673 expanded and 3600 contracted gene families in the M. delavayi genome. Significant expansion or contraction in the size of particular gene families is often associated with the adaptive divergence of related species (Zhang et al., 2016). Also, a total of 41 gene families showed significant expansion (P < 0.05), and 37 gene families showed significant contraction (P < 0.05) in M. delavayi. The significantly expanded gene families contained 312 genes, which are mainly involved in “response to light,” “response to salt stress,” “response to water deprivation,” and “calcium-mediated signaling” (Supplementary Table S6). This also agrees with the previous predication that some species from the expanded lineage II of Brassicaceae are salt-tolerant (Monihan et al., 2020).
We further examined the shared and species-specific gene families between M. delavayi and other alpine crucifers with genomes available: A. alpine (Willing et al., 2015), B. stricta, C. himalaica (Zhang et al., 2019), E. heterophyllum (Guo et al., 2018), and L. meyenii (Zhang et al., 2016). These alpine crucifers were found to develop more species-specific genes to adapt to alpine habitats. We next examined whether M. delavayi developed more such genes in addition to those shared with other alpine crucifers. We identified a total of 28,835 homologous gene families, 13,117 of which were shared by six alpine crucifers. We identified 1383 gene families specific to the M. delavayi genome (Figure 3A). GO enrichments of these genes in species-specific gene families revealed that they were mainly involved in “response to UV-B,” “response to cold,” “DNA repair,” and “cellular response to salt stress” (Figure 3B and Supplementary Table S7).

Figure 3. (A) Venn diagram showing shared and unique gene families among six species of Brassicaceae. (B) Gene ontology (GO) enrichment of M. delavayi–specific genes.
Fast-Evolving Genes in M. delavayi
In plants, tolerance to UV-B radiation and cold are critical for surviving at high altitudes (Zhang et al., 2016). Increased Ka relative to Ks in certain genes may explain the adaptive evolution of organisms at the molecular level (Qiu et al., 2012). We identified 1203 syntenic gene blocks containing 20,442 collinear gene pairs in the M. delavayi and A. thaliana genomes. A total of 327 genes under positive selection had a Ka/Ks ratio greater than 1.0. The Swiss-Port functional classification revealed that the fast-evolving genes with putative functions were related to “DNA repair,” “response to UV-B radiation,” “defense response,” and “response to cold” (Supplementary Table S8). In particular, we found that DRT101 (Mde002296.11) related to the DNA repair from UV damage evolved quickly in the M. delavayi genome (Supplementary Table S8). In the maca genome, genes related to DNA repair (DRT102) have also been found to evolve rapidly (Zhang et al., 2016). Both DRT101 and DRT102 belong to the DNA-damage-repair/toleration (DRT) genes (Supplementary Table S9), and they may encode UV-specific excision repair activities (Pang et al., 1993; Hays and Pang, 1994). The accelerated evolution of DRT and other genes may help M. delavayi adapt to the high-altitude environment.
Discussion
In this study, we performed de novo assembly of the M. delavayi genome based on an Illumina HiSeq 2500 platform. M. delavayi is the first sequenced species of the genus Megacarpaea. This reference genome provides a basis for further studying speciation based on genomic data for the genus and comparative genomics studies in the family Brassicaceae. The genome of M. delavayi was estimated at 899 Mb, and the final assembly genome was 883.81 Mb, representing about 98% of the estimated genome size (Table 1). WGD and expansion of repetitive elements in the M. delavayi genome might have led to its larger size than other species (Figure 1B and Supplementary Table S3). Based on the genome-scale single copy genes, our phylogenetic analysis clearly shows that Megacarpaea was placed in the expanded lineage II of Brassiaceae, and it was closely related to Anastatica, Iberis, and Biscutella. This finding resolved the ambiguous phylogenetic position of the genus Megacarpaea in the previous study (Nikolov et al., 2019) because of the difficulties in gene orthologous alignments (Walker et al., 2017).
Our genomic analyses suggested one species-specific WGD event in the M. delavayi genome. This WGD paralleled independently to those that occurred in the closely related species, Anastatica, Biscutella, and Iberis. These WGDs were estimated to occur within the Miocene shortly after the radiative divergences of the sampled genera. Such repeated WGDs accompanying lineage divergences together drove species diversification of the family (Kagale et al., 2014). In addition, WGDs should have also played an important role for crucifers to colonize the arid habitats because of the obvious advantages of the polyploids under selective pressure (Dong et al., 2019).
Compared with the published alpine crucifers with available genomes (Guo et al., 2018; Zeng et al., 2019; Zhang et al., 2019), we also found that M. delavayi retained numerous species-specific genes, which are involved in “response to UV-B,” “response to cold,” “DNA repair,” and “cellular response to salt stress” (Figure 3B and Supplementary Table S7). These genes derived from WGD or other ways may also play an important role for M. delavayi to adapt to a cold and UV-B stressed habitat at high altitude (Cheviron and Brumfield, 2012). In addition, fast-evolving genes in M. delavayi were also found to be involved in “DNA repair” and abiotic stresses (Supplementary Table S8). All these findings suggest that M. delavayi had developed obvious genomic changes to adapt to alpine habitats. The reference genome presented here provides an important resource for further studying molecular adaptation of this and other alpine plants to the highlands.
Data Availability Statement
The genomic sequence data of Megacarpaea delavayi in this study have been deposited in the NCBI under BioProject PRJNA630110. The assembled genome and genome annotation information have been deposited in the National Genomics Data Center (https://bigd.big.ac.cn/?lang=zh) under BioProject PRJCA002887.
Author Contributions
QH and JL designed the research. LZ and HB collected the materials and performed the genome sequencing and assembly. QY, WY, HB, TL, JJ, and LZ performed the genome annotation and evolution analyses. QY, QH, and JL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported equally by the National Key Research and Development Program of China (2017YFC0505203) and the Fundamental Research Funds for the Central Universities (2018CDDY-S02-SCU and SCU2019D013), the National High-Level Talents Special Support Plans (10 Thousand of People Plan), and 985 and 211 Projects of the Sichuan University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2020.00812/full#supplementary-material
FIGURE S1 | 17-k-mer frequency distribution of sequencing reads from M. delavayi. The size of the M. delavayi genome was estimated at 899 Mb.
FIGURE S2 | Phylogenetic tree with divergence time and expanded/contracted gene family for M. delavayi and other Brassicaceae species. The numbers above the branches are the predicted divergence times.
TABLE S1 | Sequencing statistics of the M. delavayi genome.
TABLE S2 | Evaluation M. delavayi genome assembly with BUSCO.
TABLE S3 | Numbers of annotated protein-coding genes in M. delavayi genome.
TABLE S4 | Intact LTR-RTs in M. delavayi and other Brassicaceae species.
TABLE S5 | Protein data sets used for comparative genomic analyses.
TABLE S6 | Function of significantly expanded genes in M. delavayi genome.
TABLE S7 | GO enrichment of M. delavayi specific genes.
TABLE S8 | Function of fast-evolving genes in M. delavayi genome.
TABLE S9 | DRT genes copy number in M. delavayi and L. meyenii genomes (identity > 80%).
Footnotes
References
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bowers, J. E., Chapman, B. A., Rong, J., and Paterson, A. H. (2003). Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438. doi: 10.1038/nature01521
Buchfink, B., Xie, C., and Huson, D. H. (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chen, Y., Ma, T., Zhang, L., Kang, M., Zhang, Z., Zheng, Z., et al. (2020). Genomic analyses of a “living fossil”: the endangered dove-tree. Mol. Ecol. Resour. 20, 1–14.
Cheo, T. Y., Lu, L., Yang, G., Al-Shehbaz, I., and Dorofeev, V. (2001). Megacarpaea. Flora China 8, 39–40.
Cheviron, Z. A., and Brumfield, R. T. (2012). Genomic insights into adaptation to high-altitude environments. Heredity 108, 354–361. doi: 10.1038/hdy.2011.85
Conesa, A., and Götz, S. (2008). Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics 2008:619832.
De Bie, T., Cristianini, N., Demuth, J. P., and Hahn, M. W. (2006). CAFE: a computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271. doi: 10.1093/bioinformatics/btl097
Dong, Y., Gupta, S., Sievers, R., Wargent, J. J., Wheeler, D., Putterill, J., et al. (2019). Genome draft of the Arabidopsis relative Pachycladon cheesemanii reveals novel strategies to tolerate New Zealand’s high ultraviolet B radiation environment. BMC Genomics 20:838. doi: 10.1186/s12864-019-6084-4
Edger, P. P., Heidel-Fischer, H. M., Bekaert, M., Rota, J., Glöckner, G., Platts, A. E., et al. (2015). The butterfly plant arms-race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. U.S.A. 112, 8362–8366. doi: 10.1073/pnas.1503926112
Ellinghaus, D., Kurtz, S., and Willhoeft, U. (2008). LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9:18. doi: 10.1186/1471-2105-9-18
Guo, X., Hu, Q., Hao, G., Wang, X., Zhang, D., Ma, T., et al. (2018). The genomes of two Eutrema species provide insight into plant adaptation to high altitudes. DNA Res. 25, 307–315. doi: 10.1093/dnares/dsy003
Guo, X., Liu, J., Hao, G., Zhang, L., Mao, K., Wang, X., et al. (2017). Plastome phylogeny and early diversification of Brassicaceae. BMC Genomics 18:176. doi: 10.1186/s12864-017-3555-3
Haas, B. J., Delcher, A. L., Mount, S. M., Wortman, J. R., Smith, R. K., Hannick, L. I., et al. (2003). Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666. doi: 10.1093/nar/gkg770
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Haas, B. J., Salzberg, S. L., Zhu, W., Pertea, M., Allen, J. E., Orvis, J., et al. (2008). Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9:R7.
Hays, J. B., and Pang, Q. (1994). UV-B-Inducible and Constitutive Genes that Mediate Repair and Toleration Of UV-Damaged DNA in the Plant Arabidopsis Thaliana. Berlin: Springer.
Hu, T. T., Pattyn, P., Bakker, E. G., Cao, J., Cheng, J. F., Clark, R. M., et al. (2011). The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat. Genet. 43, 476–483.
Huang, C.-H., Zhang, C., Liu, M., Hu, Y., Gao, T., Qi, J., et al. (2016). Multiple polyploidization events across asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 33, 2820–2835. doi: 10.1093/molbev/msw157
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kagale, S., Robinson, S. J., Nixon, J., Xiao, R., Huebert, T., Condie, J., et al. (2014). Polyploid evolution of the Brassicaceae during the Cenozoic era. Plant Cell 26, 2777–2791. doi: 10.1105/tpc.114.126391
Kajitani, R., Toshimoto, K., Noguchi, H., Toyoda, A., Ogura, Y., Okuno, M., et al. (2014). Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24, 1384–1395. doi: 10.1101/gr.170720.113
Kang, M., Wu, H., Yang, Q., Huang, L., Hu, Q., Ma, T., et al. (2020). A chromosome-scale genome assembly of Isatis indigotica, an important medicinal plant used in traditional Chinese medicine: an Isatis genome. Hortic. Res. 7:18.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kiefer, C., Willing, E.-M., Jiao, W.-B., Sun, H., Piednoël, M., Hümann, U., et al. (2019). Interspecies association mapping links reduced CG to TG substitution rates to the loss of gene-body methylation. Nat. plants 5, 846–855. doi: 10.1038/s41477-019-0486-9
Kumar, S., Stecher, G., Suleski, M., and Hedges, S. B. (2017). TimeTree: a resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 34, 1812–1819. doi: 10.1093/molbev/msx116
Lei, S., Xiaobo, L., Guirong, S., Yun, Y., and Bin, L. (2009). Effect of Megacarpaea delavayi Franch on digestive juice in rat with heat due to food stagnation. Chinese J. Ethnomed. Ethnopharm. 18, 1–3.
Li, H. (2015). BFC: correcting Illumina sequencing errors. Bioinformatics 31, 2885–2887. doi: 10.1093/bioinformatics/btv290
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Li, M., Tian, S., Jin, L., Zhou, G., Li, Y., Zhang, Y., et al. (2013). Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438. doi: 10.1038/ng.2811
Liu, W. S., Wei, W., and Dong, M. (2009). Clonal and genetic diversity of Carex moorcroftii on the Qinghai-Tibet plateau. Biochem. Syst. Ecol. 37, 370–377. doi: 10.1016/j.bse.2009.07.003
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18.
Majoros, W. H., Pertea, M., and Salzberg, S. L. (2004). TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879. doi: 10.1093/bioinformatics/bth315
Mandáková, T., Joly, S., Krzywinski, M., Mummenhoff, K., and Lysaka, M. A. (2010). Fast diploidization in close mesopolyploid relatives of Arabidopsis. Plant Cell 22, 2277–2290. doi: 10.1105/tpc.110.074526
Mandáková, T., Li, Z., Barker, M. S., and Lysak, M. A. (2017). Diverse genome organization following 13 independent mesopolyploid events in Brassicaceae contrasts with convergent patterns of gene retention. Plant J. 91, 3–21. doi: 10.1111/tpj.13553
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
McKain, M. R., Tang, H., McNeal, J. R., Ayyampalayam, S., Davis, J. I., DePamphilis, C. W., et al. (2016). A phylogenomic assessment of ancient polyploidy and genome evolution across the Poales. Genome Biol. Evol. 8, 1150–1164.
Monihan, S. M., Magness, C. A., Ryu, C.-H., McMahon, M. M., Beilstein, M. A., and Schumaker, K. S. (2020). Duplication and functional divergence of a calcium sensor in the Brassicaceae. J. Exp. Bot. 71, 2782–2795. doi: 10.1093/jxb/eraa031
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185.
Nikolov, L. A., Shushkov, P., Nevado, B., Gan, X., Al-Shehbaz, I. A., Filatov, D., et al. (2019). Resolving the backbone of the Brassicaceae phylogeny for investigating trait diversity. New Phytol. 222, 1638–1651. doi: 10.1111/nph.15732
Ossowski, S., Schneeberger, K., Lucas-Lledó, J. I., Warthmann, N., Clark, R. M., Shaw, R. G., et al. (2010). The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327, 92–94. doi: 10.1126/science.1180677
Ou, S., and Jiang, N. (2018). LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422. doi: 10.1104/pp.17.01310
Pang, Q., Hays, J. B., Rajagopal, I., and Schaefer, T. S. (1993). Selection of Arabidopsis cDNAs that partially correct phenotypes of Escherichia coli DNA-damage-sensitive mutants and analysis of two plant cDNAs that appear to express UV-specific dark repari activities. Plant Mol. Biol. 22, 411–426. doi: 10.1007/bf00015972
Qiu, Q., Zhang, G., Ma, T., Qian, W., Ye, Z., Cao, C., et al. (2012). The yak genome and adaptation to life at high altitude. Nat. Genet. 44, 946–949.
Shen, L., Fang, C., Wang, J., Sa, L., and Yang, L. (2009). The effect of megacarpaea delavayi franch on intestinal propulsive and gastric emptying function in mice. J. Dali Univ. 8, 8–10.
Slotte, T., Hazzouri, K. M., Ågren, J. A., Koenig, D., Maumus, F., Guo, Y. L., et al. (2013). The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat. Genet. 45, 831–835. doi: 10.1038/ng.2669
Smit, A. F. A., and Hubley, R. (2008). RepeatModeler Open-1.0. Available online at: http://www.repeatmasker.org/ (accessed November 24, 2017).
Soltis, P. S., and Soltis, D. E. (2016). Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 30, 159–165. doi: 10.1016/j.pbi.2016.03.015
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stanke, M., Keller, O., Gunduz, I., Hayes, A., Waack, S., and Morgenstern, B. (2006). AUGUSTUS: A b initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439.
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 25, 4–10.
Walker, J. F., Yang, Y., Moore, M. J., Mikenas, J., Timoneda, A., Brockington, S. F., et al. (2017). Widespread paleopolyploidy, gene tree conflict, and recalcitrant relationships among the carnivorous Caryophyllales. Am. J. Bot. 104, 858–867. doi: 10.3732/ajb.1700083
Wang, X., Wang, H., Wang, J., Sun, R., Wu, J., Liu, S., et al. (2011). The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 43, 1035–1040.
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Waterhouse, R. M., Seppey, M., Simao, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Willing, E. M., Rawat, V., Mandáková, T., Maumus, F., James, G. V., Nordström, K. J. V., et al. (2015). Genome expansion of Arabis alpina linked with retrotransposition and reduced symmetric DNA methylation. Nat. Plants 1, 1–7.
Wu, H., Ma, T., Kang, M., Ai, F., Zhang, J., Dong, G., et al. (2019). A high-quality Actinidia chinensis (kiwifruit) genome. Hortic. Res. 6, 1–9.
Wu, S., Han, B., and Jiao, Y. (2020). Genetic contribution of paleopolyploidy to adaptive evolution in angiosperms. Mol. Plant 13, 59–71. doi: 10.1016/j.molp.2019.10.012
Xing, Y., Liu, Y., Zhang, Q., Nie, X., Sun, Y., Zhang, Z., et al. (2019). Hybrid de novo genome assembly of Chinese chestnut (Castanea mollissima). Gigascience 8:giz112.
Xu, H., Luo, X., Qian, J., Pang, X., Song, J., Qian, G., et al. (2012). FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7:e52249. doi: 10.1371/journal.pone.0052249
Xu, Z., and Wang, H. (2007). LTR-FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268.
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zeng, X., Yuan, H., Dong, X., Peng, M., Jing, X., Xu, Q., et al. (2019). Genome-wide Dissection of Co-selected UV-B Responsive Pathways in the UV-B Adaptation of Qingke. Mol. Plant 13, 112–127. doi: 10.1016/j.molp.2019.10.009
Zhang, J., Tian, Y., Yan, L., Zhang, G., Wang, X., Zeng, Y., et al. (2016). Genome of plant maca (Lepidium meyenii) illuminates genomic basis for high-altitude adaptation in the central andes. Mol. Plant 9, 1066–1077. doi: 10.1016/j.molp.2016.04.016
Zhang, T., Hu, Y., Jiang, W., Fang, L., Guan, X., Chen, J., et al. (2015). Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537.
Zhang, T., Qiao, Q., Novikova, P. Y., Wang, Q., Yue, J., Guan, Y., et al. (2019). Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. U.S.A. 116, 7137–7146. doi: 10.1073/pnas.1817580116
Zhang, Z., Chen, Y., Zhang, J., Ma, X., Li, Y., Li, M., et al. (2020). Improved genome assembly provides new insights into genome evolution in a desert poplar (Populus euphratica). Mol. Ecol. Resour. 20, 1–14.
Keywords: Megacarpaea delavayi, genome sequence, alpine adaptation, whole-genome duplication, Brassicaceae
Citation: Yang Q, Bi H, Yang W, Li T, Jiang J, Zhang L, Liu J and Hu Q (2020) The Genome Sequence of Alpine Megacarpaea delavayi Identifies Species-Specific Whole-Genome Duplication. Front. Genet. 11:812. doi: 10.3389/fgene.2020.00812
Received: 30 March 2020; Accepted: 06 July 2020;
Published: 03 August 2020.
Edited by:
Xiaoming Song, North China University of Science and Technology, ChinaReviewed by:
Michael Eric Schranz, Wageningen University and Research, NetherlandsJingyin Yu, Boyce Thompson Institute, United States
Copyright © 2020 Yang, Bi, Yang, Li, Jiang, Zhang, Liu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanjun Hu, huquanjun@gmail.com
†These authors have contributed equally to this work