Contactin 1: An Important and Emerging Oncogenic Protein Promoting Cancer Progression and Metastasis


1
Department of Medicine, McMaster University, Hamilton, ON L8S 4K1, Canada
2
The Research Institute of St Joe’s Hamilton, St. Joseph’s Hospital, Hamilton, ON L8N 4A6, Canada
3
Urological Cancer Center for Research and Innovation (UCCRI), St. Joseph’s Hospital, Hamilton, ON L8N 4A6, Canada
4
Life-Tech Industry Alliance, Shenzhen 518000, China
5
Department of Surgery, McMaster University, Hamilton, ON L8S 4K1, Canada
6
Department of Oncology, McMaster University, Hamilton, ON L8S 4K1, Canada
*
Author to whom correspondence should be addressed.
Genes 2020, 11(8), 874; https://doi.org/10.3390/genes11080874
Submission received: 8 July 2020
/
Revised: 28 July 2020
/
Accepted: 28 July 2020
/
Published: 31 July 2020
(This article belongs to the Special Issue Molecular Biomarkers in Solid Tumors)
Abstract
:Even with recent progress, cancer remains the second leading cause of death, outlining a need to widen the current understanding on oncogenic factors. Accumulating evidence from recent years suggest Contactin 1 (CNTN1)’s possession of multiple oncogenic activities in a variety of cancer types. CNTN1 is a cell adhesion molecule that is dysregulated in many human carcinomas and plays important roles in cancer progression and metastases. Abnormalities in CNTN1 expression associate with cancer progression and poor prognosis. Mechanistically, CNTN1 functions in various signaling pathways frequently altered in cancer, such as the vascular endothelial growth factor C (VEGFC)-VEGF receptor 3 (VEFGR3)/fms-related tyrosine kinase 4 (Flt4) axis, phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT), Notch signaling pathway and epithelial-mesenchymal transition (EMT) process. These oncogenic events are resulted via interactions between tumor and stroma, which can be contributed by CNTN1, an adhesion protein. CNTN1 expression in breast cancer correlates with the expression of genes functioning in cancer-stroma interactions and skeletal system development. Evidence supports that CNTN1 promotes cancer-stromal interaction, resulting in activation of a complex network required for cancer progression and metastasis (bone metastasis for breast cancer). CNTN1 inhibitions has been proven to be effective in experimental models to reduce oncogenesis. In this paper, we will review CNTN1′s alterations in cancer, its main biochemical mechanisms and interactions with its relevant cancer pathways.
1. Introduction
Cell adhesion molecules (CAMs) are membrane receptors that bind to extracellular matrix molecules or receptors [1]. CAMs forms structural linkages and mediates interaction between the cell cytoskeleton and the extracellular matrix or between cells. This interaction facilitates the transduction of downstream signaling and regulate several important biological processes including cell division, migration and differentiation. The expression of CAMs are tightly regulated for refined control of cell proliferation, differentiation, mobility and survival in cellular processes. However, these processes may be hijacked and mis-regulated in tumorigenesis [1,2]. Aberrant expression of CAMs has been shown to associate with invasive phenotype of tumour cells. Metastatic dissemination is the leading cause of death for cancer patients worldwide [3]. This well-orchestrated process follows few key steps, including tumour cell invasion of the primary site, intravasation into blood and/or lymphatic vessels, dissemination into the circulation, and successful adherence and colonization to the final site of metastasis. For tumour cells to complete this metastatic cascade successfully, their ability for adaptation or plasticity needs to be enhanced or evolved, which is critically regulated through interactions with extracellular matrix (ECM) in the tumour microenvironment. Many CAMs play important roles in regulating these homotypic and heterotypic interactions [1,2,4].
Contactin 1 (CNTN1) is a neuronal cell adhesion molecule belonging to the subgroup of glycosyl phosphatidylinositol (GPI)-anchored neural cell adhesion molecules of the immunoglobulin superfamily (IgSF) [5]. Despite being primarily studied for its roles in the human nervous system, CNTN1 has been implicated in several signaling pathways altered in cancer. The aberrant activation of CNTN1 has been reported to be involved in pathological phenotypes such as cell proliferation, invasion, metastasis and poor prognosis [6,7]. On these premises, the interest of CNTN1 as a target to impede cancer progression is increasing. This review analyzes the up-to-date knowledge supporting CNTN1′s involvement in cancer progression and metastasis, and its related molecular mechanisms. In accordance with the PRISMA guidelines, we performed a systemic literature search through the PubMed database using the term “Contactin 1” and “cancer”. A total of 43 manuscripts from 2006 to 2020 were retrieved. We examined all abstracts and eliminated those: (1) studies yielding values of p > 0.05; (2) irrelevant to cancers and (3) retracted articles. We thus selected and discuss 33 articles in this review (Figure 1). The topic of CNTN1 in cancer progression has been previously reviewed [6,8], however some recent findings have been made and will be discussed.
2. The Role of CNTN1 in Neuronal Development
Of the six members of the CNTNs, Contactin 1 (CNTN1) was the first identified and the most thoroughly investigated for its function in pathological disorders [9]. CNTN1 is structurally comprised of 6 N-terminal Immunoglobulin (Ig) C2 domain, followed by 4 fibronectin type III (FNIII) repeats on the C-terminal, and a hydrophobic C-terminal amino acid sequence. This structure is common in GPI-linked proteins as it allows for lipid anchorage to the extracellular matrix (Figure 2) [9]. CNTN1 is mostly expressed in the human brain and neuronal tissues and is limitedly expressed in other tissues. CNTN1 primarily functions as an axonal glycoprotein important in the facilitation of axonal growth and the outgrowth of neurites [10,11], but also plays important roles in other neuronal developmental processes, such as differentiation and development of glial cells, myelination, synaptogenesis, and fasciculation [11,12,13,14]. The CNTN1 knockout transgenic mice shows a cerebellar phenotype in which the parallel fibers of the granule cell neurons are misoriented, concurrent with a 25% reduction on cerebellar volume, supporting its importance in axon guidance [15]. Specifically, the CNTN1 mutants show an overt ataxia phenotype by day 10 which subsequently progresses to pronounced smaller size, emaciated appearance and uncontrolled movement by day 15. The CNTN1 homozygous mutants were lethal by postnatal day 18, which may be attributable to impaired cerebellar functions, which interfere with normal feeding behavior.
CNTN1 binds with several extracellular matrix molecules (Figure 3). The association of CNTN1 with tenascin-C, tenascin-R, and receptor protein tyrosine phosphatase β (RPTPβ) promotes axonal growth and fasciculation which are pivotal in neuron-glial integration [16]. CNTN1 on the axon surface associates in trans with transmembrane protein tyrosine receptor type Z (PTPRZ) on the glial cells, in turn facilitates neurite outgrowth and glial adhesion [17,18,19,20]. CNTN1 also modulates the maturation and proliferation of oligodendrocyte precursor cells (OPCs) via its interaction with PTPRZ and Notch receptor [14,21,22]. When binding laterally in cis with the receptor protein tyrosine phosphatase α (RPTPα), CNTN1 promotes transduction of extracellular signals of tyrosine kinase FYN and influences cell mobility [23]. Interestingly, whist the interaction of CNTN1 with RPTPß substrate stimulates dorsal root ganglion (DRG) sensory axons extension [19,24], its association with Tenascin-R substrate actually inhibits cerebellar granule neuron receptor [25], suggesting the functions of CNTN1 may vary depending on cellular and molecular context. Recent study has identified an essential role of CNTN1 in radical neuronal migration and final localization of neurons in the developing mouse neocortex. The knockdown of CNTN1 via in utero electroporation resulted in significantly delayed radical migration mediated by RhoA activation, a Rho-GTPase critical for actin cytoskeletal reorganization [26]. Clinically, the expression of CNTN1 is dramatically reduced in patients with age-related memory decline [27], suggesting a role of CNTN1 in memory associated processes. A previous report also described a lethal form of congenital myopathy possibly caused by the loss of CNTN1 in the neuromuscular junction due to a familial mutation [28]. Collectively, these observations reveal that CNTN1 plays a critical role in the development of central nervous system through its adhesion functions. Adhesion is an essential process regulating tumorigenesis and cancer progression; in this regard, it is important to revisit the evidence supporting CNTN1-associated oncogenic functions.
3. Relevance of CNTN1 in Tumorigenesis
Neural cell adhesion molecules of the IgSF are complex membrane-anchored proteins that mediates multiple molecular interactions and include CNTNs, neural cell adhesion molecule (NCAM), L1, and neuron-glia related (Nr)-CAM [29]. It has become increasingly clear that changes in the adhesive properties of cancer cells, in terms of both cell–cell adhesion and adhesion to the extracellular matrix, are involved in tumor progression. Previous reports have supported that certain members of the Ig superfamily may be important in facilitating tumour invasion and metastasis [30,31]. The first CAM identified is NCAM, which contains 5 Ig-like and 2 fibronectin type III repeats with 2 major transmembrane isoforms and a GPI-linked isoform. Studies have shown NCAM to be commonly upregulated and promotes an adhesion switch during EMT that is associated with cancer invasion. Enforced expression of NCAM promotes mesenchymal-like properties, as evident by the reciprocal staining of E-cadherin in NCAM from RipTag2 mouse model of pancreatic cancer and aberrant persistence of E-cadherin expression resulted from NCAM deficiency. Furthermore, NCAM localized in membrane compartments binds and stimulates fibroblast growth factor receptor 1 (FGFR1) through its fibronectin type III (F3) domains and activates MAPK-mediated cell migration in non-neural cells. These findings imply a strong association of NCAM with EMT in human cancers. Indeed, the correlation between deregulated NCAM expression and poor prognosis has been found in different cancer types including neuroblastoma [32,33], myeloma [34,35], acute myeloid leukemia [36,37], pancreatic cancer [38,39,40], and small cell lung cancer [41].
Sharing structural and functional homology with NCAM, the role of CNTN1 outside of the nervous system was largely unexplored until recently. Northern blots analysis has shown low levels of CNTN1 transcripts in several tissues including pancreas, lung, kidney, and skeletal muscles but how CNTN1 functions in these tissues remains unexplored [42]. However, as the central nervous system was the only defective organ in CNTN1 deficient mice [15], the protein is not a major contributor to the development of other organs at least in mice. The CNTN1 gene locus is mapped on chromosome 12q11-q12 and is the only IgSF genes that is located at this position. Intriguingly, several other genes, which are mapped in positions between 12q11 and q13, have all been implicated in human cancers. These include the homeobox protein HOX3 [43], the integrin 5 subunit [44], collagen type II [45] and the proto-oncogenes erb-b3 [46] and int-1 [47]. Intriguingly, integrin 5 subunit function in the cellular signaling emanated from adhesion [48], supporting the involvement of the CNTN1 genes residing in this region in cancer adhesion. Moreover, the chromosome position 12q13 is a heritable fragile site implicated in embryonic lethality [49] and is also a frequent breakpoint in several cancers [50]. Amplifications and gains of genomic materials are frequent in this region for malignant gliomas [51] and astrocytoma [52]. The location of CNTN1 gene locus close to a tumor breakpoint provides a potential genetic evidence for CNTN1 to function in tumor formation and progression. Moreover, CNTN1 was identified as a direct downstream molecule of the vascular endothelial growth factor C (VEGFC)-VEGF receptor 3 (VEFGR3)/fms-related tyrosine kinase 4 (Flt4) axis important in lymphangiogenesis and lymphatic invasion in cancers [53,54,55]. With growing interest in the investigation of CNTN1 for its cancer related functions, CNTN1 expression was found to be upregulated in many cancers including lung [56,57,58,59], gastric [60,61], breast [62], prostate [63,64], stomach [65], thyroid [7], esophageal [66] cancers, hepatocellular carcinoma [67,68], astrocytic gliomas [69], esophageal squamous cell carcinoma (ESCC) [70], and oral squamous cell carcinoma (OSCC) [71]. Unsurprisingly, increased CNTN1 expression was found to have functional associations in promoting cancer cell progression and metastasis. In these subsections below, we will discuss in details current studies that have investigated CNTN1′s role in human cancers (Table 1).
3.1. Lung Cancer
The role of CNTN1 as a metastasis-promoting oncogene was described by Su et al. in a genome wide cDNA microarray analysis to search for potential regulatory genes involved in cancer invasion and metastasis [53]. CNTN1 was discovered as an immediate downstream effector of the vascular endothelial growth factor (VEGF)C-VEFGR3/Flt4 that induced invasion and metastasis in lung cancer via the Src/p38 MAPK-mediated C/EBP signaling pathway (Figure 4). The activation of VEGF-C/Flt4 axis robustly upregulated CNTN1, leading to rearrangement of F-actin containing microfilaments bundles important in cell motility, enhanced cell invasion in vitro, and higher frequency of lung metastasis formation in vivo. The expression of CNTN1 was significantly correlated with Flt expression, tumor stage, lymph node metastasis, and patient survival in lung adenocarcinoma patients. The vascular endothelial growth factor (VEGF)/VEGF-receptor signaling pathway is integrally involved with growth, invasion, and metastasis of carcinomas through promoting angiogenesis and lymphangiogenesis [55]. During metastasis into lymph node, newly formed lymphatic capillaries, or pre-existing afferent lymphatic vessels allow passage for dissemination of tumor cells. The VEGFC-VEFGR3/Flt4 is a lymphatic specific biochemical axis with extracellular VEGFC as ligands, cell membrane VEGFR3 (Flt4) as receptor, and extracellular or intracellular pathway-related molecules as executors. The VEGFR3 (Flt4) mediate dissemination of several tumors and was found to be overexpressed in prostate [74], lung [75], colorectal [76], and head and neck cancers [77]. As well, VEGFR3 (Flt4) expression was found to correlate with different stages of cervical carcinogenesis [78]. Accumulating evidence suggest that the up and downstream effectors of the VEGFC-VEFGR3/Flt4 axis form a complicated biochemical network which work in synergy to facilitate tumor lymphangiogenesis and lymphatic metastasis. In this regard, it is reasonable to speculate that CNTN1 may be involved as an executor to facilitate this process.
CNTN1′s actions in this aspect may be mediated via facilitation of epithelial-to-mesenchymal transition (EMT). Epithelial cells depend on a fine-tuned and tightly regulated EMT for conversion into mesenchymal state during development [79]. Cells undergoing EMT adopt properties such as enhanced migratory and invasive abilities, increased apoptosis resistance, and extended production of extracellular matrix elements, all of which promotes cancer metastasis. In tumor cells, local cancer epithelial cells take over the evolutionarily conserved EMT process to lose their polarity and cell-cell contact while undergoing a dramatic cytoskeletal remodeling to acquire an invasive, well-defined mesenchymal phenotype [80], including the common loss of E-cadherin and overexpression of mesenchymal proteins (N-cadherin, vimentin, and fibronectin) [79,81]. Yan et al. discovered that while the knockdown of CNTN1 did not influence lung cancer cell proliferation, it significantly reduced cancer cell’s invasive abilities both in vitro and in xenograft model [58]. Specifically, CNTN1 fulfils this metastasis-promoting role in part through inhibition of E-cadherin. Although this suppression is likely mediated by the upregulation of transcriptional factors SIP1 and Slug, there’s also evidence that CNTN1 may downregulate E-cadherin at the gene level by inhibition of the pleckstrin homology (PH) domain and leucine-rich repeat protein phosphatase 2 (PHLPP2) instead of phosphatase and tensin homolog (PTEN), in turn resulting in AKT activation (Figure 4) [58]. AKT activity is upregulated by both upstream and downstream phosphatases, PTEN and PHLPP [82]. The activation of AKT reduces E-cadherin and constitutes a major component of EMT in promoting cancer cell invasion and metastasis.
Intriguingly, CNTN1 is also important in promoting chemo-resistant properties in lung cancer cells via induction of EMT. BCT-100 is an anti-cancer agent effective in treating arginine auxotrophic tumors including small cell lung cancer (SCLC) [62]. In a gene chip assay analyzing BCT-100-resistant cells in comparison to parent cells, CNTN1 was robustly upregulated and induced an EMT phenotype in resistant cells via targeting of the AKT pathways. Further functional analysis demonstrated that CNTN1 silencing re-sensitized resistant cells to BCT-100 treatment as well as attenuated the EMT phenotypes in resistant cell lines [57]. Similarly, CNTN1 expression was elevated in multidrug resistance (MDR) A549/cisplatin (A549/DDP) cells compared to its progenitor A549 lung cancer cells, and the silencing of CNTN1 rendered both cells higher cisplatin sensitivity and upregulated cisplatin-induced apoptosis, leading to inhibition of tumor invasion and metastasis [59]. This enhanced chemoresistance of lung cancer cells induced by CNTN1 overexpression was also found to be attributable to PI3K/AKT activated EMT enhancement (Figure 4). Indeed, CNTN1 downregulation successfully hindered cisplatin resistance and malignant progression via partly inactivating the EMT process by regulating the PI3K/AKT pathway. This finding was corroborated clinically by the positive association between CNTN1 expression and lymphatic invasion in non-small-cell lung carcinoma (NSCLC) patients that have received adjuvant cisplatin- or carboplatin-based treatments following surgery [72]. The tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) was also found to upregulate the expression of CNTN1, leading to enhanced lung cancer cells invasion via the activation of α7 nicotinic acetylcholine receptor (α7 nAChR) downstream of the AKT and extracellular signal-regulated kinase (ERK) signaling pathway [56]. The involvement of CNTN1 in promoting EMT and AKT suggest a potential involvement in its facilitation of cancer stem cell-like characteristics such as migration and anoikis resistance in cancer cells.
3.2. Gastric and Upper Gastrointestinal Cancers
Following the discovery of CNTN1′s oncogenic role in lung cancer, it has become increasingly clear that its role in the induction of invasion and metastasis may be a shared commonality in other cancers as well. In vitro studies found that the knockdown of Flt4 in human MKN45 gastric cancer cells with short hairpin RNA directly downregulated CNTN1 expression [60], which validates the role of CNTN1 in the VEGFC-VEFGR3/Flt4 axis. Compared to adjacent normal gastric mucosal tissues, both CNTN1 mRNA and protein expression were significantly upregulated in primary lesions and its expression level was positively correlated with VEFGC and Flt4 expression as well as lymphatic invasion, lymph node metastasis, TNM (tumor, node, metastasis) stage and worse prognosis in gastric cancer patients [60]. The induction of CNTN1 by the VEGFC-VEFGR3/Flt4 signalling axis is a result of the VEGFC-facilitated binding of CCAAT enhancer binding protein α (CEBPA) with the CNTN1 promoter [53] (Figure 4). This association was also validated in esophageal squamous cell carcinoma (ESCC) [66]. Recent study has shown that a reduction in CNTN1 expression alone was sufficient to reverse the enhanced migration ability of esophageal cancer cells attributable to ectopic VEFGC expression in vitro. As well, ESCC tissues showed significant upregulation of CNTN1 expression compared to adjacent non-tumor tissues and this increased expression was positively correlated with ESCC stage, lymph node metastasis and lymphatic invasion, supporting CNTN1′s role in esophageal cancer progression [70]. In a study investigating oral squamous cell carcinoma (OSCC), Flt-4 stimulation or inhibition directly resulted in upregulated or downregulated expression of CNTN1 respectively [71]. In vitro cell proliferation and migration assays further demonstrated that this association was accompanied by enhanced or inhibited cell proliferation and migration. The VEGFC-mediated lymphangiogenesis is suggested to be maintained via a paracrine mechanism in which the Flt4 expressed on the lymphatic endothelial cells is activated via its association with tumor cell-derived VEGFC [54]. Shigetomi et al. demonstrated that CNTN1 expression was directly regulated by Flt4 stimulation or inhibition, and this alteration was robustly associated with enhanced or reduced OSCC cell proliferation and migration correspondingly [54]. This finding suggests that the VEGFC-VEFGR3/Flt4 axis in tumor microenvironment may promote cell proliferation and migration via upregulating both VEGFC and CNTN1 in an autocrine manner, in turn contributing to cancer progression through the development of lymphatic metastasis. This process likely works in conjunction with its primary paracrine mechanism responsible for lymphangiogenesis in the lymphatic endothelial cells. Similar to ESCC, the upregulation of CNTN1 expression in OSCC patients was associated with poor overall survival (OS) [71]. The OSCC cells with CNTN1 knocked-down exhibits significant reduction in invasion but not proliferative properties [71], which is in line with previous findings demonstrating that CNTN1 may promote progression of cancer cells through exclusive activation of metastatic pathways.
3.3. Prostate Cancer
Accumulating evidence supports a pivotal role of cancer stem cells (CSC) in tumor progression and metastasis [83,84,85]. Prostate cancer stem cells (PCSC) have been identified as a major player underlying castration-resistant prostate cancer (PCSC) and associate with bone metastasis and poor prognosis [86,87]. Yan et al. was the first to describe a contribution of CNTN1 in prostate cancer stem cell like (PCSC)-derived tumor initiation [64]. The expression of CNTN1 was upregulated in PCSCs compared to non-stem prostate cancer cells with a concurrent downregulation of E-cadherin. This may provide a novel perspective for CNTN1′s role in CSCs in facilitating the acquisition of a more invasive cell phenotype. These observations, on the other hand, shed light on a potential mechanism leading to CNTN1 expression in cancers, i.e., the plasticity of PCSCs is likely a contributor. Disruption of the epithelial homeostasis with EMT is a major venue contributing to prostate cancer cells becoming invasive and metastasize [81]. As well, ectopic CNTN1 overexpression promoted cellular invasion in vitro and enhanced xenograft formation in vivo. Similarly, CNTN1 overexpression in PC cells produced more and larger lung nodules in a lung metastasis model compared to empty vector (EV) controls. Analysis of gene expression of CNTN1-knockdown in PCSCs showed predominantly affected genes to function in tumorigenesis, suggesting potential CNTN1 modulation of the expressions of tumorigenesis-relevant genes. Clinically, while CNTN1 expression was negative or low in normal prostate glands, it was upregulated in advanced prostate carcinomas and are clearly present in both primary prostate tumors and matched lymph node and bone metastasis.
This association was corroborated by a recent study in which elevated CNTN1 expression was found to positively associate with larger tumor size, advanced tumour stage, higher risk of metastasis and shorter overall survival (OS) in PC patients following radical prostatectomy [63]. Further analysis showed that silencing or knockdown of CNTN1 expression resulted in suppression of proliferation, invasion and migration capabilities in PC cells concurrent with a reduced PI3K/AKT signaling. CNTN1 inhibition also increased expression of E-cadherin while reduced N-cadherin and Vimentin, suggesting an inhibition of the EMT process pivotal in cancer metastases (Figure 4). Furthermore, docetaxel (Dox)-resistant PC cells exhibited upregulated CNTN1 expression and an EMT phenotype compared to parental cells [73]. Silencing of CNTN1 with short hairpin RNA (shRNA) inhibited cellular malignant phenotype and reduced expression of pluripotent markers such as CD44, octamer-binding transcription factor 4 (OCT-4), and Nanog. CNTN1 silencing sensitized Dox-resistant PC cells and inhibited PI3K/AKT signaling in vivo. These observations are in accordance with elevated CNTN1 expression found in drug resistant SCLC cells compared with progenitor cells [57], indicating that CNTN1 may function to promote a more malignant and cancer stem cell-like characteristics with higher migration ability and potentially anoikis resistance in cancer cells.
3.4. Breast Cancer
In a study investigating breast cancer (BC), ectopic CNTN1 overexpression enhanced cell proliferation, invasion, migration as well as cell cycle progression from G1 to S phase in breast cancer cells in vitro [88]. Notably, CNTN1 overexpression also enhanced breast cancer xenograft tumor growth in vivo in nude mice. However, a potential relationship of CNTN1 with primary BC has not been thoroughly investigated. To address this issue, we have taken advantage of the availability of numerous BC genetic databases; one of them is bc-GenExMiner 4.5 [89,90,91], which contains 10,716 BC tissues with gene expression determined by DNA microarray and 4712 BC samples profiled for gene expression using RNA-sequencing (RNA-seq). The dataset contains a total of n = 15428 BCs from multiple independent studies (cohorts; n = 57, 54 DNA microarray and three RNA-seq studies); importantly, data has been filtered by experts in the field for cross cohort analyses.
Based on gene expression, breast cancers are classified as five intrinsic subtypes: luminal A and B (ER+), human epidermal growth factor receptor 2 (HER2)-enriched (HER2-E), normal-like, basal-like, and claudin-low (the latter two being triple negative) [92,93,94,95]. Both HER2-E and basal-like tumors are aggressive subtypes [96]. Among the 5 subtypes, we noticed a significant higher level of CNTN1 in HER2-E in a population consisting of 50 DNA microarray studies (n = 9254) and in the population of three RNA-seq investigation (n = 4712) (Figure 5). The levels of CNTN1 expression vary among BC subtypes with different aggressiveness. For instance, Luminal B is a more aggressive BC sub-type compared to Luminal A but with reduced CNTN1 expression (Figure 5). However, HER2-E BCs are more aggressive compared to Luminal and Normal-like BCs [96] and express a significant higher level of CNTN1 than these sub-types of BCs (Figure 5). While these analyses reveal factors in addition to CNTN1 contributing to BC aggressiveness, evidence (Figure 5) supports an association of CNTN1 with this BC characteristics. Using both populations (DNA microarray and RNA-seq), we have analyzed CNTN1-correlated gene expression with correlations (r ≥ 0.4) determined by Pearson correlation and performed Gene Ontology (GO) enrichment analysis. In both positively correlated gene populations, enrichment in the terms of extracellular matrix organization (GO:0030198) and cell adhesion (GO:0007155) in Biological Process (BP) was observed in BC (All patients, Table 2). We further analyzed GO enrichment based on the intrinsic subtypes of BC and found robust enrichments in both GO terms (GO:0030198 and GO:0007155) in all BC intrinsic subtypes (Table 2). These analyses provide appealing evidence supporting the oncogenic function of CNTN1 in promoting BC via enhancing BC cell adhesion and interaction with extracellular matrix. Adhesion to bone marrow stromal components are critical for homing and surviving in bone marrow and thus metastatic outgrowth of bone metastasis [97,98]. Bone is a common site of BC metastasis [97,98]; in this regard, the enrichment of GO:0001501/skeletal system development in genes that are positively associated with CNTN1 expression in BC (Table 2) supports CNTN1′s role in facilitating BC bone metastasis. This possibility is supported by CNTN1-mediated promotion of prostate cancer metastasis [64] as prostate cancer predominantly metastasizes to the bone [99]. Collectively, our analyses suggest important functions of CNTN1 in promotion of BC progression and metastasis in part through enhancing BC cell adhesion.
3.5. Other Cancers
In hepatocellular carcinoma (HCC), CNTN1 expression was markedly increased in HCC tumors and this upregulation was correlated with tumor size and TNM stage [68]. As well, CNTN1 expression was found to be an independent prognostic factor for OS (hazard ratio 2.383, 95% confidence interval 1.262–4.503; p = 0.007) and DFS (disease free survival) (hazard ratio 2.356, 95% confidence interval 1.370–4.049; p = 0.002) in HCC patients. RET/PTC3 (Ret proto-oncogene and Ret-activating protein ELE1) rearrangement is the most frequent genetic alteration in thyroid cancer and most functions of RET are mediated through pathways including ERK, JNK, and PI3K/AKT [100,101,102,103]. CNTN1 was discovered as a potential RET-PTC3 downstream gene in GEO profile database and RET inhibitor regorafenib successfully downregulated CNTN1 expression in thyroid cancer cells [7]. In a study exploring the global gene and miRNA expression profiles of neuroblastoma cells after bortezomib treatment, CNTN1 was found to be downregulated along with RET, further confirming this relationship [104]. Interestingly, in a study exploring the therapeutic efficacy of Ganoderma lucidum extract (GLE), a potentially effective tumor growth inhibitor, CNTN1 was identified to be a part of the hub mRNA down-expressed in GLE-treated Hepa1–6-bearing C57BL/6 mice, mediated by miRNA mmu-miR23a-5p [105].
CNTN1 expression was found to be elevated in thyroid tumor tissues in comparison to its pre-cancer counterparts, and this alteration was particularly evident in the junctional zones where normal glandular structure and tumorous structure meets. This upregulation of CNTN1 was positively correlated with tumor size, TNM and tumor stage. Functionally, CNTN1 silencing in vitro restrained thyroid cancer cell migration and invasion abilities.
In a similar manner, significant correlation of CNTN1 expression with tumor size and TNM stage was also observed in stomach cancer [65]. While normal astrocytes did not express CNTN1, glioblastomas showed an overexpression of CNTN1 protein and a positive association was identified between increasing malignancy grade and CNTN1 expression [69]. Recent evidence identified CNTN1 as a novel melanoma-associate biomarker [106]. This is consistent with CNTN1′s role as an activator of Notch signaling and the association of Notch1 activation with a more metastatic phenotype in primary melanoma cells (Figure 4) [107]. The Notch signaling pathway is highly conserved, and its dysregulation is associated with many cancers [108]. Accumulating evidence have suggested Notch to be a key regulator in cancer stem cells (CSC) [109]. CNTN1 was identified as a ligand for NOTCH1 and activates Notch signaling by the release of notch intracellular domain (NCID) [14]. The knockdown of CNTN1 suppresses cellular proliferation and invasion in thyroid cancer and also inhibits the expression of cyclin D1 (CCDN1) [7], a Notch target gene, suggesting CNTN1 may participates in the tumor proliferation and metastasis in part via regulating the Notch pathway.
By taking advantage of the recently established GEPIA2 program with RNA sequencing data from The Cancer Genome Atlas (TCGA) and Genotype-Tissue Expression (GTEx) projects, we performed survival analyses with respect to CNTN1 expression across all 33 TCGA cancer types [110]. Our initial screen revealed a significant association between elevated CNTN1 and reduced overall survival (OS) in all cancer types (Figure 6a). With that, we performed detailed OS analysis for all cancer types, significant association between elevated CNTN1 with reduced survival was identified in bladder urothelial carcinoma (BLCA) (Figure 6b), brain lower grade glioma (LGG) (Figure 6c) and stomach adenocarcinoma (STAD) (Figure 6d). While these analyses support a positive association between high CNTN1 expression and poor OS in cancers, this association might be cancer-type specific at least at the level of mRNA; high CNTN1 expression displays a reverse association with poor OS in LGG (Figure 6c).
Mounting evidence in the past decade have supported CNTN1′s unique role in promoting tumor progression and metastasis in cancers (Table 1). Although detailed molecular mechanisms underpinning CNTN1′s oncogenic role remains incompletely understood, it is likely CNTN1 facilitate cancer migration and invasion through a combination of these aforementioned mechanisms (Figure 4).
4. Perspectives
In the last decade, CNTN1 has emerged as a promising oncogene in multiple cancer types. However, its oncogenic potential has not been thoroughly investigated, which limits our recognition of CNTN1′s utility in cancer diagnosis and therapy. In support of its therapeutic potential, a set of xenograft models reinforces CNTN1′s functionality in promoting tumorigenesis. To further investigate the contributions of CNTN1 in tumorigenesis and progression, a more physiologically relevant model that recapitulate the heterogeneity and tumour microenvironment would be a good step forward to advance current research. Considering CNTN1′s role in facilitating cell invasion and metastasis, it may be of interest to generate CNTN1-relevant conditional knock-in transgenic models to further explore the causal link between CNTN1 with cancer progression and metastasis. For example, a Cre-LoxP system can be explored in which mice carrying the transgene insertion of a strong translational and transcriptional termination (STOP) sequence flanked by loxP or FRT sites between the promoter sequence and CNTN1 which can be crossed with mice carrying tissue-specific promoters [111]. When Cre or FLP recombinase are expressed and present, the STOP cassette will be removed, allowing CNTN1 expression in the specific tissue of interest. This precise mediation of gene expression may allow a more informative investigation in the functionality of CNTN1 in carcinomas.
The involvement of CNTN1 in cancer formation and progression is supported by its common amplification in cancers and the association of this upregulation with the clinical features of cancers. The expression level of CNTN1 showed a clear association with clinicopathological parameters and poor prognosis in many cancers including lung [58], gastric [60], prostate [63], astrocytic gliomas [69], stomach [65], thyroid [7] cancers, hepatocellular carcinoma (HCC) [68] and oral squamous cell carcinoma (OSCC) [54]. As well, CNTN1 was discovered to be a novel biomarker for melanoma [106], glioblastoma [112], and colorectal cancer [113]. High CNTN1 expression was associated with biochemical recurrence following radical proctectomy in PC [64]. However, a competing endogenous RNA network study reported downregulation of CNTN1 in metastatic PC compared to primary PC [114]. In a high-throughput analysis from The Cancer Genome Atlas (TCGA) database, CNTN1 was identified as one of six pivotal prognostic EMT-related genes (ERGs) to affiliate with pro-oncogenic pathways and show a significant prognostic value in gastric cancer through OS analysis [61]. Similarly, an in-silico analysis of differentially expressed genes with functional protein products of NSCLC in five independent databases identified CNTN1 among with six other protein to associate with shorter OS and recurrence-free survival for predicted high-risk groups of NSCLC [115].
The upregulation of CNTN1, its association with worse clinical features, and its functionality in promoting cancer progression suggest CNTN1 being a potential target of cancer therapy. CNTN1′s physiological role as a neuronal cell adhesion molecule was likely explored by tumorigenesis. In view of the critical aspect of communications between cancer and stroma, proper adhesion is critical for cancer evolution or progression [97,98], targeting CNTN1 might thus be a useful option. However, it is important to note that while several drugs targeting cell adhesion molecules (e.g., V- or1-integrins) have shown promising outcomes in suppressing tumor growth in preclinical models, many of them have not significantly improved the disease-free or OS in patients [116]. Nonetheless, some encouraging findings have been made. For example, the anti-v antibody Intetumumab showed a non-significant trend towards improvements in OS compared to dacarbazine alone in a randomized, phase II study for patients with stage IV melanoma [117]. As well, the pan-V inhibitor abituzumab reduced the cumulative incidence of bone lesion progression compared to placebo arm in a randomized phase II trial for patients with metastatic castration-resistant PC, despite not significantly extending progression-free survival (PFS) [118]. Nonetheless, these clinical trials illustrate that directly targeting cell adhesion molecules alone may not be sufficient to bring survival advantages to patients, suggesting a need for combinational therapies involving cell adhesion-based treatment.
A potential combination is with the emerging immunotherapy. In this regard, studies have investigated the potential of using immunotherapies in conjunction with integrin inhibitors to improve therapeutic responses. Preclinical mouse models have showed that combined administration of a fusion protein, consisting of a Fc domain and integrin targeting peptide, with albumin/IL-2 or anti-PD-1 immunotherapy was able to significantly enhance anti-tumor immunity and improve survival [119]. CNTN1 is an attractive target of cancer therapy. CNTN1 physiological expression in the central nerve system and its upregulation in multiple cancer types may qualify it as a tumor-associated antigen (TAA). TAAs are well-regarded targets for developing immunotherapy [120]. CNTN1 was identified as a TAA along with other four TAAs (CRKII, CFL1, NME2, and TKT) in IDTH1-mutant lower grade gliomas through a proteomic and immunologic approach [121]. CNTN1 triggered the activation of endogenous T cells in a substantial proportion for up to 56% of patients with astrocytic and/or oligodendroglial IDH1-mutant lower grade gliomas, but none in healthy donors. Furthermore, the extracellular exposure of CNTN1 is an appealing feature for developing immunotherapies. It is thus intriguing to envisage the possibility of immunotargeting of CNTN1 with a concurrent inhibition of cancer cell adhesion.
Likely, mechanisms leading to CNTN1 upregulation in cancers are complex. For instance, while the VEGFC/Flt4 pathway unregulated CNTN1 expression in lung cancer [53], CNTN1 upregulation in prostate cancer is an outcome of the conversion of non-CSC PC cells to PCSC and CNTN1 in turn contributes to PCSC-derived metastasis [64,122]. The connection of CNTN1 to PCSC is appealing. CSCs play a crucial role in cancer recurrence, metastasis and acquisition of resistance for chemotherapy and radiotherapy [83]. The maintenance of CSCs in its undifferentiated stage largely relies on the stem cell niche of structural moieties such as E-cadherin and stemness promoting pathways including Wnt Notch, transforming growth factor (TGF)- and nuclear factor (NF)-Kb [123,124,125]. Collectively, future research should focus on the connections of CNTN1 with CSCs. While detailed mechanisms governing CNTN’s oncogenic functions require further investigations, its physiological role in neuron development via CNTN’s adhesion properties suggests this theme being possibly hijacked by tumorigenesis. This potential is in agreement with the robust enrichment of genes that are positively correlated with CNTN1 expression in the GO terms of “extracellular matrix organization” and “cell adhesion” (Table 2). The potential mechanisms underlying this adhesion modification remains unknown. Those of interaction proteins in neuron development (Figure 3) may shed light on the mechanisms responsible CNTN1-derived modulation of cancer adhesion. As CNTN1 lacks of a intracellular region (Figure 2 and Figure 4), it remains a possibility that CNTN1′s interaction with proteins functioning in tumorigenesis may include those residing in the lipid bilayer of cell membrane. Regardless the nature of CNTN1 binding proteins, identification of these is an appealing avenue of future investigation.
5. Conclusions
In recent decades, drastic improvement in strategies for detection and treatment of cancer patients have contributed to a marked decrease of cancer mortality worldwide. In the concept of “molecular targeted therapy”, correcting for alterations that occur in cancer progression and metastasis remains the fundamental goal. Changes in the intercellular adhesive properties of cancer cells as a consequence of altered CNTN1 expression certainly play a role in tumorigenesis. Positioned at the crossroad of pathways associated with cancer invasion and metastasis, CNTN1 represents a promising target for cancer therapy. Indeed, silencing or inhibition of CNTN1 in experimental models have already highlighted its suitability as a potential target in lung, gastric, prostate, thyroid cancers, and oral squamous cell carcinoma. Like all successful molecular targeted therapy, an effective CNTN1-targeted therapy in cancer patients would benefit from a rational selection of delivery contexts. Although CNTN1 only show limited expression in normal tissues, it is hard to know whether targeting CNTN1 in a tissue specific manner may have adverse clinical outcomes.
Author Contributions
Conceptualization, Y.G. and D.T.; material preparation, Y.G., T.L., A.K. and P.M.; writing—original draft preparation, Y.G. and D.T.; writing—review, all authors; writing editing—Y.G. and D.T.; supervision, D.T. All authors have read and agreed to the published version of the manuscript.
Funding
D.T. is supported by grants from Cancer Research Society, Canadian Cancer Society (grant #: 319412) and CIHR and by funds from Hamilton Urologic Oncology Research Center (HUORC). Y.G. is supported by Studentship provided by Ontario Graduate Scholarships and Research Institute of St Joe’s Hamilton.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Makrilia, N.; Kollias, A.; Syrigos, L.M.K. Cell Adhesion Molecules: Role and clinical significance in cancer. Cancer Investig. 2009, 27, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Multitasking in tumor progression: Signaling functions of cell adhesion molecules. Ann. N. Y. Acad. Sci. 2004, 1014, 58–66. [Google Scholar] [CrossRef]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Syrigos, K.; Karayiannakis, A.J. Adhesion molecules as targets for the treatment of neoplastic diseases. Curr. Pharm. Des. 2006, 12, 2849–2861. [Google Scholar] [CrossRef]
- Low, M.G. Glycosyl-phosphatidylinositol: A versatile anchor for cell surface proteins 1. FASEB J. 1989, 3, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-H.; Yu, J.-W.; Jiang, B.-J. Contactin 1: A potential therapeutic target and biomarker in gastric cancer. World J. Gastroenterol. 2015, 21, 9707–9716. [Google Scholar] [CrossRef]
- Shi, K.; Xu, N.; Yang, C.; Wang, L.; Pan, W.; Zheng, C.; Fan, L. Contactin 1 as a potential biomarker promotes cell proliferation and invasion in thyroid cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 12473–12481. [Google Scholar]
- Zhang, R.; Xie, L.; Liu, C.; Yang, H.; Lin, H.; Zhang, Q.; Tang, W.; Ji, F.; Sun, S. Contactin-1: A promising progression biomarker and therapeutic target of carcinoma. Minerva Med. 2017, 108, 193–195. [Google Scholar]
- Gennarini, G.; Bizzoca, A.; Picocci, S.; Puzzo, D.; Corsi, P.; Furley, A.J. The role of Gpi-anchored axonal glycoproteins in neural development and neurological disorders. Mol. Cell. Neurosci. 2017, 81, 49–63. [Google Scholar] [CrossRef]
- Bizzoca, A.; Corsi, P.; Gennarini, G. The mouse F3/contactin glycoprotein. Cell Adhes. Migr. 2009, 3, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Bizzocaa, A.; Corsia, P.; Polizzia, A.; Pintoa, M.F.; Xenaki, D. F3/Contactin acts as a modulator of neurogenesis during cerebral cortex development. Dev. Biol. 2012, 365, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Çolakoğlu, G.; Bergstrom-Tyrberg, U.; Berglund, E.O.; Ranscht, B. Contactin-1 regulates myelination and nodal/paranodal domain organization in the central nervous system. Proc. Natl. Acad. Sci. USA 2014, 111, E394–E403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dityatev, A.; Bukalo, O.; Schachner, M. Modulation of synaptic transmission and plasticity by cell adhesion and repulsion molecules. Neuron Glia Boil. 2008, 4, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.-D.; Ang, B.-T.; Karsak, M.; Hu, W.-P.; Cui, X.-Y.; Duka, T.; Takeda, Y.; Chia, W.; Sankar, N.; Ng, Y.-K.; et al. F3/contactin acts as a functional ligand for notch during oligodendrocyte maturation. Cell 2003, 115, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Berglund, E.O.; Murai, K.K.; Fredette, B.; Sekerková, G.; Marturano, B.; Weber, L.; Mugnaini, E.; Ranscht, B. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron 1999, 24, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, Y.; Watanabe, K. Contactins: Emerging key roles in the development and function of the nervous system. Cell Adh. Migr. 2009, 3, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Bouyain, S.; Watkins, D.J. The protein tyrosine phosphatases PTPRZ and PTPRG bind to distinct members of the contactin family of neural recognition molecules. Proc. Natl. Acad. Sci. USA 2010, 107, 2443–2448. [Google Scholar] [CrossRef] [Green Version]
- Peles, E.; Nativ, M.; Campbell, P.L.; Sakurai, T.; Martinez, R.; Lev, S.; O Clary, D.; Schilling, J.; Barnea, G.; Plowman, G.D.; et al. The carbonic anhydrase domain of receptor tyrosine phosphatase beta is a functional ligand for the axonal cell recognition molecule contactin. Cell 1995, 82, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, T.; Lustig, M.; Nativ, M.; Hemperly, J.J.; Schlessinger, J.; Peles, E.; Grumet, M. Induction of neurite outgrowth through contactin and nr-CAM by extracellular regions of glial receptor tyrosine phosphatase β. J. Cell Boil. 1997, 136, 907–918. [Google Scholar] [CrossRef]
- Parent, A.-S.; Mungenast, A.E.; Lomniczi, A.; Sandau, U.S.; Peles, E.; Bosch, M.A.; Rønnekleiv, O.K.; Ojeda, S.R. A contactin–receptor-like protein tyrosine phosphatase β complex mediates adhesive communication between astroglial cells and gonadotrophin-releasing hormone neurones. J. Neuroendocr. 2007, 19, 847–859. [Google Scholar] [CrossRef]
- Lamprianou, S.; Chatzopoulou, E.; Thomas, J.-L.; Bouyain, S.; Harroch, S. A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17498–17503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.-D.; Ma, Q.-H.; Gennarini, G.; Xiao, Z.-C. Cross-talk between F3/contactin and notch at axoglial interface: A role in oligodendrocyte development. Dev. Neurosci. 2006, 28, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Umemori, H.; Satot, S.; Yagi, T.; Aizawal, S.; Yamamoto, T. Initial events of myelination involve Fyn tyrosine kinase signalling. Nature 1994, 367, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. The role of nr. CAM in neural development and disorders—Beyond a simple glue in the brain. Mol. Cell. Neurosci. 2012, 49, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Pesheva, P.; Gennarini, G.; Goridis, C.; Schachner, M. The F3/11 cell adhesion molecule mediates the repulsion of neurons by the extracellular matrix glycoprotein J1-160/180. Neuron 1993, 10, 69–82. [Google Scholar] [CrossRef]
- Chen, Y.A.; Lu, I.L.; Tsai, J.W. Contactin-1/F3 regulates neuronal migration and morphogenesis through modulating rhoa activity. Front Mol. Neurosci. 2018, 11, 422. [Google Scholar] [CrossRef]
- Shimazaki, K.; Hosoya, H.; Takeda, Y.; Kobayashi, S.; Watanabe, K. Age-related decline of F3/contactin in rat hippocampus. Neurosci. Lett. 1998, 245, 117–120. [Google Scholar] [CrossRef]
- Compton, A.G.; Albrecht, D.E.; Seto, J.; Cooper, S.T.; Ilkovski, B.; Jones, K.J.; Challis, D.; Mowat, D.; Ranscht, B.; Bahlo, M.; et al. Mutations in Contactin-1, a Neural adhesion and neuromuscular junction protein, cause a familial form of lethal congenital myopathy. Am. J. Hum. Genet. 2008, 83, 714–724. [Google Scholar] [CrossRef] [Green Version]
- Falk, J.; Bonnon, C.A.; Girault, J.; Faivre-Sarrailh, C. F3/contactin, a neuronal cell adhesion molecule implicated in axogenesis and myelination. Boil. Cell 2002, 94, 327–334. [Google Scholar] [CrossRef]
- Prag, S.A.; Lepekhin, E.; Kolkova, K.; Hartmann-Petersen, R.; Kawa, A.; Walmod, P.S.; Belman, V.; Gallagher, H.C.; Berezin, V.; Bock, E.; et al. NCAM regulates cell motility. J. Cell Sci. 2002, 115, 283–292. [Google Scholar]
- Lehembre, F.; Yilmaz, M.; Wicki, A.; Schomber, T.; Strittmatter, K.; Ziegler, D.; Kren, A.; Went, P.; Derksen, P.W.B.; Berns, A.; et al. NCAM-induced focal adhesion assembly: A functional switch upon loss of E-cadherin. EMBO J. 2008, 27, 2603–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaheta, R.A.; Hundemer, M.; Mayer, G.; Vogel, J.-U.; Kornhuber, B.; Činátl, J.; Markus, B.H.; Driever, P.H.; Cinatl, J., Jr. Expression Level of Neural Cell Adhesion Molecule (NCAM) Inversely Correlates with the Ability of Neuroblastoma Cells to Adhere to Endothelium In Vitro. Cell Commun. Adhes. 2002, 9, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Daher, F.H.; Michaelis, M.; Hasenberg, C.; Weich, E.M.; Jonas, D.; Kotchetkov, R.; Doerr, H.W.; Cinatl, J. Chemoresistance induces enhanced adhesion and transendothelial penetration of neuroblastoma cells by down-regulating NCAM surface expression. BMC Cancer 2006, 6, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Santón, A.; Bellas, C. Neural cell adhesion molecule expression in plasma cells in bone marrow biopsies and aspirates allows discrimination between multiple myeloma, monoclonal gammopathy of uncertain significance and polyclonal plasmacytosis. Histopathology 2004, 44, 375–380. [Google Scholar] [CrossRef]
- Bataille, R.; Jégo, G.; Robillard, N.; Barillé-Nion, S.; Harousseau, J.-L.; Moreau, P.; Amiot, M.; Pellat-Deceunynck, C. The phenotype of normal, reactive and malignant plasma cells. Identification of “many and multiple myelomas” and of new targets for myeloma therapy. Haematologica 2006, 91, 1234–1240. [Google Scholar]
- Raspadori, D.; Damiani, D.; Lenoci, M.; Rondelli, D.; Testoni, N.; Nardi, G.; Sestigiani, C.; Mariotti, C.; Birtolo, S.; Tozzi, M.; et al. CD56 antigenic expression in acute myeloid leukemia identifies patients with poor clinical prognosis. Leukemia 2001, 15, 1161–1164. [Google Scholar] [CrossRef]
- Yang, D.-H.; Lee, J.-J.; Mun, Y.-C.; Shin, H.-J.; Kim, Y.-K.; Cho, S.-H.; Chung, I.-J.; Seong, C.-M.; Kim, H.-J. Predictable prognostic factor of CD56 expression in patients with acute myeloid leukemia with t(8:21) after high dose cytarabine or allogeneic hematopoietic stem cell transplantation. Am. J. Hematol. 2007, 82, 1–5. [Google Scholar] [CrossRef]
- Kameda, K.; Shimada, H.; Ishikawa, T.; Takimoto, A.; Momiyama, N.; Hasegawa, S.; Misuta, K.; Nakano, A.; Nagashima, Y.; Ichikawa, Y. Expression of highly polysialylated neural cell adhesion molecule in pancreatic cancer neural invasive lesion. Cancer Lett. 1999, 137, 201–207. [Google Scholar] [CrossRef]
- Tezel, E.; Kawase, Y.; Takeda, S.; Oshima, K.; Nakao, A. Expression of Neural Cell Adhesion Molecule in Pancreatic Cancer. Pancreas 2001, 22, 122–125. [Google Scholar] [CrossRef]
- Naito, Y.; Kinoshita, H.; Okabe, Y.; Kawahara, R.; Sakai, T.; Suga, H.; Arikawa, S.; Oshima, K.; Kojiro, M. CD56 (NCAM) expression in pancreatic carcinoma and the surrounding pancreatic tissue. Kurume Med. J. 2006, 53, 59–62. [Google Scholar] [CrossRef] [Green Version]
- Miyahara, R.; Tanaka, F.; Nakagawa, T.; Matsuoka, K.; Isii, K.; Wada, H. Expression of neural cell adhesion molecules (polysialylated form of neural cell adhesion molecule and L1-cell adhesion molecule) on resected small cell lung cancer specimens: In relation to proliferation state. J. Surg. Oncol. 2001, 77, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Berglund, E.O.; Ranscht, B. Molecular Cloning and in Situ Localization of the Human Contactin Gene (CNTN1) on Chromosome 12q11-q12. Genomics 1994, 21, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Acampora, D.; D’Esposito, M.; Faiella, A.; Pannese, M.; Migliaccio, E.; Morelli, F.; Stornaiuolo, A.; Nigro, V.; Simeone, A.; Boncinelli, E. The human HOX gene family. Nucleic Acids Res. 1989, 17, 10385–10402. [Google Scholar] [CrossRef]
- Sosnoski, D.M.; Emanuel, B.S.; Hawkins, A.L.; Van Tuinen, P.; Ledbetter, D.H.; Nussbaum, R.L.; Kaos, F.T.; Schwartz, E.; Phillips, D.; Bennett, J.S. Chromosomal localization of the genes for the vitronectin and fibronectin receptors alpha subunits and for platelet glycoproteins IIb and IIIa. J. Clin. Investig. 1988, 81, 1993–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arheden, K.; Mandahl, N.; Heim, S.; Mitelman, F. In situ hybridization localizes the human type II alpha 1 collagen gene (COL2A1) to 12q13. Hereditas 2008, 110, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Kraus, M.H.; Issing, W.; Miki, T.; Popescu, N.C.; Aaronson, S.A. Isolation and characterization of ERBB3, a third member of the ERBB/epidermal growth factor receptor family: Evidence for overexpression in a subset of human mammary tumors. Proc. Natl. Acad. Sci. USA 1989, 86, 9193–9197. [Google Scholar] [CrossRef] [Green Version]
- Arheden, K.; Mandahl, N.; Strömbeck, B.; Isaksson, M.; Mitelman, F. Chromosome localization of the human oncogene INT1 to 12q13 by in situ hybridization. Cytogenet Cell Genet. 1988, 47, 86–87. [Google Scholar] [CrossRef]
- Desgrosellier, S.J.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Stetten, G.; Sroka, B.; Corson, V.L.; Opitz, J.M.; Reynolds, J.F.; Norbury-Glaser, M. Fragile site in chromosome 12 in a patient with two miscarriages. Am. J. Med Genet. 1988, 31, 521–525. [Google Scholar] [CrossRef]
- Mitelman, F. Catalog of Chromosome Abberrations in Cancer, 4th ed.; Wiley-Liss: New York, NY, USA, 1991. [Google Scholar]
- Reifenberger, G.; Reifenberger, J.; Ichimura, K.; Collins, V.P. Amplification at 12q13–14 in human malignant gliomas is frequently accompanied by loss of heterozygosity at loci proximal and distal to the amplification site. Cancer Res. 1995, 55, 731–734. [Google Scholar]
- Warr, T.J.; Ward, S.; Burrows, J.; Harding, B.; Wilkins, P.; Harkness, W.; Hayward, R.; Darling, J.; Thomas, D. Identification of extensive genomic loss and gain by comparative genomic hybridisation in malignant astrocytoma in children and young adults. Genes Chromosom. Cancer 2001, 31, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Su, J.-L.; Yang, P.-C.; Shih, J.-Y.; Yang, C.-Y.; Wei, L.-H.; Hsieh, C.-Y.; Chou, C.-H.; Jeng, Y.-M.; Wang, M.-Y.; Chang, K.-J.; et al. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell 2006, 9, 209–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigetomi, S.; Imanishi, Y.; Shibata, K.; Sakai, N.; Sakamoto, K.; Fujii, R.; Habu, N.; Otsuka, K.; Sato, Y.; Watanabe, Y.; et al. VEGF-C/Flt-4 axis in tumor cells contributes to the progression of oral squamous cell carcinoma via upregulating VEGF-C itself and contactin-1 in an autocrine manner. Am. J. Cancer Res. 2018, 8, 2046–2063. [Google Scholar] [PubMed]
- Wang, J.; Huang, Y.; Zhang, J.; Wei, Y.; Mahoud, S.; Bakheet, A.M.H.; Tang, J. Pathway-related molecules of VEGFC/D-VEGFR3/NRP2 axis in tumor lymphangiogenesis and lymphatic metastasis. Clin. Chim. Acta 2016, 461, 165–171. [Google Scholar] [CrossRef]
- Hung, Y.-H.; Hung, W.-C. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) enhances invasiveness of lung cancer cells by up-regulating contactin-1 via the α7 nicotinic acetylcholine receptor/ERK signaling pathway. Chem. Interact. 2009, 179, 154–159. [Google Scholar] [CrossRef]
- Xu, S.; Lam, S.-K.; Cheng, P.N.-M.; Ho, J. Contactin 1 modulates pegylated arginase resistance in small cell lung cancer through induction of epithelial-mesenchymal transition. Sci. Rep. 2019, 9, 12030. [Google Scholar] [CrossRef]
- Yan, J.; Wong, N.; Hung, C.L.; Chen, W.X.-Y.; Tang, D. Contactin-1 reduces e-cadherin expression via activating akt in lung cancer. PLoS ONE 2013, 8, e65463. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Sun, S.; Ji, F.; Liu, C.; Lin, H.; Xie, L.; Yang, H.; Tang, W.; Zhou, Y.; Xu, J.; et al. CNTN-1 enhances chemoresistance in human lung adenocarcinoma through induction of epithelial-mesenchymal transition by targeting the PI3K/Akt pathway. Cell. Physiol. Biochem. 2017, 43, 465–480. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.-W.; Wu, S.-H.; Lu, R.-Q.; Wu, J.-G.; Ni, X.-C.; Zhou, G.-C.; Jiang, H.-G.; Zheng, L.-H.; Li, X.-Q.; Du, G.-Y.; et al. Expression and significances of contactin-1 in human gastric cancer. Gastroenterol. Res. Pr. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zhou, S.; Liu, B. Identification and validation of an individualized emt-related prognostic risk score formula in gastric adenocarcinoma patients. BioMed Res. Int. 2020, 2020, 7082408. [Google Scholar] [CrossRef]
- Xu, S.; Lam, S.-K.; Cheng, P.N.-M.; Ho, J. Recombinant human arginase induces apoptosis through oxidative stress and cell cycle arrest in small cell lung cancer. Cancer Sci. 2018, 109, 3471–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Yang, X.; Zhao, T.; Du, H.; Wang, T.; Zhong, S.; Yang, B.; Li, H. Upregulation of contactin-1 expression promotes prostate cancer progression. Oncol. Lett. 2019, 19, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Ojo, D.; Kapoor, A.; Lin, X.; Pinthus, J.H.; Aziz, T.; Bismar, T.A.; Wei, F.; Wong, N.; De Melo, J.; et al. Neural cell adhesion protein CNTN1 promotes the metastatic progression of prostate cancer. Cancer Res. 2016, 76, 1603–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-C.; Zhao, J.; Hu, C.-E.; Gan, J.; Zhang, W.; Huang, G. Comprehensive analysis of vascular endothelial growth factor-c related factors in stomach cancer. Asian Pac. J. Cancer Prev. 2014, 15, 1925–1929. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Zhou, J.; Zhu, H.; Xie, L.; Wang, F.; Liu, B.; Shen, W.; Ye, W.; Xiang, B.; Zhu, X.; et al. VEGF-C promotes the development of esophageal cancer via regulating CNTN-1 expression. Cytokine 2011, 55, 8–17. [Google Scholar] [CrossRef]
- Tsai, K.-H.; Hsien, H.-H.; Chen, L.-M.; Ting, W.-J.; Yang, Y.-S.; Kuo, C.-H.; Tsai, C.-H.; Tsai, F.J.; Tsai, H.J.; Huang, E.-J. Rhubarb inhibits hepatocellular carcinoma cell metastasis via GSK-3-β activation to enhance protein degradation and attenuate nuclear translocation of β-catenin. Food Chem. 2013, 138, 278–285. [Google Scholar] [CrossRef]
- Li, G.-Y.; Huang, M.; Pan, T.-T.; Jia, W.-D. Expression and prognostic significance of contactin 1 in human hepatocellular carcinoma. OncoTargets Ther. 2016, 9, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Eckerich, C.; Zapf, S.; Ulbricht, U.; Müller, S.; Fillbrandt, R.; Westphal, M.; Lamszus, K. Contactin is expressed in human astrocytic gliomas and mediates repulsive effects. Glia 2005, 53, 1–12. [Google Scholar] [CrossRef]
- Liu, P.; Chen, S.; Wu, W.; Liu, B.; Shen, W.; Wang, F.; He, X.; Zhang, S. Contactin-1 (CNTN-1) Overexpression is Correlated with Advanced Clinical Stage and Lymph Node Metastasis in Oesophageal Squamous Cell Carcinomas. Jpn. J. Clin. Oncol. 2012, 42, 612–618. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-M.; Cao, W.; Ye, D.; Ren, G.-X.; Wu, Y.-N.; Guo, W. Contactin 1 (CNTN1) expression associates with regional lymph node metastasis and is a novel predictor of prognosis in patients with oral squamous cell carcinoma. Mol. Med. Rep. 2012, 6, 265–270. [Google Scholar] [CrossRef]
- Zhang, R.; Yao, W.; Qian, P.; Li, Y.; Jiang, C.; Ao, Z.; Qian, G.; Wang, C.; Wu, G.; Li, J.; et al. Increased sensitivity of human lung adenocarcinoma cells to cisplatin associated with downregulated contactin-1. Biomed. Pharmacother. 2015, 71, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, Y.; Li, C.; Xu, P.; Gao, Y.; Xu, Y. CNTN-1 promotes docetaxel resistance and epithelial-to-mesenchymal transition via the PI3K/Akt signaling pathway in prostate cancer. Arch. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Kaushal, V.; Mukunyadzi, P.; A Dennis, R.; Siegel, E.R.; E Johnson, D.; Kohli, M. Stage-specific characterization of the vascular endothelial growth factor axis in prostate cancer: Expression of lymphangiogenic markers is associated with advanced-stage disease. Clin. Cancer Res. 2005, 11, 584–593. [Google Scholar] [PubMed]
- Li, Q.; Dong, X.; Gu, W.; Qiu, X.; Wang, E. Clinical significance of co-expression of VEGF-C and VEGFR-3 in non-small cell lung cancer. Chin. Med J. 2003, 116, 727–730. [Google Scholar] [PubMed]
- Witte, D.; Thomas, A.; Ali, N.; Carlson, N.; Younes, M. Expression of the vascular endothelial growth factor receptor-3 (VEGFR-3) and its ligand VEGF-C in human colorectal adenocarcinoma. Anticancer. Res. 2002, 22, 1463–1466. [Google Scholar]
- Neuchrist, C.; Erovic, B.M.; Handisurya, A.; Fischer, M.B.; Steiner, G.E.; Hollemann, D.; Gedlicka, C.; Saaristo, A.; Burian, M. Vascular endothelial growth factor C and vascular endothelial growth factor receptor 3 expression in squamous cell carcinomas of the head and neck. Head Neck 2003, 25, 464–474. [Google Scholar] [CrossRef]
- Van Trappen, P.O.; Steele, D.; Lowe, D.G.; Baithun, S.; Beasley, N.; Thiele, W.; Weich, H.; Krishnan, J.; Shepherd, J.H.; Pepper, M.S.; et al. Expression of vascular endothelial growth factor (VEGF)-C and VEGF-D, and their receptor VEGFR-3, during different stages of cervical carcinogenesis. J. Pathol. 2003, 201, 544–554. [Google Scholar] [CrossRef]
- Zhang, Y.; Weinberg, R.A. Epithelial-to-mesenchymal transition in cancer: Complexity and opportunities. Front. Med. 2018, 12, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Ksiazkiewicz, M.; Markiewicz, A.; Zaczek, A.J. Epithelial-mesenchymal transition: A hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiolohy 2012, 79, 195–208. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Boil. 2014, 15, 178–196. [Google Scholar] [CrossRef] [Green Version]
- Molina, J.R.; Agarwal, N.K.; Morales, F.C.; Hayashi, Y.; Aldape, K.D.; Cote, G.; Georgescu, M.-M. PTEN, NHERF1 and PHLPP form a tumor suppressor network that is disabled in glioblastoma. Oncogene 2011, 31, 1264–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Eun, K.; Ham, S.W.; Kim, H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017, 50, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Colombel, M.; Eaton, C.L.; Hamdy, F.; Ricci, E.; Van Der Pluijm, G.; Cecchini, M.; Mege-Lechevallier, F.; Clézardin, P.; Thalmann, G. Increased expression of putative cancer stem cell markers in primary prostate cancer is associated with progression of bone metastases. Prostate 2011, 72, 713–720. [Google Scholar] [CrossRef]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nature 2013, 15, 274–283. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; He, S.; Geng, J.; Song, Z.-J.; Han, P.-H.; Qin, J.; Zhao, Z.; Song, Y.-C.; Wang, H.-X.; Dang, C.-X. Overexpression of Contactin 1 promotes growth, migration and invasion in Hs578T breast cancer cells. BMC Cell Boil. 2018, 19, 5. [Google Scholar] [CrossRef] [Green Version]
- Jézéquel, P.; Campone, M.; Gouraud, W.; Guérin-Charbonnel, C.; Leux, C.; Ricolleau, G.; Campion, L. bc-GenExMiner: An easy-to-use online platform for gene prognostic analyses in breast cancer. Breast Cancer Res. Treat. 2011, 131, 765–775. [Google Scholar] [CrossRef]
- Jézéquel, P.; Frénel, J.-S.; Campion, L.; Guérin-Charbonnel, C.; Gouraud, W.; Ricolleau, G.; Campone, M. bc-GenExMiner 3.0: New mining module computes breast cancer gene expression correlation analyses. Database 2013, 2013, bas060. [Google Scholar] [CrossRef]
- Breast Cancer Gene-Expression Miner v4.5. 22 June 2020. Available online: http://bcgenex.ico.unicancer.fr (accessed on 15 June 2020).
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; A Rees, C.; Pollack, J.R.; Ross, U.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar]
- Haakensen, V.D.; Lingjærde, O.C.; Lüders, T.; Riis, M.L.; Prat, A.; Troester, M.A.; Holmen, M.M.; Frantzen, J.O.; Romundstad, L.; Navjord, D.; et al. Gene expression profiles of breast biopsies from healthy women identify a group with claudin-low features. BMC Med Genom. 2011, 4, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Oprea-Ilies, G.M.; Krishnamurti, U. New developments in breast cancer and their impact on daily practice in pathology. Arch. Pathol. Lab. Med. 2017, 141, 490–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yersal, Ö.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Esposito, M.; Kang, Y. Bone metastasis and the metastatic niche. J. Mol. Med. 2015, 93, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, M.; Guise, T.A.; Kang, Y. The Biology of Bone Metastasis. Cold Spring Harb. Perspect. Med. 2018, 8, a031252. [Google Scholar] [CrossRef]
- Bagnall, P. Diagnosis and treatment of prostate cancer. Nurs. Times 2014, 110, 12–15. [Google Scholar]
- Schweppe, R.E.; Kerege, A.A.; Sharma, V.; Poczobutt, J.M.; Gutierrez-Hartmann, A.; Grzywa, R.L.; Haugen, B.R. Distinct genetic alterations in the mitogen-activated protein kinase pathway dictate sensitivity of thyroid cancer cells to mitogen-activated protein kinase kinase 1/2 inhibition. Thyroid 2009, 19, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Grubbs, E.G.; Ng, P.K.-S.; Bui, J.; Busaidy, N.L.; Chen, K.; Lee, J.E.; Lu, X.; Lu, H.; Meric-Bernstam, F.; Mills, G.B.; et al. RET fusion as a novel driver of medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2014, 100, 788–793. [Google Scholar] [CrossRef] [Green Version]
- Jeong, W.-J.; Mo, J.H.; Park, M.W.; Choi, I.J.; An, S.-Y.; Jeon, E.-H.; Ahn, S.-H. Sunitinib inhibits papillary thyroid carcinoma with RET/PTC rearrangement but not BRAF mutation. Cancer Boil. Ther. 2011, 12, 458–465. [Google Scholar] [CrossRef] [Green Version]
- Shiozaki, A.; Shen-Tu, G.; Bai, X.; Iitaka, D.; De Falco, V.; Santoro, M.; Keshavjee, S.; Liu, M. XB130 mediates cancer cell proliferation and survival through multiple signaling events downstream of akt. PLoS ONE 2012, 7, e43646. [Google Scholar] [CrossRef] [PubMed]
- Łuczkowska, K.; Rogińska, D.; Litwińska, Z.; Paczkowska, E.; Schmidt, C.A.; Machaliński, B. Molecular Mechanisms of Bortezomib Action: Novel Evidence for the miRNA–mRNA Interaction Involvement. Int. J. Mol. Sci. 2020, 21, 350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Zhao, R.; Ni, H.; Zhao, K.; He, Y.; Fang, S.; Chen, Q.-L. Molecule mechanisms of Ganoderma lucidum treated hepatocellular carcinoma based on the transcriptional profiles and miRNA-target network. Biomed. Pharmacother. 2020, 125, 110028. [Google Scholar] [CrossRef]
- Mauerer, A.; Roesch, A.; Hafner, C.; Stempfl, T.; Wild, P.; Meyer, S.; Landthaler, M.; Vogt, T. Identification of new genes associated with melanoma. Exp. Dermatol. 2011, 20, 502–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinnix, C.C.; Lee, J.T.; Liu, Z.-J.; McDaid, R.; Balint, K.; Beverly, L.J.; Brafford, P.A.; Xiao, M.; Himes, B.; Zabierowski, S.E.; et al. Active Notch1 confers a transformed phenotype to primary human melanocytes. Cancer Res. 2009, 69, 5312–5320. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Teoh, S.L. Notch signalling pathways and their importance in the treatment of cancers. Curr. Drug Targets 2017, 18, 1. [Google Scholar]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Tratar, U.L.; Horvat, S.; Cemazar, M. Transgenic mouse models in cancer research. Front. Oncol. 2018, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nord, H.; Hartmann, C.; Andersson, R.; Menzel, U.; Pfeifer, S.; Piotrowski, A.; Bogdan, A.; Kloc, W.; Sandgren, J.; Olofsson, T.; et al. Characterization of novel and complex genomic aberrations in glioblastoma using a 32K BAC array. Neuro-Oncology 2009, 11, 803–818. [Google Scholar] [CrossRef]
- Kok-Sin, T.; Mokhtar, N.M.; Hassan, N.Z.A.; Sagap, I.; Rose, I.M.; Harun, R.; Jamal, R. Identification of diagnostic markers in colorectal cancer via integrative epigenomics and genomics data. Oncol. Rep. 2015, 34, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Tao, W.; He, T.; Li, L.-Y. Prediction of potential prognostic biomarkers in metastatic prostate cancer based on a competing endogenous RNA regulatory network. Res. Sq. 2020. preprint. [Google Scholar]
- Cury, S.S.; De Moraes, D.; Freire, P.P.; De Oliveira, G.; Marques, D.V.P.; Fernandez, G.J.; Dal-Pai-Silva, M.; Hasimoto, É.N.; Reis, P.; Rogatto, S.R.; et al. Tumor transcriptome reveals high expression of IL-8 in non-small cell lung cancer patients with low pectoralis muscle area and reduced survival. Cancers 2019, 11, 1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjunpää, H.; Asens, M.L.; Guenther, C.; Fagerholm, S.C. Cell adhesion molecules and their roles and regulation in the immune and tumor microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [Green Version]
- O’Day, S.; on the behalf of the CNTO 95 Investigators; Pavlick, A.; Loquai, C.; Lawson, D.; Gutzmer, R.; Richards, J.; Schadendorf, D.; Schadendorf, D.; Thompson, J.A.; et al. A randomised, phase II study of intetumumab, an anti-αv-integrin mAb, alone and with dacarbazine in stage IV melanoma. Br. J. Cancer 2011, 105, 346–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Le Moulec, S.; Gimmi, C.; Bruns, R.; Straub, J.; Miller, K. Differential effect on bone lesions of targeting integrins: Randomized phase II trial of abituzumab in patients with metastatic castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 3192–3200. [Google Scholar] [CrossRef] [Green Version]
- Kwan, B.H.; Zhu, E.F.; Tzeng, A.; Sugito, H.R.; Eltahir, A.; Ma, B.; Delaney, M.K.; Murphy, P.A.; Kauke, M.J.; Angelini, A.; et al. Integrin-targeted cancer immunotherapy elicits protective adaptive immune responses. J. Exp. Med. 2017, 214, 1679–1690. [Google Scholar] [CrossRef]
- Peres, L.D.P.; Da Luz, F.A.C.; Pultz, B.D.A.; Brigido, P.C.; De Araujo, R.A.; Goulart, L.R.; Silva, M. Peptide vaccines in breast cancer: The immunological basis for clinical response. Biotechnol. Adv. 2015, 33, 1868–1877. [Google Scholar] [CrossRef]
- Dettling, S.; Stamova, S.; Warta, R.; Schnölzer, M.; Rapp, C.; Rathinasamy, A.; Reuss, D.E.; Pocha, K.; Roesch, S.; Jungk, C.; et al. Identification of CRKII, CFL1, CNTN1, NME2, and TKT as novel and frequent T-cell targets in human IDH-mutant Gglioma. Clin. Cancer Res. 2018, 24, 2951–2962. [Google Scholar] [CrossRef] [Green Version]
- Mei, W.; Lin, X.; Kapoor, A.; Gu, Y.; Zhao, K.; Tang, D. The contributions of prostate cancer stem cells in prostate cancer initiation and metastasis. Cancers 2019, 11, 434. [Google Scholar] [CrossRef] [Green Version]
- Nie, D.; Boral, D. Cancer stem cells and niche mircoenvironments. Front. Biosci. 2012, 4, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcellos-Hoff, M.H.; Lyden, D.; Wang, T.C. The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer 2013, 13, 511–518. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Systemic literature searching conditions and selection of articles for review.

Figure 2.
Schematic representation of the Contactin 1 (CNTN1) structure. CNTN1 is a 1020 amino acids (AA) protein comprised of six immunoglobulin (Ig)-like repeats followed by four fibronectin (FN) type III-like domains and is connected by a glycosylphosphatidylinositol (GPI)-anchor to the plasma membrane.
Figure 2.
Schematic representation of the Contactin 1 (CNTN1) structure. CNTN1 is a 1020 amino acids (AA) protein comprised of six immunoglobulin (Ig)-like repeats followed by four fibronectin (FN) type III-like domains and is connected by a glycosylphosphatidylinositol (GPI)-anchor to the plasma membrane.

Figure 3.
Schematic summary of traditional extracellular interactions of CNTN1.

Figure 4.
Graphic illustration of the currently known mechanisms underlying CNTN1-induced migration and invasion of cancer cells. CNTN1 acts as a downstream effector of the VEGFC-VEFGR3/Flt4 axis, leading to further activation of the CCAAT enhancer binding protein α (CEBPA) facilitated SRC/p38 mitogen-activated protein kinase (MAPK) signaling. CNTN1 activates Notch signaling via interacting with NOTCH1 and notch intracellular domain (NCID). CNTN1 inhibits E-cadherin in part via transcriptional regulation of SIP1 and Slug as well as AKT activation. The inhibition of E-cadherin and upregulation of Vimentin and N-cadherin are characteristic events of EMT.
Figure 4.
Graphic illustration of the currently known mechanisms underlying CNTN1-induced migration and invasion of cancer cells. CNTN1 acts as a downstream effector of the VEGFC-VEFGR3/Flt4 axis, leading to further activation of the CCAAT enhancer binding protein α (CEBPA) facilitated SRC/p38 mitogen-activated protein kinase (MAPK) signaling. CNTN1 activates Notch signaling via interacting with NOTCH1 and notch intracellular domain (NCID). CNTN1 inhibits E-cadherin in part via transcriptional regulation of SIP1 and Slug as well as AKT activation. The inhibition of E-cadherin and upregulation of Vimentin and N-cadherin are characteristic events of EMT.

Figure 5.
Box and whisker plot of CNTN1 expression examined by bc-GenExMiner 4.5 [91] from RNA-seq (A) and DNA microarray (B) separated by breast cancer subtypes. Global significant difference between groups was assessed by Welch’s test, followed by Dunnett–Tukey–Kramer’s test for pairwise comparison; **** p > 0.0001.
Figure 5.
Box and whisker plot of CNTN1 expression examined by bc-GenExMiner 4.5 [91] from RNA-seq (A) and DNA microarray (B) separated by breast cancer subtypes. Global significant difference between groups was assessed by Welch’s test, followed by Dunnett–Tukey–Kramer’s test for pairwise comparison; **** p > 0.0001.

Figure 6.
CNTN1 associates with alterations in overall survival (OS). Survival analyzes were performed on all 33 The Cancer Genome Atlas (TCGA) cancer types organized in the Gene Expression Profiling Interactive Analysis (GEPIA2) website. OS with respect to CNTN1 expression was shown for all 33 TCGA cancer types (a), BLCA (b), LGG (c), and STAD (d). BLCA: bladder urothelial carcinoma; LGG: brain lower grade glioma; STAD: stomach adenocarcinoma. Group cut-off was set at 50% and hazard ratio was calculated based on Cox Proportional-Hazards Model.
Figure 6.
CNTN1 associates with alterations in overall survival (OS). Survival analyzes were performed on all 33 The Cancer Genome Atlas (TCGA) cancer types organized in the Gene Expression Profiling Interactive Analysis (GEPIA2) website. OS with respect to CNTN1 expression was shown for all 33 TCGA cancer types (a), BLCA (b), LGG (c), and STAD (d). BLCA: bladder urothelial carcinoma; LGG: brain lower grade glioma; STAD: stomach adenocarcinoma. Group cut-off was set at 50% and hazard ratio was calculated based on Cox Proportional-Hazards Model.



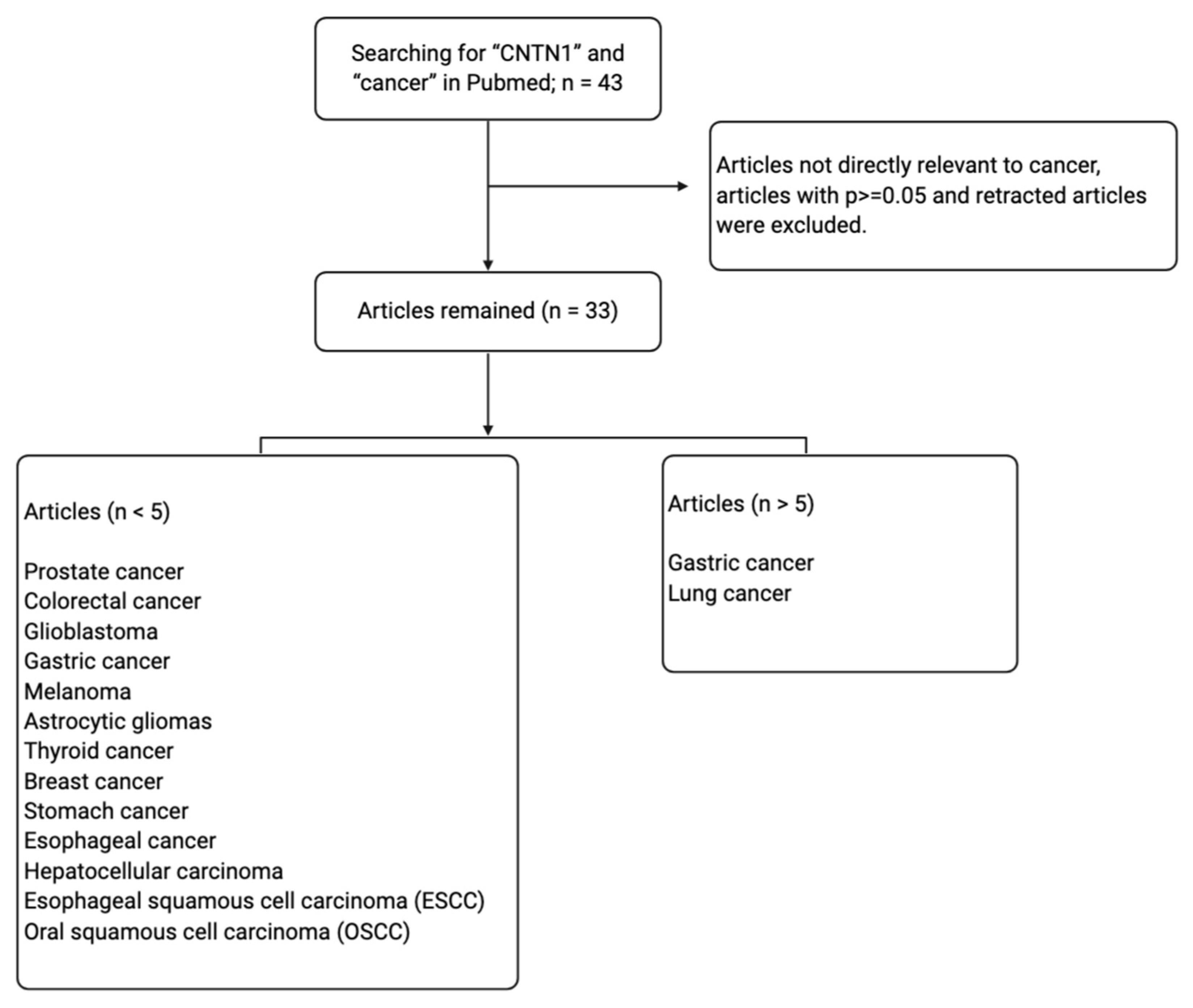{kind=link}
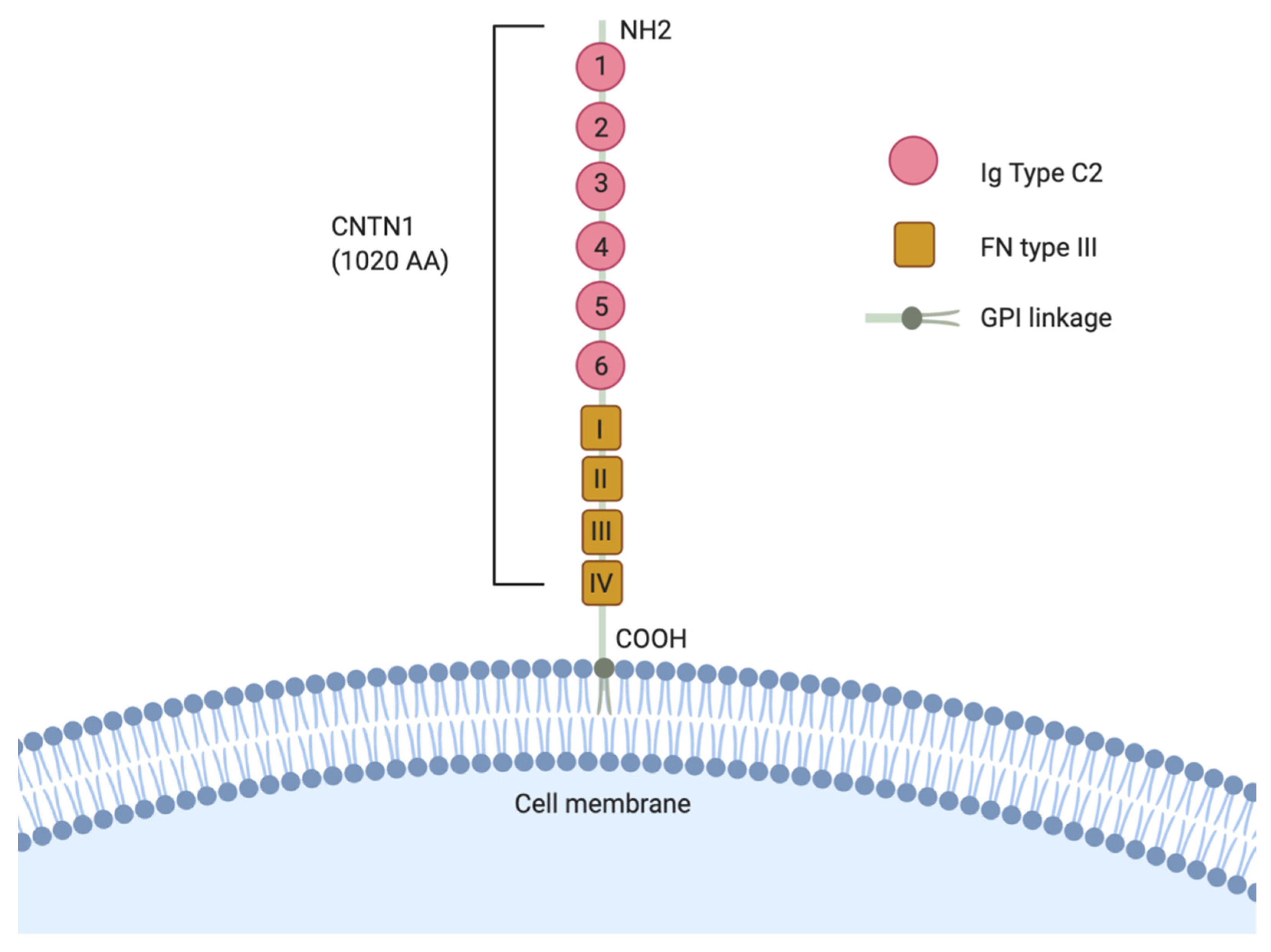{kind=link}
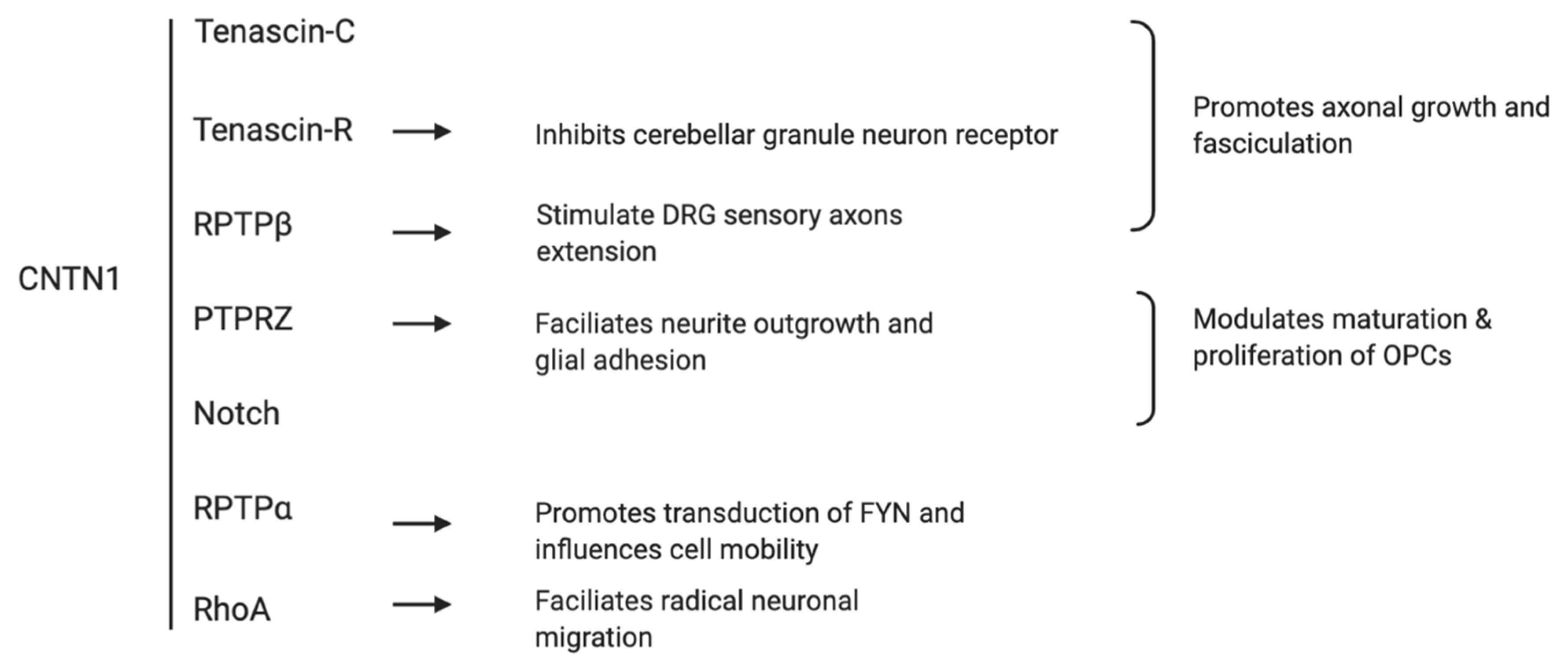{kind=link}
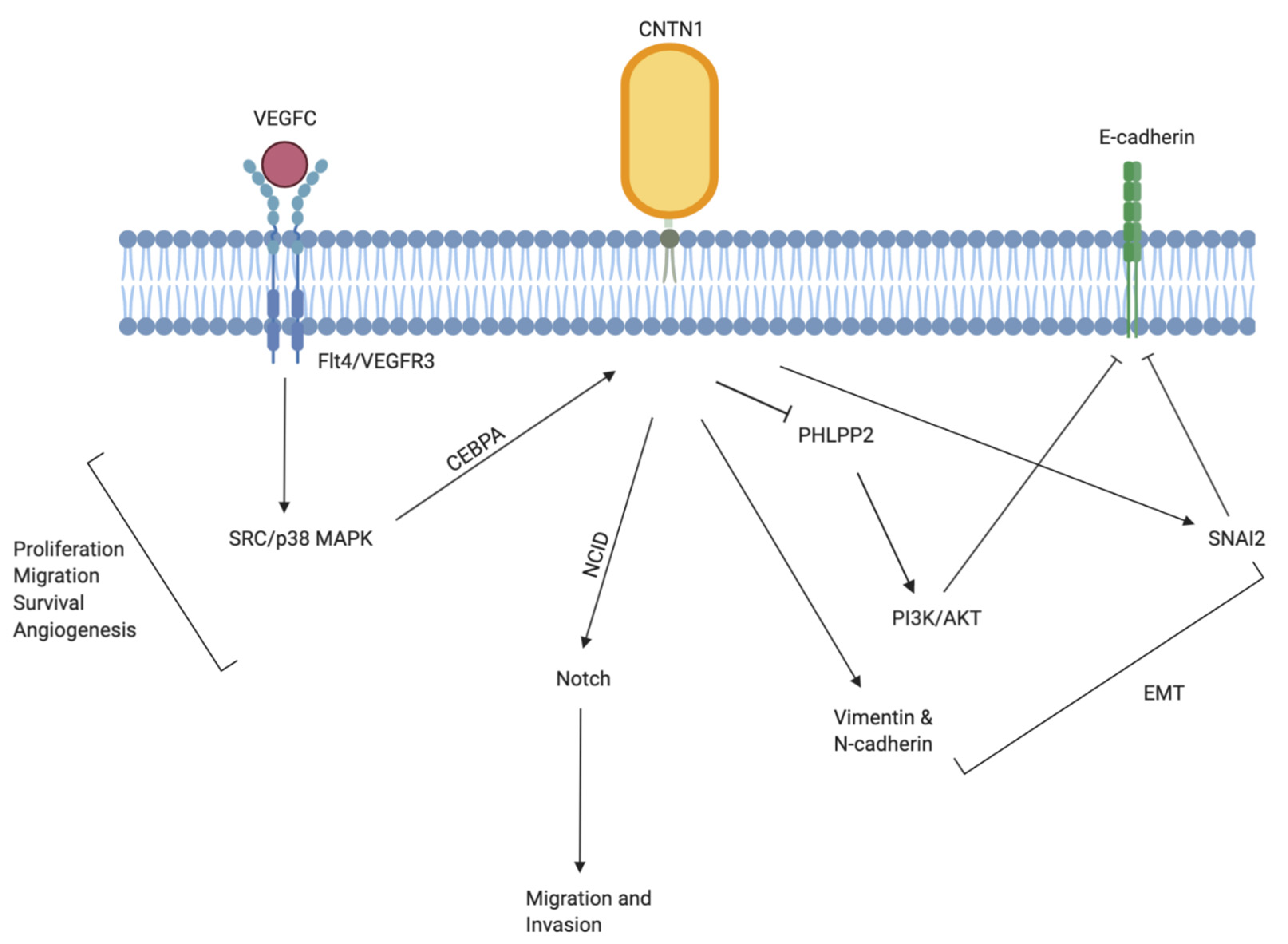{kind=link}
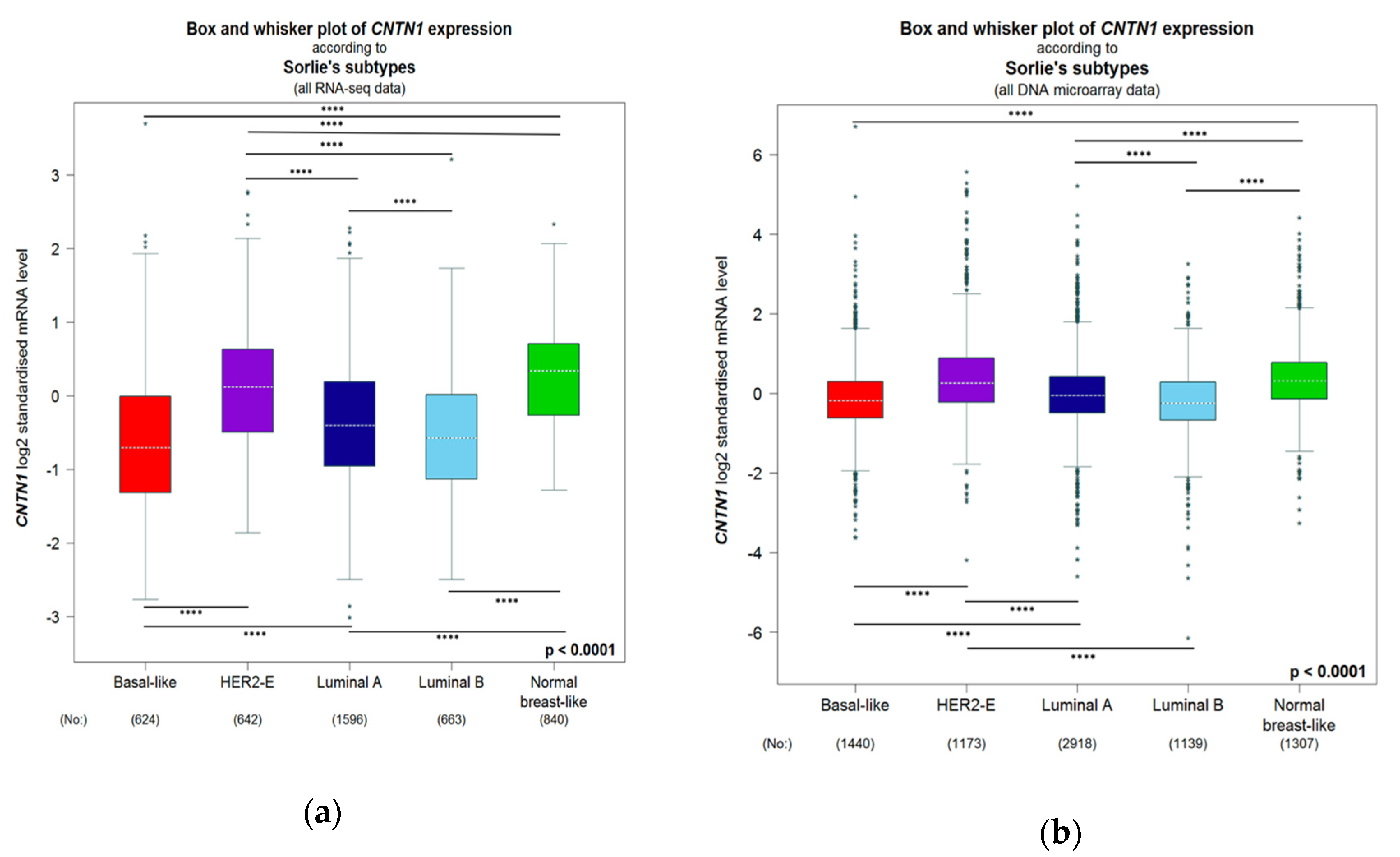{kind=link}
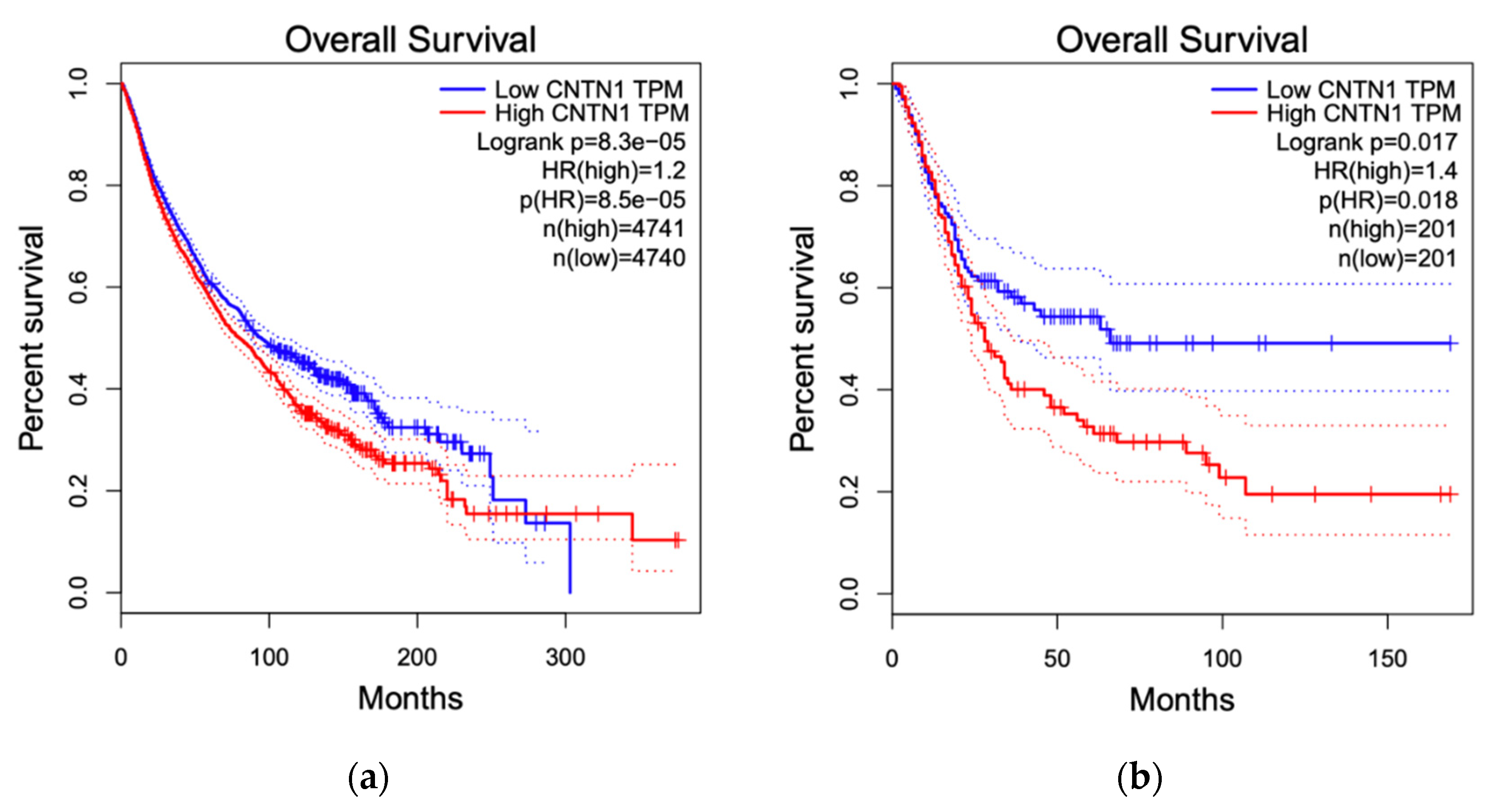{kind=link}
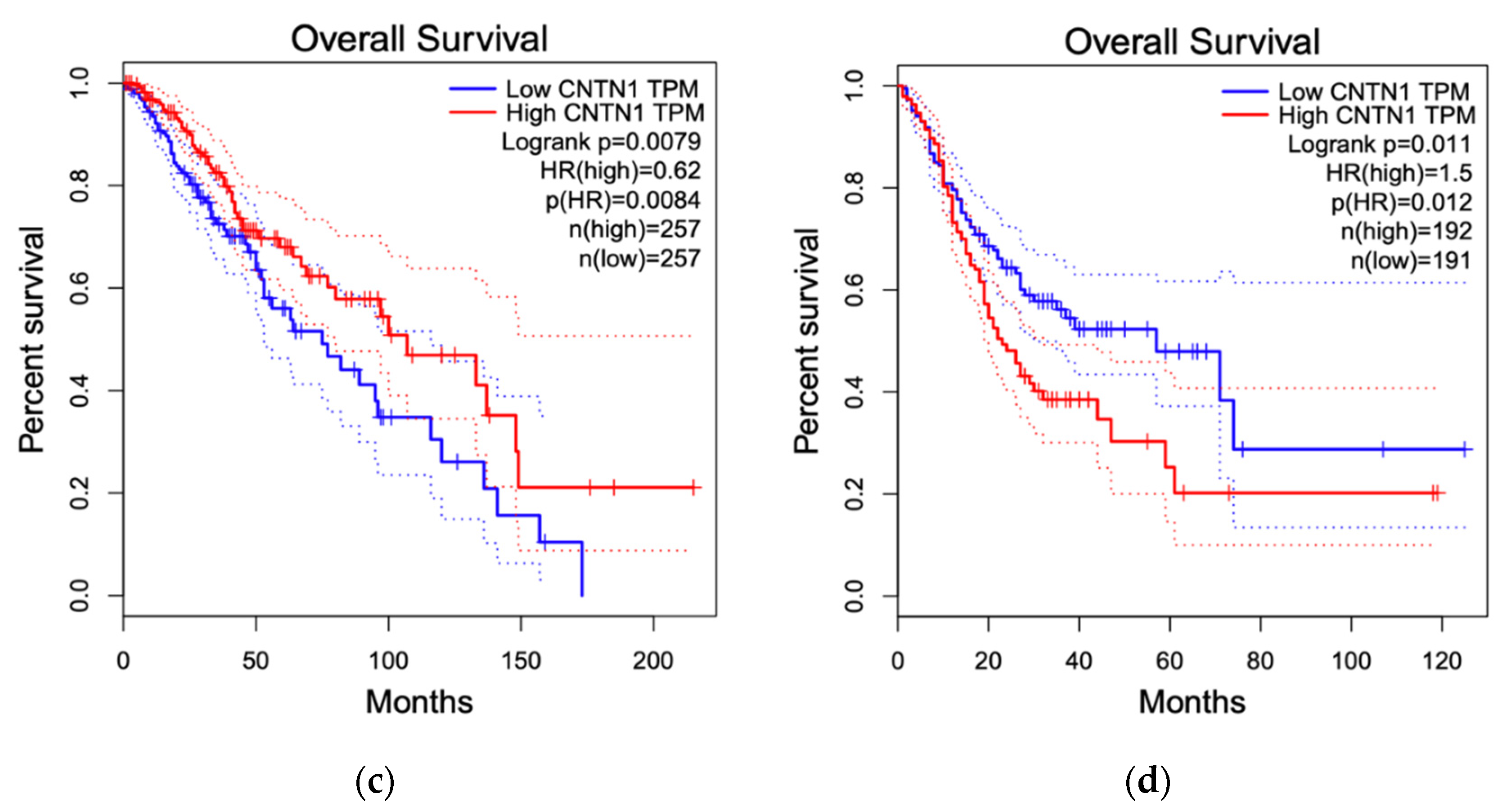{kind=link}
Table 1.
Changes in CNTN1 expression in cancer and correlation with tumor progression.
| Tumor Type | CNTN1 Expression in Cancer and Major Findings | References |
|---|---|---|
| Lung cancer |
| [58] |
| [59] | |
| [56] | |
| [57] | |
| [53] | |
| [72] | |
| Gastric adenocarcinoma (GAC) |
| [61] |
| [60] | |
| Oesophageal squamous cell carcinoma (ESCC) |
| [70] |
| [66] | |
| Oral squamous cell carcinoma (OSCC) |
| [54] |
| [71] | |
| Hepatocellular carcinoma (HCC) |
| [68] |
| Astrocytic Gliomas |
| [69] |
| Breast cancer |
| [62] |
| Thyroid cancer |
| [7] |
| Stomach cancer |
| [65] |
| Prostate cancer (PC) |
| [64] |
| [63] | |
| [73] |
Table 2.
GO enrichment of CNTN1-positively correlated genes.
| Go ID | Term | p-Value | %Target List | Associated Genes |
|---|---|---|---|---|
| All patients | ||||
| GO:0030198 | extracellular matrix organization | −5 | 14.63 | CCDC80, COL8A2, ECM2, FBN1, ITGA11, LOX |
| GO:0007155 | cell adhesion | −5 | 17.07 | CCN4, CDH11, CNTN1, COL12A1, ITGA11, OMD, SPON1 |
| Basal-like | ||||
| GO:0030198 | extracellular matrix organization | −15 | 14.08 | ADAM12, APBB2, CCDC80, COL3A1, COL6A3, CRISPLD2, DCN, ECM2, FBLN1, FBN1, JAM2, JAM3, LAMA2, MATN3, MMP16, MMP2, NDNF, POSTN, SMOC2, VCAN |
| GO:0001501 | skeletal system development | −8 | 7.75 | CDH11, CHRD, COL3A1, EVC, EBN1, IGF2, MATN3, MMP16, PTH1R, TLL1, VCAN |
| GO:0007155 | cell adhesion | −8 | 12.68 | ADAM12, CDH11, CNTN1, COL6A3, EDIL3, FAP, FEZ1, JCAD, LAMA2, NUAK1, OMD, PCDH7, POSTN, SEMA5A, SPOCK1, SVEP1, THBS2, VCAN |
| Her2-E | ||||
| GO:0030198 | extracellular matrix organization | −19 | 21.74 | ADAM12, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, COL8A1, COL8A2, CRISPLD2, DCN, DDR2, ECM2, FBN1, JAM3, LOX, LUM, MMP2, RECK, SPARC, VCAN |
| GO:0001501 | skeletal system development | −7 | 8.7 | CDH11, COL1A2, COL1A2, COL3A1, COL5A2, EVC, FBN1, VCAN |
| GO:0007155 | cell adhesion | −7 | 15.22 | ADAM12, CCN4, CDH11, CNTN1, COL5A1, COL8A1, DDR2, EDIL3, FAP, OMD, PCDH7, SPON1, THBS2, VCAN |
| Luminal A | ||||
| GO:0030198 | extracellular matrix organization | −38 | 9.83 | ADAM12, ADAM19, BGN, CCDC80, COL10A1, COL11A1, COL14A1, COL16A1, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, COL5A3, COL6A1, COL6A2, COL6A3, COL7A1, COL8A1, COL8A2, COMP, CRISPLD2, CDN, DDR2, ECM2, EGFL6, ELN, FBLN1, FBLN2, FBLN5, FBN1, FN1, FOXF2, ITGA11, ITGB1, JAM2, JAM3, LAMA1, LAMA2, LAMA4, LAMB1, LAMC1, LOX, LOXL1, LUM, MATN3, MFAP2, MFAP5, MMP11, MMP13MMP14, MMP16, MMP19, MMP2, MMP27, NID1, NID2, PDGFRA, POSTN, PXDN, RECK, SH3PXD2A, SPARC, VCAN |
| GO:0001501 | skeletal system development | −14 | 11.67 | ALX4, ARSE, BMP1, BMP7, CDH11, COL10A1, COL1A1, COL1A2, COL3A1, COL6A2, COMP, EVC, FBN1, FRZB, GJA5, GLI2, HOXA11, IGF1, IGF2, MATN3, MMP14, MMP16, NKX3–2, PTH1R, RASSF2, SH3PXD2B, SHOX2, TLL1, VCAN |
| GO:0007155 | cell adhesion | −29 | 3.53 | ADAM12, ADAM23, BOC, CCN2, CCN4, CDH11, CERCAM, CNTN1, COL12A1, COL16A1, COL5A1, COL5A3, COL6A1, COL6A2, COL6A3, COL6A6, COL7A1, COL8A1, CXCL12, CYP1B1, DDR2, DPP4, DPT, EGFL6, EMILIN1, ENTPD1, FAP, FAT1, FEZ1, FN1, GAS6, GPNMB, ISLRITGA11, ITGB1, ITGBL1, JCAD, LAMA1, LAMA2, LAMA4, LAMB1, LAMC1, LOXL2, LSAMP, MFAP4, MFGE8, MXRA8, NID2, NRP2, NTM, NUAK1, OMD, PCDH18, PCDH7, PCDHB12, PCDHB18P, PCDHGA12, PCDHGC3, PLXNC1, POSTN, PPFIBP1, PRKCA, PTK7, ROBO1, SEMA5A, SPOCK1, SPON1, SPON2, SRPX, SSPN, TGFB1I1, THBS2, TLN2, TNFAIP6, VCAN, VCL |
| Luminal B | ||||
| GO:0030198 | extracellular matrix organization | −38 | 11.07 | ADAM12, BGN, CCDC80, COL10A1, COL11A1, COL14A1, COL16A1, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, COL5A3, COL6A1, COL6A2, COL6A3, COL7A1, COL8A1, COL8A2, COMP, CRISPLD2, DCN, DDR2, ECM2, ELN, FBLN1, FBLN2, FBLN5, FBN1, FN1, FOXF2, HSPG2, ITGA11, JAM3, LAMA1, LAMA2, LAMA4, LAMB1, LAMC1, LOX, LOXL1, LUM, MFAP2, MFAP2, MFAP5, MMP11, MMP13, MMP14, MMP19, MMP2, NDNF, NID1, NID2, PDGFRA, POSTN, PXDN, RECK, SH3PXD2A, SPARC, VCAN |
| GO:0001501 | skeletal system development | −17 | 5.44 | ARSE, BMP1, CDH11, COL10A1, COL1A1, COL1A2, COL3A1, COL5A2, COMP, DLX5, EN1, EVC, FBN1, FRZB, GJA5, GLI2, HOXA10, HOXA11, HOXA4, IGF2, MMP14, NKX3–2, PAX1, PRELP, PTH1R, SH3PXD2B, SHOX2, TLL1, VCAN |
| GO:0007155 | cell adhesion | −25 | 12.01 | ADAM12, CCN4, CDH11, CNTN1, COL5A1, COL8A1, DDR2, EDIL3, FAP, OMD, PCDH7, SPON1, THBS2, VCAN |
| Normal-like | ||||
| GO:0030198 | extracellular matrix organization | −5 | 11.48 | COL14A1, COL8A2, DDR2, JAM3, LAMA2, MMP16, RECK |
| GO:0007155 | cell adhesion | −4 | 11.48 | CNTN1, DDR2, LAMA2, PCDH7, SEMA5A, SPON1, SSPN |
Results were generated using the breast cancer genetic data and tools provided by bc-GenExMiner 4.5. The RNA-seq data was used. All patients: all breast cancers are included. Basal-like, Her2-E, Luminal A, Luminal B, and Normal-like are intrinsic subtypes of breast cancer.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gu, Y.; Li, T.; Kapoor, A.; Major, P.; Tang, D. Contactin 1: An Important and Emerging Oncogenic Protein Promoting Cancer Progression and Metastasis. Genes 2020, 11, 874. https://doi.org/10.3390/genes11080874
AMA Style
Gu Y, Li T, Kapoor A, Major P, Tang D. Contactin 1: An Important and Emerging Oncogenic Protein Promoting Cancer Progression and Metastasis. Genes. 2020; 11(8):874. https://doi.org/10.3390/genes11080874
Chicago/Turabian StyleGu, Yan, Taosha Li, Anil Kapoor, Pierre Major, and Damu Tang. 2020. "Contactin 1: An Important and Emerging Oncogenic Protein Promoting Cancer Progression and Metastasis" Genes 11, no. 8: 874. https://doi.org/10.3390/genes11080874
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.