2.1. Chemical Synthesis
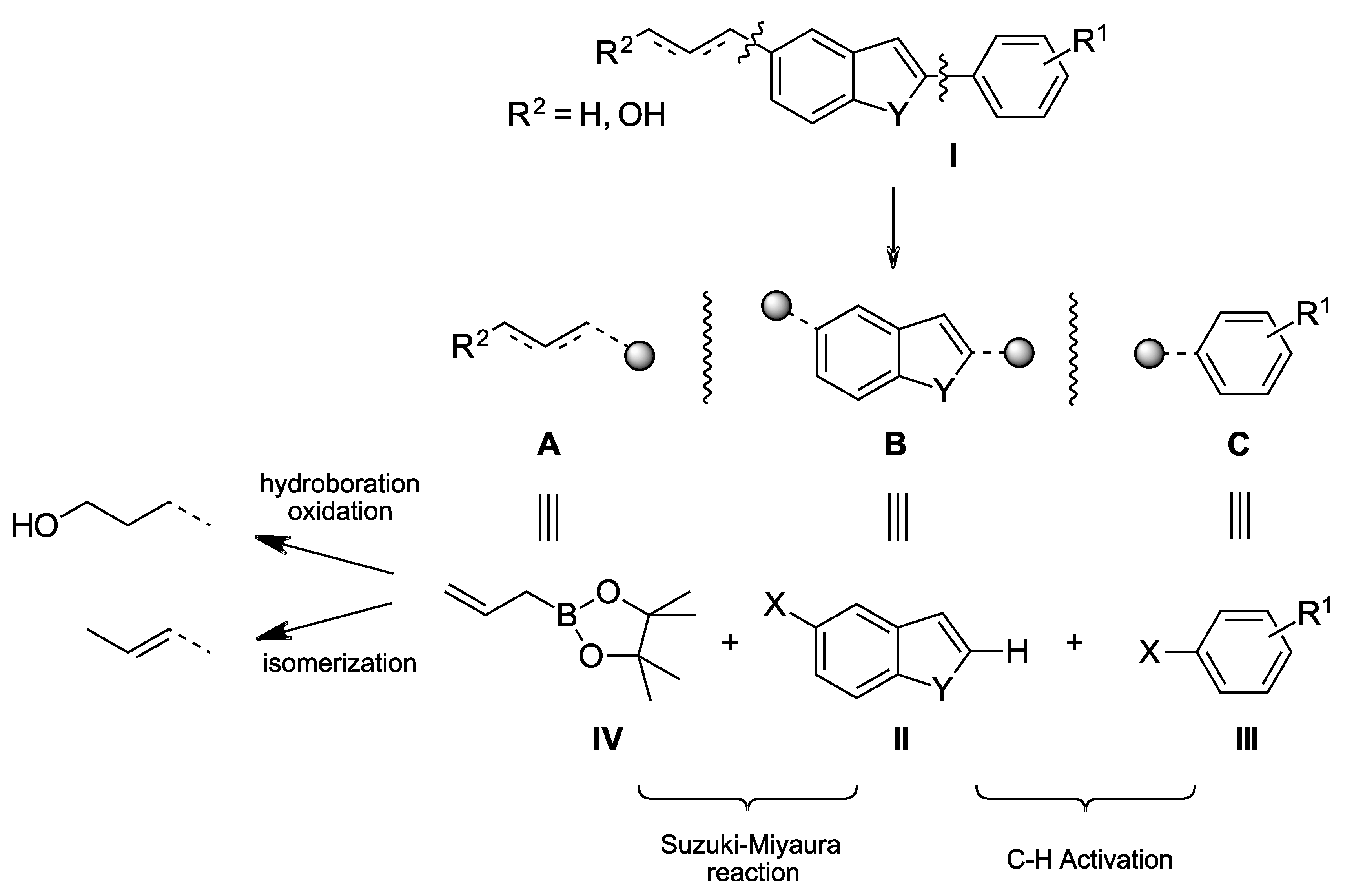
2.1.1. Synthesis of 5-Allylbenzo[b]furan (14)
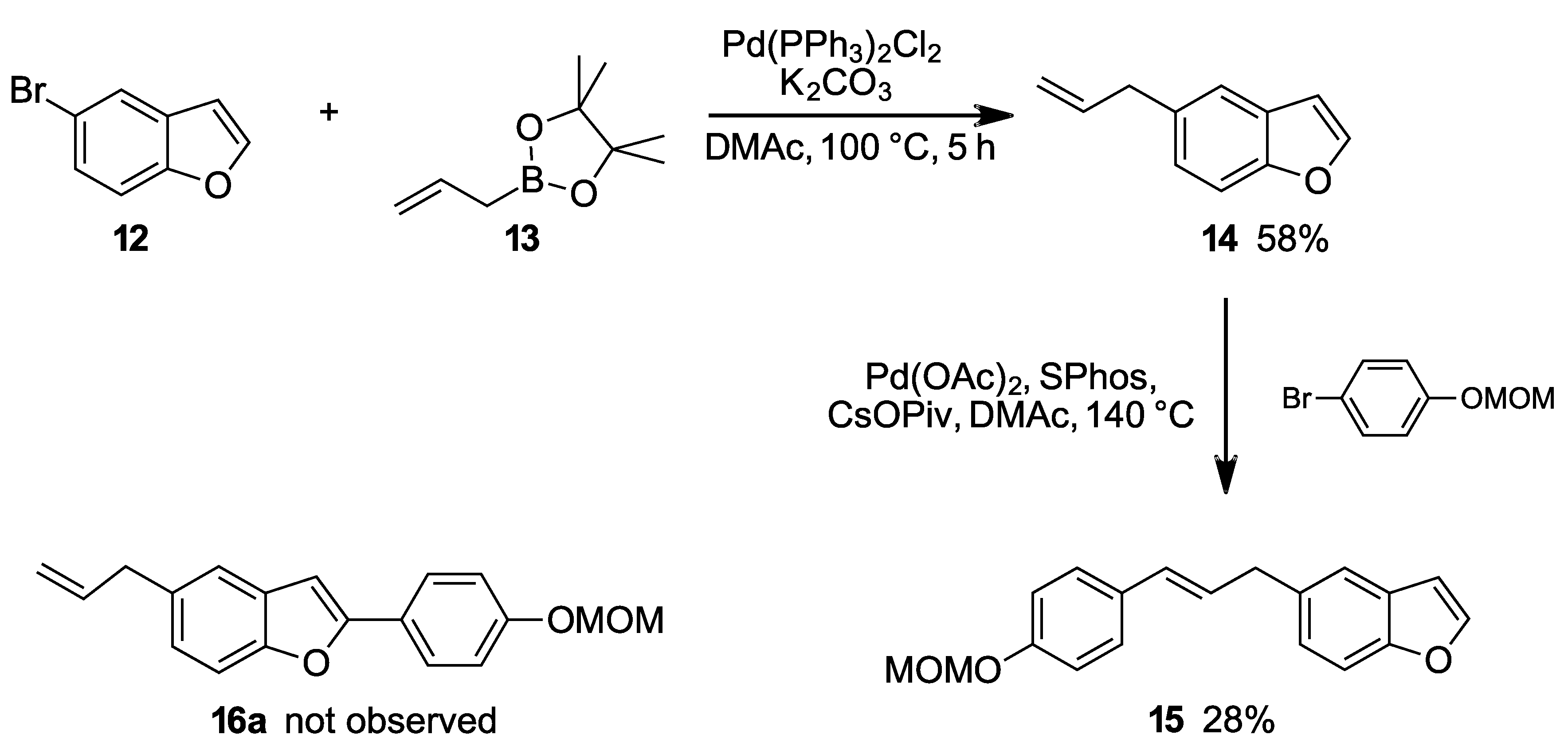
From 5-bromobenzo[b]furan 12: Compound 12 (98 mg, 0.5 mmol), allyl-B(pin) (140 µL, 0.75 mmol, 1.5 equiv.), K2CO3 (138 mg, 1.0 mmol, 2.0 equiv.) and Pd(PPh3)2Cl2 (18 mg, 0.025 mmol, 5 mol %) was mixed in 1 mL DMAc at 100 °C and stirred for 24 h. The reaction mixture was then cooled to room temperature and filtered through celite, washed with EtOAc. The product was washed with saturated aq. NH4Cl and a last time with 10 mL brine then dried over Na2SO4 and the solvent was evaporated under reduced pressure to obtain coupling product 14 (67 mg, colorless oil), 85% yield.
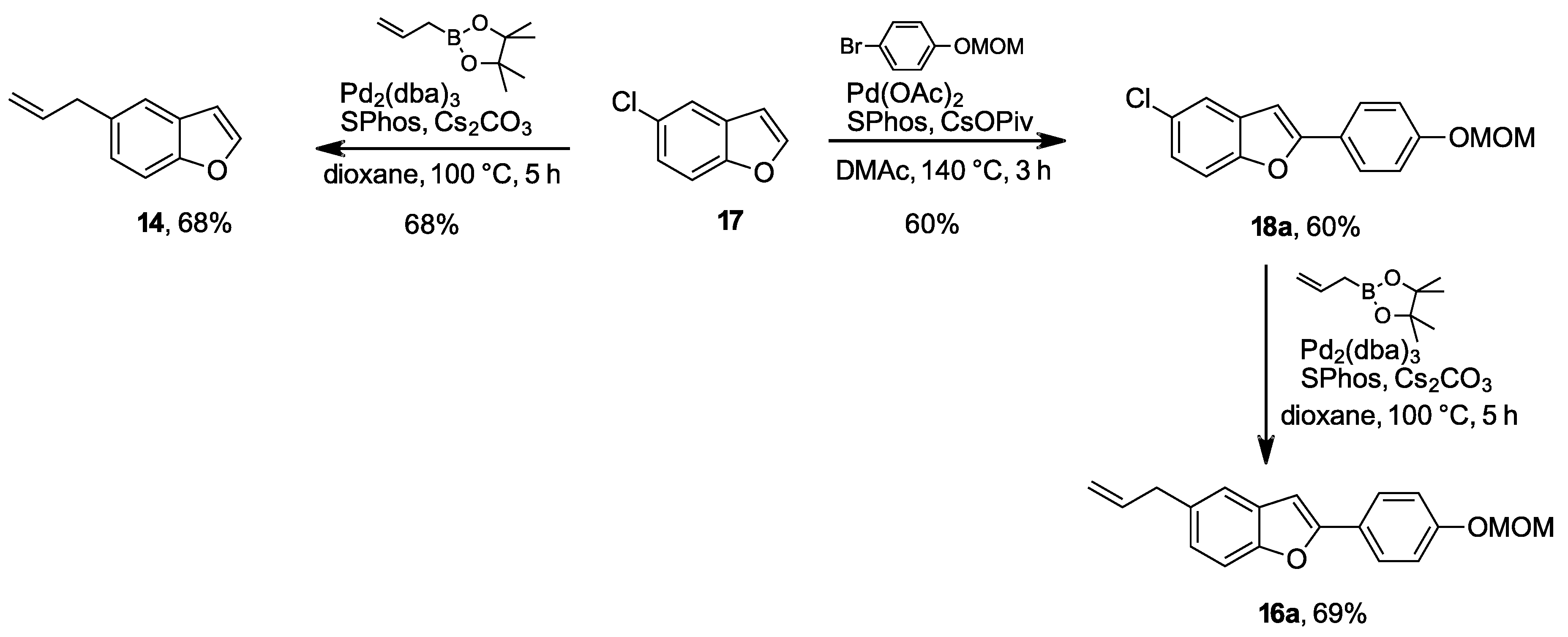
From 5-chlorobenzo[b]furan 17: Compound 17 (76 mg, 0.5 mmol), allyl-B(pin) (126 mg, 140 µL, 0.75 mmol, 1.5 equiv.), Cs2CO3 (326 mg, 1.0 mmol, 2.0 equiv.), SPhos (21 mg, 0.05 mmol, 10 mol %) and Pd2(dba)3 (23 mg, 0.025 mmol, 5 mol %) was mixed in 2 mL DMAc and at 100 °C and stirred for 6 h. The reaction mixture was then cooled to room temperature and filtered through celite and washed with EtOAc. The product was washed with saturated aq. NH4Cl and a last time with 10 mL brine then dried over Na2SO4 and the solvent was evaporated under reduced pressure to obtain coupling product 14 (54 mg, colorless oil), 68% yield.
1H-NMR (200 MHz, CDCl3) δ (ppm) 3.51 (d, J = 6.7 Hz, 2H), 5.08–5.17 (m, 2H), 5.95–6.15 (m, 1H), 6.73–6.75 (m, 1H), 7.16 (d, J = 8.6 Hz, 1H), 7.44–7.48 (m, 2H), 7.62 (d, J = 2.1 Hz, 1H). 13C-NMR (50 MHz, CDCl3) δ (ppm) 40.1, 106.4, 111.1, 115.6, 120.7, 125.1, 127.6, 134.5, 138.0, 145.1, 153.7. MS analyst, m/z (Int.) 158(100), 157(46), 129(82), 128(50), 115(18), 102(10), 89(10), 77(22), 63(15).
2.1.2. General Procedure of C-H Activation Reaction on 5-Chlorobenzo[b]furan (17)
Procedure A: Benzo-fused starting material (1.0 mmol, 1.0 equiv.), aryl bromide (1.5 mmol, 1.5 equiv.), cesium pivalate (350 mg, 1.5 mmol, 1.5 equiv.), Pd(OAc)2 (9 mg, 0.04 mmol, 0.04 equiv.) and SPhos (16.4 mg, 0.08 mmol, 0.08 equiv.) was mixed in 2 mL of degassed DMAc. The mixture was stirred at 140 °C for 24 h in argon atmosphere. The reaction mixture was cooled to room temperature and then diluted with 15 mL diethyl ether or ethyl acetate (depending on the polarity of the product) and filtered through a pad of celite. The organic phase was washed with saturated NH4Cl solution, once with brine and then dried over Na2SO4. The solvent was removed under reduced pressure. Purification was performed on silica gel eluting with LP or LP/EtOAc mixtures (depending on the polarity of the product).
2.1.3. 5-Chloro-2-(4-(methoxymethoxy)phenyl)benzo[b]furan (18a)
Prepared according to the general procedure A. mp 138–141 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.50 (s, 3H), 5.22 (s, 2H), 6.82 (s, 1H), 7.09–7.13 (m, 2H), 7.19 (dd, J = 8.7, 2.1 Hz, 1H), 7.39 (d, J = 8.7 Hz, 1H), 7.50 (d, J = 2.1 Hz, 1H), 7.76 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 56.1, 94.3, 99.5, 111.9, 116.5 (2C), 120.1, 123.8, 123.9, 126.5 (2C), 128.4, 130.8, 153.1, 157.4, 157.9. HR-MS analyst [M + H]+ m/z (predicted) = 289.0631, m/z (measured) = 289.0636, difference = −1.90 ppm.
2.1.4. 5-Chloro-2-(4-methoxyphenyl)benzo[b]furan (18c)
Prepared according to the general procedure A. mp 144–146 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 3.86 (s, 3H), 6.81 (s, 1H), 6.95–7.00 (m, 2H), 7.18 (dd, J = 8.7, 2.1 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 2.1 Hz, 1H), 7.76–7.80 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 55.4, 99.2, 111.9, 114.3 (2C), 120.1, 122.8, 123.8, 126.6 (2C), 128.3, 130.9, 153.1, 157.5, 160.3.
2.1.5. 5-Chloro-2-(3,5-dimethoxyphenyl)benzo[b]furan (18d)
Prepared according to the general procedure A. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.86 (s, 6H), 6.48 (t, J = 2.3 Hz, 1H), 6.93 (d, J = 0.8 Hz, 1H), 6.99 (d, J = 2.3 Hz, 2H), 7.23 (dd, J = 8.7, 2.1 Hz, 1H), 7.42 (d, J = 8.7 Hz, 1H), 7.53 (d, J = 2.1 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 55.5 (2C), 101.3, 101.4 (2C), 103.2, 112.1, 120.5, 124.5, 128.5, 130.5, 131.7, 153.2, 157.2, 161.2 (2C).
2.1.6. 5-Chloro-2-phenylbenzo[b]furan (18e)
Prepared according to the general procedure A. mp 125–128 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 6.96 (s, 1H), 7.20–7.55 (m, 6H), 7.81–7.87 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 100.8, 112.1, 120.4, 124.4, 125.0 (2C), 128.5, 128.8 (2C), 129.0, 130.0, 130.6, 153.2, 157.4.
2.1.7. 5-Chloro-2-(4-fluorophenyl)benzo[b]furan (18f)
Prepared according to the general procedure A. mp 119–122 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 6.86 (s, 1H), 7.09–7.24 (m, 3H), 7.38–7.52 (m, 2H), 7.76–7.83 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 100.5, 112.1, 116.0 (d, 2JC-F = 21.9 Hz), 120.4, 124.4, 126.3 (d, 4JC-F = 3.4 Hz), 126.9 (d, 3JC-F = 8.3 Hz, 2C), 128.6, 130.5, 153.2, 156.5, 163.1 (d, 1JC-F = 247.9 Hz).
2.1.8. 5-Chloro-2-(4-(difluoromethyl)phenyl)benzo[b]furan (18g)
Prepared according to the general procedure A. mp 121–124 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 6.69 (t, JH-F = 56.4 Hz, 1H), 7.02 (s, 1H), 7.26 (dd, J = 8.7, 2.1 Hz, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.55–7.62 (m, 3H), 7.90–7.93 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 102.1, 112.2, 114.4 (t, 1JC-F = 237.6 Hz), 120.7, 125.0, 125.2 (2C), 126.1 (t, 3JC-F = 6.1 Hz), 128.7, 130.3, 132.2, 134.6 (t, 2JC-F = 22.5 Hz), 153.4, 156.1. HR-MS analyst [M + H]+ m/z (predicted) = 279.0388, m/z (measured) = 279.0392, difference = −1.62 ppm.
2.1.9. 5-Chloro-2-(2-chlorophenyl)benzo[b]furan (18h)
Prepared according to the general procedure A. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 7.26–7.54 (m, 6H), 7.61 (d, J = 1.9 Hz, 1H), 8.04 (dd, J = 7.7, 1.8 Hz, 1H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 106.8, 112.0, 120.9, 125.1, 127.0, 128.4, 128.5, 129.0, 129.5, 130.4, 130.9, 131.5, 152.5, 153.4.
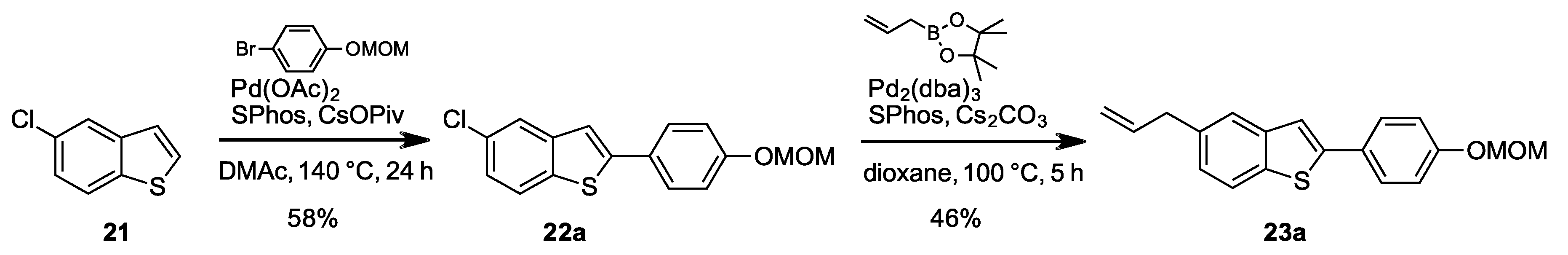
2.1.10. 5-Chloro-2-(4-(methoxymethoxy)phenyl)benzo[b]thiophene (22a)
Prepared according to the general procedure A. mp 175–177 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.51 (s, 3H), 5.22 (s, 2H), 7.09–7.12 (m, 2H), 7.25 (dd, J = 8.7, 2.0 Hz, 1H), 7.34 (s, 1H), 7.60–7.63 (m, 2H), 7.69–7.71 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 56.1, 94.4, 116.7 (2C), 117.7, 122.8, 123.2, 124.4, 127.7, 127.8 (2C), 130.7, 137.3, 142.0, 146.1, 157.7. HR-MS analyst [M + H]+ m/z (predicted) = 305.0403, m/z (measured) = 305.0396, difference = 2.02 ppm.
2.1.11. 5-Chloro-2-(4-methoxyphenyl)benzo[b]thiophene (22c)
Prepared according to the general procedure A. mp 163–165 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 3.86 (s, 3H), 6.96 (d, J = 8.8 Hz, 2H), 7.21–7.26 (m, 1H), 7.34 (s, 1H), 7.63 (d, J = 8.7 Hz, 2H), 7.68–7.72 (m, 2H). 13C-NMR (100 MHz, benzene-d6) δ (ppm) = 54.6, 114.5 (2C), 118.6, 122.3, 123.4, 124.5, 127.7, 127.9 (2C), 130.6, 137.4, 141.3, 146.4, 160.1. HR-MS analyst [M + H]+ m/z (predicted) = 275.0297, m/z (measured) = 275.0300, difference = −1.04 ppm.
2.1.12. General Procedure of the Suzuki-Miyaura Coupling of Chloro Benzo-Fused Derivatives
Procedure B: 5-chloro benzo-fused (1.0 mmol, 1.0 equiv.), allylboronic acid pinacol ester (1.5 mmol, 1.5 equiv.), cesium carbonate (486 mg, 1.5 mmol, 1.5 equiv.), Pd2(dba)3 (45 mg, 0.05 mmol, 0.05 equiv.) and SPhos (41 mg, 0.1 mmol, 0.1 equiv.) was mixed in 2 mL of dried dioxane. The mixture was stirred at 100 °C for 5 h in argon atmosphere. The reaction mixture was cooled to room temperature and then diluted with 15 mL diethyl ether or ethyl acetate (depending on the polarity of the product) and filtered through a pad of celite. The organic phase was washed with saturated NH4Cl solution, once with brine and then dried over Na2SO4. The solvent was removed under reduced pressure. Purification was performed on silica gel eluting with LP or LP/EtOAc mixtures (depending on the polarity of the product).
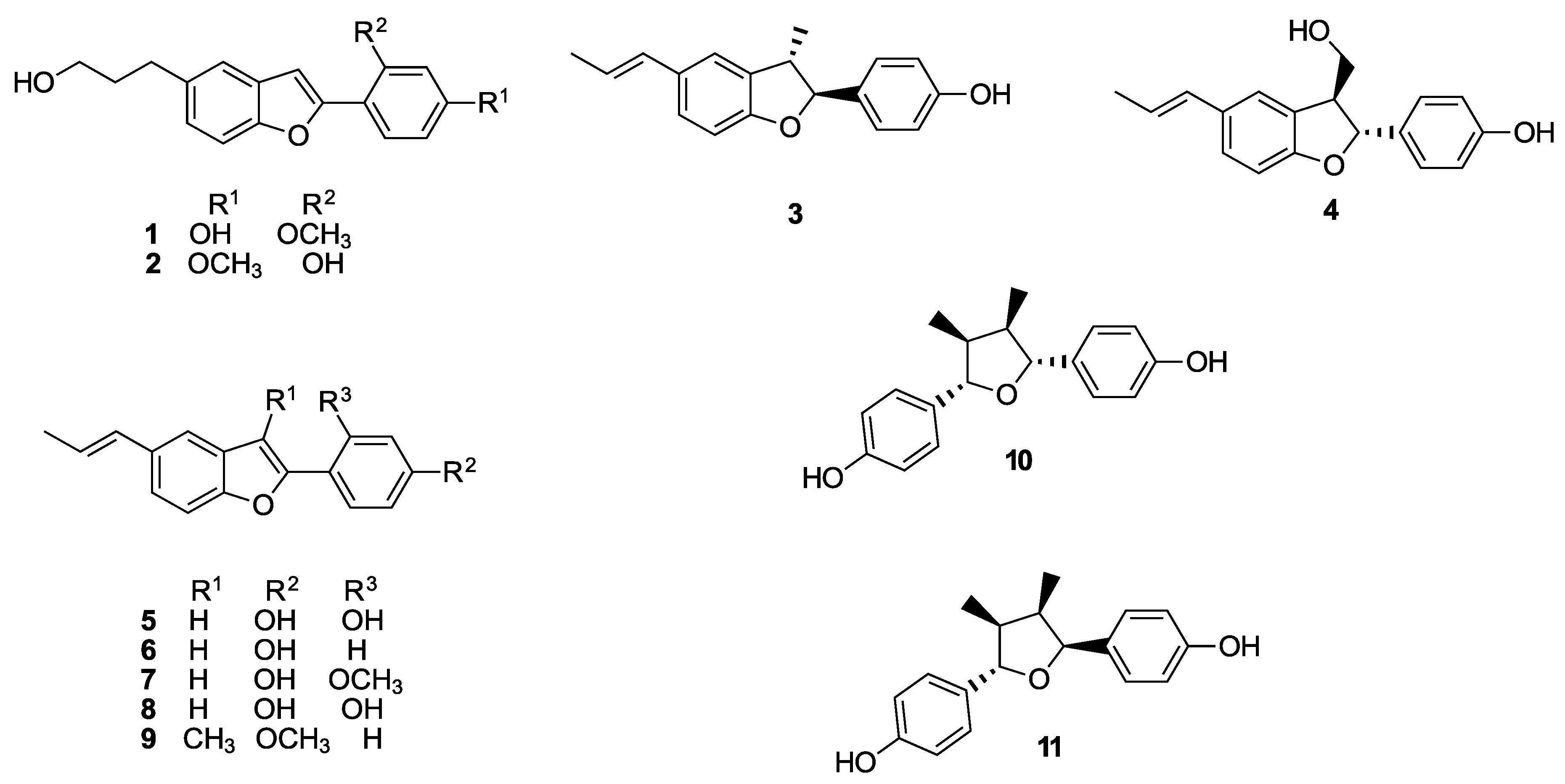
2.1.13. 5-Allyl-2-(4-(methoxymethoxy)phenyl)benzo[b]furan (16a)
Prepared according to the general procedure B. mp 126–129 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.47–3.51 (m, 5H), 5.07–5.23 (m, 4H), 6.04 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 6.85 (s, 1H), 7.07–7.14 (m, 3H), 7.36–7.44 (m, 2H), 7.76–7.81 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 40.2, 56.1, 94.4, 99.9, 110.8, 115.5, 116.5 (2C), 120.2, 124.5, 124.7, 126.3 (2C), 129.6, 134.6, 138.1, 153.6, 156.1, 157.5. HR-MS analyst [M + H]+ m/z (predicted) = 295.1329, m/z (measured) = 295.1323, difference = 2.1 ppm.
2.1.14. 5-Allyl-2-(4-methoxyphenyl)benzo[b]furan (16c)
Prepared according to the general procedure B. mp 130–132 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.49 (d, J = 6.6 Hz, 2H), 3.86 (s, 3H), 5.07–5.17 (m, 2H), 6.05 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 6.84 (s, 1H), 6.93–7.00 (m, 2H), 7.09 (dd, J = 8.4, 1.7 Hz, 1H), 7.36–7.45 (m, 2H), 7.77–7.82 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 40.2, 55.4, 99.6, 110.7, 114.3 (2C), 115.5, 120.2, 123.5, 124.6, 126.4 (2C), 129.7, 134.6, 138.1, 153.5, 156.3, 160.0. HR-MS analyst [M + H]+ m/z (predicted) = 265.1223, m/z (measured) = 265.1221, difference = 0.93 ppm.
2.1.15. 5-Allyl-2-(3,5-dimethoxyphenyl)benzo[b]furan (16d)
Prepared according to the general procedure B. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.37 (d, J = 6.7 Hz, 2H), 3.76 (s, 6H), 4.98–5.03 (m, 2H), 5.92 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 6.37 (t, J = 2.3 Hz, 1H), 6.84 (d, J = 0.8 Hz, 1H), 6.91 (d, J = 2.3 Hz, 2H), 7.01 (dd, J = 8.4, 1.7 Hz, 1H), 7.27–7.34 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 40.2, 55.5, 101.2, 101.8, 103.0 (2C), 110.9, 115.6, 120.5, 125.3, 129.3, 132.3, 134.8, 138.0, 153.7, 155.0, 161.1 (2C). HR-MS analyst [M + H]+ m/z (predicted) = 295.1329, m/z (measured) = 295.1328, difference = 0.17 ppm.
2.1.16. 5-Allyl-2-phenylbenzo[b]furan (16e)
Prepared according to the general procedure B. mp 118–120 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 3.37 (d, J = 6.5 Hz, 2H), 4.96–5.04 (m, 2H), 5.92 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 6.83 (s, 1H), 7.00 (d, J = 8.4 Hz, 1H), 7.21–7.35 (m, 5H), 7.72–7.75 (d, J = 7.2 Hz, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 40.2, 101.2, 110.9, 115.6, 120.5, 124.9 (2C), 125.1, 128.5, 128.8 (2C), 129.5, 130.6, 134.7, 138.1, 153.8, 156.2. HR-MS analyst [M + H]+ m/z (predicted) = 235.1117, m/z (measured) = 235.1119, difference = −0.8 ppm.
2.1.17. 5-Allyl-2-(4-fluorophenyl)benzo[b]furan (16f)
Prepared according to the general procedure B. mp 113–115 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.50 (d, J = 6.7 Hz, 2H), 5.09–5.16 (m, 2H), 6.05 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 6.90 (s, 1H), 7.11–7.16 (m, 3H), 7.39–7.45 (m, 2H), 7.81–7.84 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 40.2, 100.9, 110.9, 115.6, 115.9 (d, 2JC-F = 21.9 Hz), 120.5, 125.2, 126.7 (d, 3JC-F = 8.5 Hz), 126.9 (d, 4JC-F = 3.0), 129.4, 134.8, 138.0, 153.7, 155.3, 162.9 (d, 1JC-F = 248.7). HR-MS analyst [M + H]+ m/z (predicted) = 253.1023, m/z (measured) = 253.1034, difference = −4.4 ppm.
2.1.18. 5-Allyl-2-(4-(difluoromethyl)phenyl)benzo[b]furan (16g)
Prepared according to the general procedure B. mp 116–118 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.50 (d, J = 6.6 Hz, 2H), 5.08–5.17 (m, 2H), 6.04 (ddt, J = 17.0, 10.4, 6.7 Hz, 1H), 6.68 (t, JH-F = 56.4 Hz, 1H), 7.04 (s, 1H), 7.15 (dd, J = 8.4, 1.6 Hz, 1H), 7.41–7.60 (m, 4H), 7.90–7.94 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 40.1, 102.6, 111.0, 114.5 (t, 1JC-F = 238.8 Hz), 115.7, 120.7, 125.0 (2C), 125.8, 126.1 (t, 3JC-F = 6.1 Hz), 129.2, 132.8, 134.1 (t, 2JC-F = 22.4 Hz), 135.0, 137.9, 153.9, 154.9. HR-MS analyst [M + H]+ m/z (predicted) = 285.1091, m/z (measured) = 285.1088, difference = 1.08 ppm.
2.1.19. 5-Allyl-2-(2-chlorophenyl)benzo[b]furan (16h)
Prepared according to the general procedure B. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 3.57 (d, J = 6.2 Hz, 2H), 4.90–5.03 (m, 2H), 5.95 (ddt, J = 17.0, 10.4, 6.7 Hz, 1H), 6.74 (d, J = 0.7 Hz, 1H), 7.17–7.67 (m, 6H), 7.76 (d, J = 8.1. Hz, 1H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 38.3, 104.5, 112.1, 116.3, 120.5, 124.4, 126.6, 129.1, 129.2, 129.5 (2C), 130.4, 130.7, 136.8, 138.0, 153.0, 157.1. HR-MS analyst [M + H]+ m/z (predicted) = 269.0733, m/z (measured) = 269.0730, difference = 1.11 ppm.
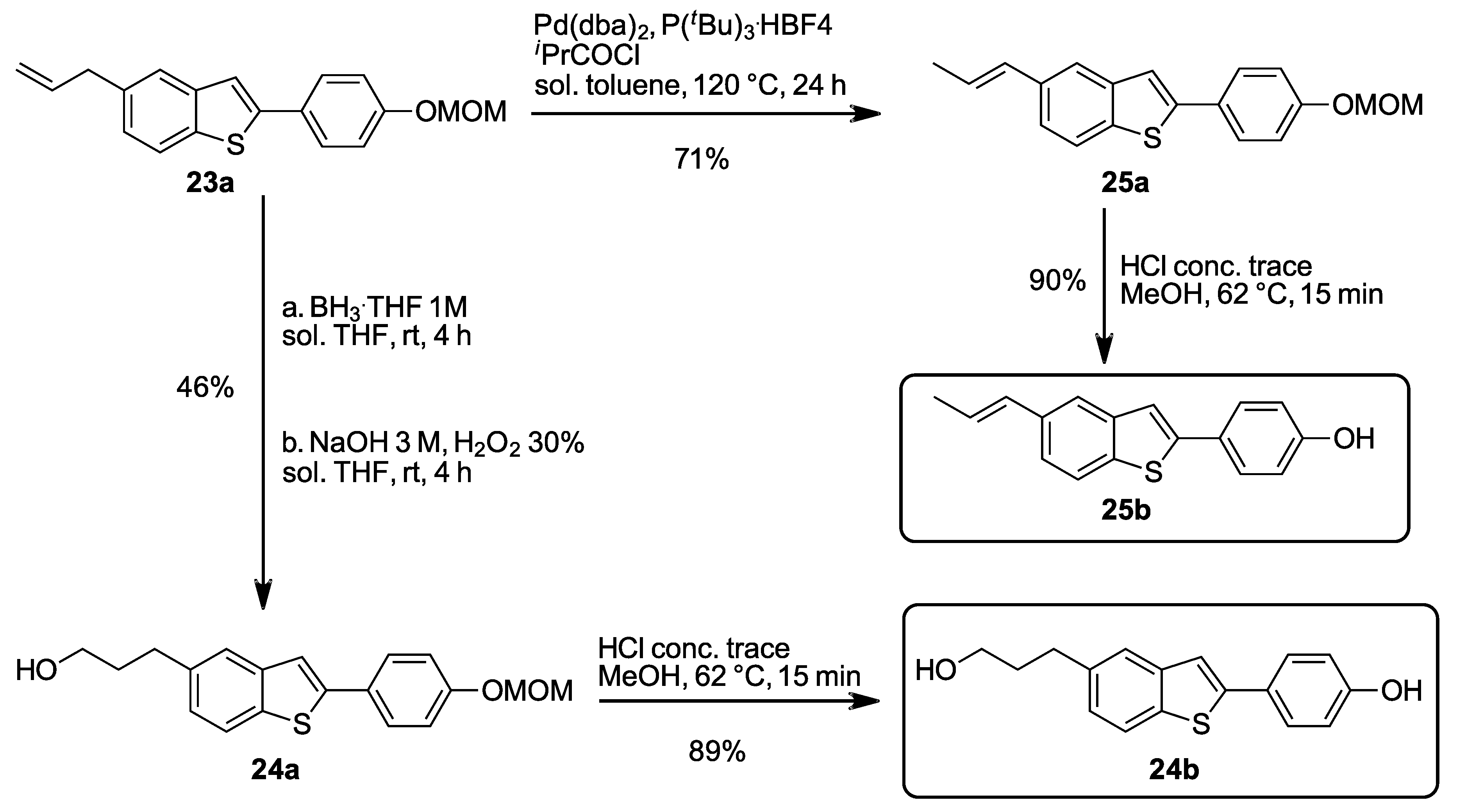
2.1.20. 5-Allyl-2-(4-(methoxymethoxy)phenyl)benzo[b]thiophene (23a)
Prepared according to the general procedure B. mp 164–166 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 3.46–3.48 (m, 5H), 5.07–5.13 (m, 2H), 5.19 (s, 2H), 6.01 (ddt, J = 16.8, 10.1, 6.7 Hz, 1H), 7.06–7.13 (m, 3H), 7.35 (s, 1H), 7.53 (d, J = 0.7 Hz, 1H), 7.59–7.61 (m, 2H), 7.70 (d, J = 8.2 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 40.2, 56.1, 94.4, 115.8, 116.7 (2C), 118.4, 122.1, 123.0, 125.3, 127.7 (2C), 128.3, 136.5, 137.2, 137.7, 141.3, 144.3, 157.4. HR-MS analyst [M + H]+ m/z (predicted) = 311.1100, m/z (measured) = 311.1108, difference = −2.62 ppm.
2.1.21. 5-Allyl-2-(4-methoxyphenyl)benzo[b]thiophene (23c)
Prepared according to the general procedure B. mp 146–147 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 3.49 (d, J = 6.7 Hz, 2H), 3.80 (s, 3H), 5.06–5.16 (m, 2H), 6.02 (ddt, J = 16.8, 10.2, 6.7 Hz, 1H), 6.90–6.98 (m, 2H), 7.13 (dd, J = 8.2, 1.6 Hz, 1H), 7.36 (s, 1H), 7.54 (s, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.71 (d, J = 8.2 Hz, 1H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 40.2, 55.4, 114.3 (2C), 115.8, 118.0, 122.0, 122.9, 125.1, 127.2, 127.7 (2C), 136.4, 137.1, 137.7, 141.3, 144.4, 159.8. HR-MS analyst [M + H]+ m/z (predicted) = 281.0995, m/z (measured) = 281.1000, difference = −1.98 ppm.
2.1.22. General Procedure for Hydroboration-Oxidation on Allyl Benzo-Fused Heterocycles
Procedure C: 5-allyl benzo-fused derivatives (0.5 mmol, 1.0 equiv.) was dissolved in 0.5 mL dry THF then the solution was cooled to 0 °C. A 1M solution of BH3.THF (0.5 mL, 0.5 mmol, 1.0 equiv.) was added slowly. Afterwards, the reaction solution was warmed to room temperature and stirred for 24 h. On the other hand, a solution of 3M NaOH and H2O2 30% was mixed in ratio of 2:3 and then cooled to 0 °C. After 24 h of reaction time, the reaction was cooled again to 0 °C and then the prepared solution of NaOH and H2O2 (1.20 mL, 1.2 mmol NaOH and 7.8 mmol H2O2) was added slowly. The reaction mixture was stirred at room temperature for 4 more hours, then diluted with 5 mL diethyl ether. The organic phase was washed with a saturated NH4Cl solution for 3 times, once with brine and dried over Na2SO4. The solvent was removed under reduced pressure. Purification was performed on silica gel eluting with LP/EtOAc mixtures.
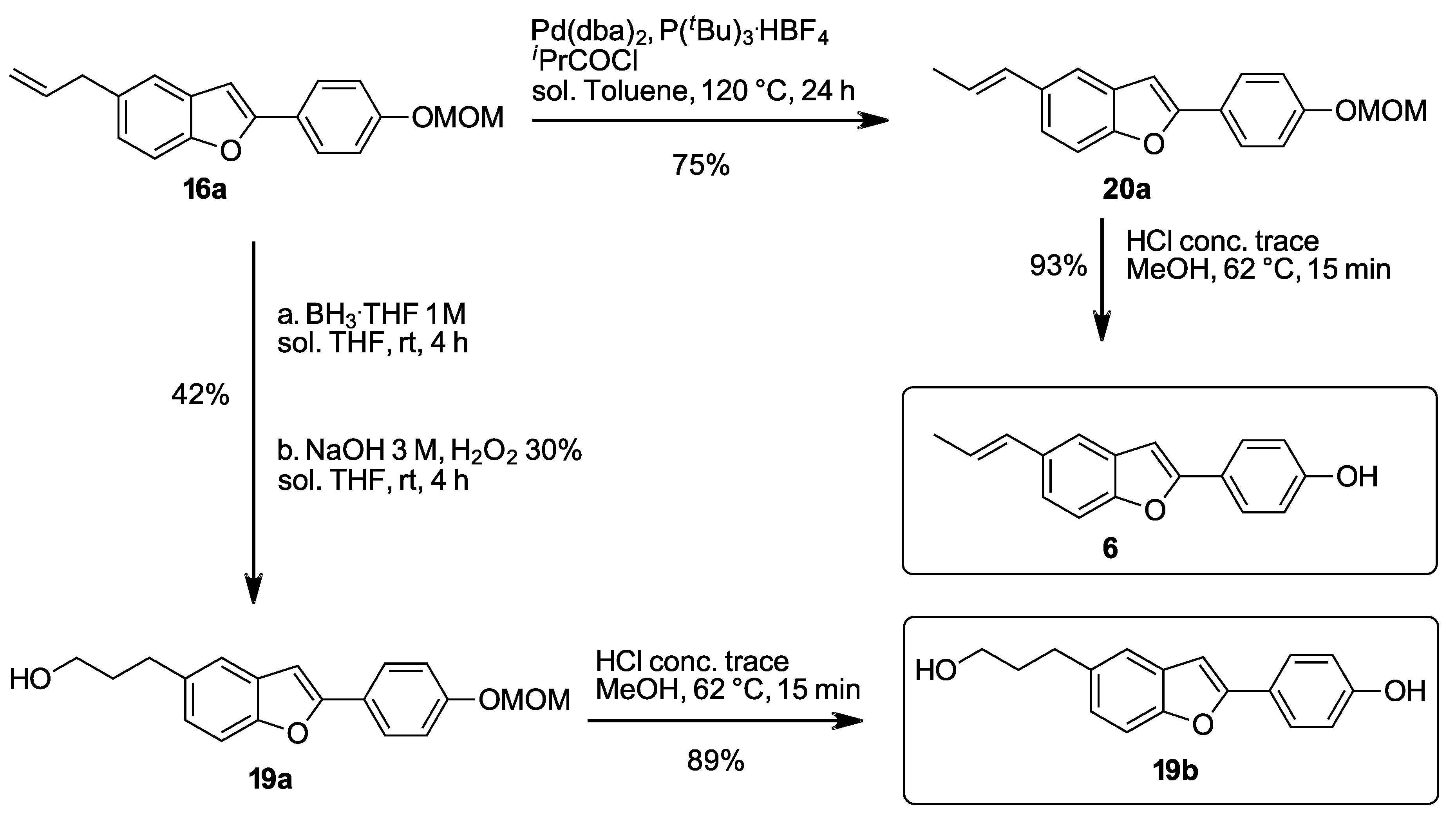
2.1.23. 3-(2-(4-(Methoxymethoxy)phenyl)benzo[b]furan-5-yl)propan-1-ol (19a)
Prepared according to the general procedure C. mp 155–156 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.33 (s, 1H), 1.88–2.01 (m, 2H), 2.81 (t, J = 7.3 Hz, 2H), 3.51 (s, 3H), 3.70 (t, J = 6.4 Hz, 2H), 5.23 (s, 2H), 6.85 (s, 1H), 7.08–7.13 (m, 3H), 7.38–7.43 (m, 2H), 7.78 (d, J = 8.7 Hz, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 32.0, 34.8, 56.1, 62.3, 94.4, 99.8 110.7, 116.5 (2C), 120.0, 124.4, 124.5, 126.3 (2C), 129.6, 136.3, 153.4, 156.1, 157.5. HR-MS analyst [M + H]+ m/z (predicted) = 313.1434, m/z (measured) = 313.1431, difference = 0.93 ppm.
2.1.24. 3-(2-(4-Methoxyphenyl)benzo[b]furan-5-yl)propan-1-ol (19c)
Prepared according to the general procedure C. mp 150–152 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.41 (s, 1H), 1.94 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.80 (t, J = 7.2 Hz, 2H), 3.70 (t, J = 6.4 Hz, 2H), 3.86 (s, 3H), 6.83 (d, J = 0.8 Hz, 1H), 6.95–7.00 (m, 2H), 7.09 (dd, J = 8.4, 1.8 Hz, 1H), 7.36–7.41 (m, 2H), 7.76–7.80 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 32.0, 34.8, 55.4, 62.3, 99.5, 110.7, 114.2 (2C), 119.9, 123.5, 124.4, 126.4 (2C), 129.7, 136.3, 153.4, 156.3, 159.9. HR-MS analyst [M + H]+ m/z (predicted) = 283.1329, m/z (measured) = 283.1339, difference = −3.63 ppm.
2.1.25. 3-(2-(3,5-Dimethoxyphenyl)benzo[b]furan-5-yl)propan-1-ol (19d)
Prepared according to the general procedure C. mp 66–68 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.43 (s, 1H), 1.94 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.80 (t, J = 7.4 Hz, 2H), 3.70 (t, J = 6.4 Hz, 2H), 3.87 (s, 6H), 6.47 (t, J = 2.2 Hz, 1H), 6.95 (s, 1H), 7.01 (d, J = 2.2 Hz, 2H), 7.12 (dd, J = 8.4, 1.6 Hz, 1H), 7.39–7.44 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 32.0, 34.7, 55.5 (2C), 62.2, 101.0, 101.7, 103.0 (2C), 110.9, 120.3, 125.1, 129.5, 132.3, 136.5, 153.5, 155.9, 161.1 (2C). HR-MS analyst [M + H]+ m/z (predicted) = 313.1434, m/z (measured) = 313.1430, difference = 1.32 ppm.
2.1.26. 3-(2-Phenylbenzo[b]furan-5-yl)propan-1-ol (19e)
Prepared according to the general procedure C. mp 128–129 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.39 (s, 1H), 1.95 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.81 (t, J = 7.6 Hz, 2H), 3.71 (t, J = 6.4 Hz, 2H), 6.97 (d, J = 0.8 Hz, 1H), 7.13 (dd, J = 8.4, 1.8 Hz, 1H), 7.33–7.47 (m, 5H), 7.84–7.87 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 32.0, 34.8, 62.3, 101.1, 110.9, 120.2, 124.9 (2C), 125.0, 128.5, 128.8 (2C), 129.4, 130.6, 136.5, 153.6, 156.2. HR-MS analyst [M + H]+ m/z (predicted) = 253.1223, m/z (measured) = 253.1220, difference = 1.05 ppm.
2.1.27. 3-(2-(4-Fluorophenyl)benzo[b]furan-5-yl)propan-1-ol (19f)
Prepared according to the general procedure C. mp 124–126 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.41 (s, 1H), 1.94 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.81 (t, J = 7.6 Hz, 2H), 3.72 (t, J = 6.4 Hz, 2H), 6.89 (d, J = 0.4 Hz, 1H), 7.11–7.15 (m, 3H), 7.33 (m, 2H), 7.80–7.84 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 32.0, 34.8, 62.3, 100.9, 110.9, 115.9 (d, 2JC-F = 22.0 Hz, 2C), 120.2, 125.0, 126.7 (d, 3JC-F = 8.2 Hz, 2C), 126.9 (d, 4JC-F = 3.4 Hz), 129.4, 136.6, 153.6, 155.3, 162.9 (d, 1JC-F = 248.6 Hz). HR-MS analyst [M + H]+ m/z (predicted) = 295.1329, m/z (measured) = 295.1327, difference = 0.69 ppm.
2.1.28. 3-(2-(4-(Difluoromethyl)phenyl)benzo[b]furan-5-yl)propan-1-ol (19g)
Prepared according to the general procedure C. mp 127–129 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.59 (s, 1H), 1.95 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.82 (t, J = 7.6 Hz, 2H), 3.71 (t, J = 6.4 Hz, 2H), 6.68 (t, J = 56.4 Hz, 1H), 7.04 (s, 1H), 7.16 (dd, J = 8.4, 1.8 Hz, 1H), 7.41–7.45 (m, 2H), 7.58 (d, J = 8.2 Hz, 2H), 7.92 (d, J = 8.5 Hz, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 32.0, 34.7, 62.2, 102.5, 111.0, 114.5 (t, 1JC-F = 238.8 Hz), 120.5, 125.0 (2C), 125.6, 126.1 (t, 3JC-F = 6.1 Hz), 129.1, 132.8, 134.1 (t, 2JC-F = 22.4 Hz), 136.8, 153.8, 154.9. HR-MS analyst [M + H]+ m/z (predicted) = 303.1191, m/z (measured) = 303.1185, difference = 2.17 ppm.
2.1.29. 3-(2-(2-Chlorophenyl)benzo[b]furan-5-yl)propan-1-ol (19h)
Prepared according to the general procedure C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.39 (s, 1H), 1.95 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.81 (t, J = 7.6 Hz, 2H), 3.71 (t, J = 6.4 Hz, 2H), 6.61 (d, J = 1.9 Hz, 1H), 7.26–7.54 (m, 6H), 7.83 (dd, J = 7.7, 1.8 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 31.8, 36.0, 64.2, 104.1, 113.3, 120.4, 124.0, 127.4, 127.8, 128.4, 129.1, 129.1, 131.9, 132.5, 134.3, 151.8, 153.5. HR-MS analyst [M + H]+ m/z (predicted) = 287.0833, m/z (measured) = 287.0826, difference = 2.71 ppm.
2.1.30. 3-(2-(4-(Methoxymethoxy)phenyl)benzo[b]thiophen-5-yl)propan-1-ol (24a)
Prepared according to the general procedure C. mp 170–172 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.44 (s, 1H), 1.95 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.82 (t, J = 7.4 Hz, 2H), 3.50 (s, 3H), 3.71 (t, J = 6.4 Hz, 2H), 5.22 (s, 2H), 7.07–7.16 (m, 3H), 7.38 (s, 1H), 7.57 (d, J = 0.9 Hz, 1H), 7.57–7.64 (m, 2H), 7.72 (d, J = 8.2 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 32.0, 34.5, 56.1, 62.2, 94.4, 116.6 (2C), 118.3, 122.1, 122.8, 125.1, 127.7 (2C), 128.3, 137.0, 138.2, 141.2, 144.3, 157.4. HR-MS analyst [M + H]+ m/z (predicted) = 329.1206, m/z (measured) = 329.1211, difference = −1.91 ppm.
2.1.31. 3-(2-(4-Methoxyphenyl)benzo[b]thiophen-5-yl)propan-1-ol (24c)
Prepared according to the general procedure C. mp 151–153 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.29 (s, 1H), 1.96 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H, H-2″), 2.82 (t, J = 7.4 Hz, 2H, H-1″), 3.71 (t, J = 6.4 Hz, 2H), 3.85 (s, 3H), 6.94–6.96 (m, 2H), 7.08–7.16 (m, 1H), 7.36–7.38 (m, 1H), 7.57 (s, 1H), 7.62–7.64 (m, 2H), 7.71 (d, J = 8.2 Hz, 1H). 13C-NMR (50 MHz, C′DCl3) δ (ppm) = 32.0, 34.5, 55.4, 62.3 (C-3″), 114.4 (2C), 118.0, 122.1, 122.7, 125.0, 127.71, 127.72 (2C), 136.9, 138.2, 141.2, 144.4, 159.8. HR-MS analyst [M + H]+ m/z (predicted) = 299.1100, m/z (measured) = 299.1072, difference = 9.57 ppm.
2.1.32. General Procedure for Isomerization on Allyl Benzo-Fused Heterocycles
Procedure D: 5-allyl benzo-fused derivatives (0.5 mmol, 1.0 equiv.), Pd(dba)2 (5.8 mg, 0.01 mmol, 0.02 equiv.), P(tBu3).HBF4 (5.8 mg, 0.02 mmol, 0.04 equiv.) and iPrCOCl (10 µL, 10.6 mg, 0.1 mmol, 0.2 equiv.) was mixed in 1 mL of degassed DMAc. The mixture was stirred at 100 °C for 6 h in argon atmosphere. The reaction mixture was cooled to room temperature and then diluted with 15 mL ethylacetate and filtered through a pad of celite. The organic phase was washed with a saturated NH4Cl solution for 3 times, once with brine and dried over Na2SO4. The solvent was removed under reduced pressure. Purification was performed on silica gel eluting with LP or LP/EtOAc mixtures (depending on the polarity of the product).
2.1.33. (E)-2-(4-(Methoxymethoxy)phenyl)-5-(prop-1-en-1-yl)benzo[b]furan (20a)
Prepared according to the general procedure D. mp 151–153 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.90 (dd, J = 6.6, 1.5 Hz, 3H), 3.50 (s, 3H), 5.22 (s, 2H), 6.20 (dq, J = 15.6, 6.5 Hz, 1H), 6.48 (dd, J = 15.7, 1.3 Hz, 1H), 6.85 (s, 1H), 7.09–7.12 (m, 2H), 7.25–7.27 (m, 1H), 7.39–7.47 (m, 2H), 7.76–7.79 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 18.5, 56.1, 94.4, 100.1, 110.9, 116.5 (2C), 117.7, 122.1, 124.4, 124.5, 126.3 (2C), 129.7, 131.2, 133.2, 154.1, 156.3, 157.6. HR-MS analyst [M + H]+ m/z (predicted) = 295.1329, m/z (measured) = 295.1328, difference = 0.22 ppm.
2.1.34. (E)-2-(4-Methoxyphenyl)-5-(prop-1-en-1-yl)benzo[b]furan (20c)
Prepared according to the general procedure D. mp 145–147 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.91 (dd, J = 6.6, 1.5 Hz, 3H), 3.86 (s, 3H), 6.22 (dq, J = 15.6, 6.5 Hz, 1H), 6.50 (dd, J = 15.7, 1.3 Hz, 1H), 6.83 (d, J = 0.6 Hz, 1H), 6.96–6.99 (m, 2H), 7.25–7.28 (m, 1H), 7.40–7.47 (m, 2H), 7.77–7.79 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 18.5, 55.4, 99.7, 110.9, 114.3 (2C), 117.7, 122.0, 123.4, 124.3, 126.4 (2C), 129.8, 131.2, 133.2, 154.1, 156.4, 160.0. HR-MS analyst [M + H]+ m/z (predicted) = 265.1223, m/z (measured) = 265.1223, difference = −0.01 ppm.
2.1.35. (E)-2-(3,5-Dimethoxyphenyl)-5-(prop-1-en-1-yl)benzo[b]furan (20d)
Prepared according to the general procedure D. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.91 (d, J = 6.4 Hz, 3H), 3.88 (s, 6H), 6.22 (dq, J = 15.9, 6.5 Hz, 1H), 6.47–6.55 (m, 2H), 6.97–7.03 (m, 3H), 7.27–7.50 (m, 3H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 18.5, 55.5 (2C), 101.0, 101.9, 102.9 (2C), 111.0, 118.0, 122.6, 124.6, 129.4, 131.1, 132.2, 133.3, 154.1, 156.1, 161.1 (2C). HR-MS analyst [M + H]+ m/z (predicted) = 313.1434, m/z (measured) = 313.1430, difference = 1.32 ppm.
2.1.36. (E)-2-Phenyl-5-(prop-1-en-1-yl)benzo[b]furan (20e)
Prepared according to the general procedure D. mp 126–128 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.92 (dd, J = 6.4, 1.8 Hz, 3H), 6.22 (dq, J = 15.9, 6.5 Hz, 1H), 6.51 (d, J = 15.9 Hz, 1H), 7.00 (s, 1H), 7.27–7.52 (m, 6H), 7.87 (m, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) =18.5, 101.3, 111.0, 118.0, 122.5, 124.5, 124.9 (2C), 128.5, 128.8 (2C), 129.5, 130.5, 131.1, 133.3, 154.2, 156.3. HR-MS analyst [M + H]+ m/z (predicted) = 235.1117, m/z (measured) = 235.1123, difference = −2.28 ppm.
2.1.37. (E)-2-(4-Fluorophenyl)-5-(prop-1-en-1-yl)benzo[b]furan (20f)
Prepared according to the general procedure D. mp 123–125 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.91 (dd, J = 6.6, 1.5 Hz, 3H), 6.22 (dq, J = 15.6, 6.5 Hz, 1H), 6.50 (dd, J = 15.7, 1.3 Hz, 1H), 6.89 (s, 1H), 7.11–7.16 (m, 2H), 7.29–7.31 (dd, J = 8.6, 1.7 Hz, 1H), 7.41–7.49 (m, 2H), 7.80–7.83 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 18.5, 101.1, 111.0, 115.9 (d, 2JC-F = 21.8 Hz), 118.0, 122.6, 124.6, 126.7 (d, 3JC-F = 8.3 Hz), 126.8 (d, 4JC-F = 3.2 Hz), 129.5, 133.4, 136.5, 154.2, 155.4, 161.3 (d, 1JC-F = 247.2 Hz, 1C). HR-MS analyst [M + H]+ m/z (predicted) = 253.1023, m/z (measured) = 253.1027, difference = −1.41 ppm.
2.1.38. (E)-2-(4-(Difluoromethyl)phenyl)-5-(prop-1-en-1-yl)benzo[b]furan (20g)
Prepared according to the general procedure D. mp 125–128 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.91 (dd, J = 6.6, 1.5 Hz, 3H), 6.23 (dq, J = 15.6, 6.5 Hz, 1H), 6.50 (dd, J = 15.7, 1.3 Hz, 1H), 6.68 (t, JH-F = 56.4 Hz, 1H), 7.05 (s, 1H), 7.32–7.34 (m, 2H), 7.51–7.59 (m, 3H), 7.92 (d, J = 8.4 Hz, 2H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 18.5, 102.7, 111.2, 114.5 (t, 1JC-F = 237.3 Hz), 118.2, 123.1, 124.8, 125.0 (2C), 126.1 (t, 3JC-F = 6.2 Hz), 129.2, 131.0, 132.7, 133.5, 134.1 (t, 2JC-F = 22.4 Hz), 154.4, 155.1. HR-MS analyst [M + H]+ m/z (predicted) = 285.1091, m/z (measured) = 285.1084, difference = 2.13 ppm.
2.1.39. (E)-2-(2-Chlorophenyl)-5-(prop-1-en-1-yl)benzo[b]furan (20h)
Prepared according to the general procedure D. mp 66–68 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.94 (d, J = 6.5 Hz, 3H), 6.33 (dq, J = 15.6, 6.5 Hz, 1H), 6.55 (dd, J = 15.7, 1.3 Hz, 1H), 6.67–6.77 (m, 2H), 7.42–7.54 (m, 5H), 7.77–7.83 (m, 1H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 18.7, 105.6, 112.1, 120.4, 124.4, 125.1, 127.0, 127.4, 128.3, 128.5, 128.9, 129.0, 129.1, 130.0, 130.6, 153.0, 153.3. HR-MS analyst [M + H]+ m/z (predicted) = 269.0728, m/z (measured) = 269.0730, difference = −0.88 ppm.
2.1.40. (E)-2-(4-(Methoxymethoxy)phenyl)-5-(prop-1-en-1-yl)benzo[b]thiophene (25a)
Prepared according to the general procedure D. mp 173–174 °C. 1H-NMR (400 MHz, CDCl3) δ (ppm) = 1.91 (dd, J = 6.6, 1.5 Hz, 3H), 3.50 (s, 3H), 5.21 (s, 2H), 6.28 (dq, J = 15.6, 6.5 Hz, 1H), 6.50 (dd, J = 15.8, 1.5 Hz, 1H), 7.08–7.10 (m, 2H), 7.32 (dd, J = 8.4, 2.6 Hz, 1H), 7.39 (s, 1H), 7.61–7.64 (m, 3H), 7.70 (d, J = 8.4 Hz, 1H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 18.5, 56.1, 94.4, 116.6 (2C), 118.5, 120.6, 122.1 (2C), 125.3, 127.7 (2C), 128.2, 131.1, 134.7, 137.7, 141.3, 144.4, 157.4. HR-MS analyst [M + H]+ m/z (predicted) = 310.1022, m/z (measured) = 310.1025, difference = −1.01 ppm.
2.1.41. (E)-2-(4-Methoxyphenyl)-5-(prop-1-en-1-yl)benzo[b]thiophene (25c)
Prepared according to the general procedure D. mp 154–156 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.93 (d, J = 6.5 Hz, 3H), 3.86 (s, 3H), 6.29 (dq, J = 15.6, 6.5 Hz, 1H), 6.51 (d, J = 15.6 Hz, 1H), 6.96 (d, J = 8.6 Hz, 2H), 7.27–7.39 (m, 2H), 7.62–7.74 (m, 4H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 18.5, 55.4, 114.3 (2C), 118.2, 120.6, 122.0, 122.1, 125.2, 127.1, 127.7 (2C), 131.1, 134.6, 137.6, 141.3, 144.5, 159.8. HR-MS analyst [M + H]+ m/z (predicted) = 281.0995, m/z (measured) = 281.1004, difference = −3.48 ppm.
2.1.42. General Procedure for De-Protection of MOM Group
Procedure D: MOM protected substrate (0.1 mmol, 1.0 equiv.) was dissolved in 0.5 mL MeOH. A 1N solution of HCl (50 µL, 0.05 mol, 0.5 equiv.) was added subsequently. The mixture was stirred at 65 °C for 30 min. The reaction mixture was cooled to room temperature and then diluted with 15 mL Et2O. The organic phase was washed with a saturated NH4Cl solution for 3 times, once with brine and dried over Na2SO4. The solvent was removed under reduced pressure. Purification was performed on silica gel eluting with LP/EtOAc 3:1 to obtain the desired product.
2.1.43. 4-(5-Allylbenzo[b]furan-2-yl)phenol (16b)
Prepared according to the general procedure E. mp 165–168 °C. 1H-NMR (400 MHz, acetone-d6) δ (ppm) = 3.33 (d, J = 6.7 Hz, 2H, H-1″), 4.88–4.99 (m, 2H, H-3″), 5.88 (ddt, J = 16.8, 10.4, 6.7 Hz, 1H, H-2″), 6.82–6.84 (m, 2H), 6.89 (s, 1H), 6.96 (dd, J = 8.5, 1.2 Hz, 1H), 7.26–7.31 (m, 2H), 7.63–7.65 (m, 2H), 8.60 (s, 1H, ArOH). 13C-NMR (100 MHz, acetone-d6) δ (ppm) = 39.8 (C-1″), 99.1, 110.4, 114.7, 115.8 (2C), 120.2, 122.2, 124.5, 126.4 (2C), 129.9, 134.8, 138.3, 153.4, 156.6, 158.2. HR-MS analyst [M + H]+ m/z (predicted) = 251.1067, m/z (measured) = 251.1062, difference = 1.82 ppm.
2.1.44. 4-(5-(3-Hydroxypropyl)benzo[b]furan-2-yl)phenol (19b)
Prepared according to the general procedure E. mp 194–196 °C. 1H-NMR (200 MHz, acetone-d6) δ (ppm) = 1.89 (ddt, J = 12.9, 8.9, 6.4 Hz, 2H), 2.80 (t, J = 7.2 Hz, 2H), 2.95 (s, 1H), 3.62 (t, J = 6.4 Hz, 2H), 6.95–6.97 (m, 3H), 7.10–7.13 (m, 1H), 7.39–7.40 (m, 2H), 7.75–7.77 (m, 2H), 8.66 (s, 1H). 13C-NMR (50 MHz, acetone-d6) δ (ppm) = 31.9, 35.1, 61.0, 99.1, 110.3, 115.7 (2C), 119.9, 122.2, 124.4, 126.3 (2C), 129.7, 137.0, 153.2, 156.5, 158.0. HR-MS analyst [M + H]+ m/z (predicted) = 269.1172, m/z (measured) = 269.1183, difference = −3.87 ppm.
2.1.45. (E)-4-(5-(Prop-1-en-1-yl)benzo[b]furan-2-yl)phenol (6)
Prepared according to the general procedure E. mp 198–199 °C. 1H-NMR (200 MHz, acetone-d6) δ (ppm) = 1.87 (dd, J = 6.6, 1.2 Hz, 3H), 6.26 (dq, J = 15.6, 6.6 Hz, 1H), 6.51 (d, J = 15.8 Hz, 1H), 6.98–7.02 (m, 3H), 7.32 (dd, J = 8.6, 1.1 Hz, 1H), 7.44 (d, J = 8.5 Hz, 1H), 7.54 (s, 1H), 7.78–7.80 (m, 2H), 8.76 (s, 1H). 13C-NMR (50 MHz, acetone-d6) δ (ppm) = 17.7, 99.3, 110.6, 115.8 (2C), 117.7, 121.9, 122.1, 123.9, 126.5 (2C), 130.0, 131.2, 133.3, 153.9, 156.8, 158.2. HR-MS analyst [M + H]+ m/z (predicted) = 251.1067, m/z (measured) = 251.1057, difference = 3.71 ppm.
2.1.46. 4-(5-(3-Hydroxypropyl)benzo[b]thiophen-2-yl)phenol (24b)
Prepared according to the general procedure E. mp 217–219 °C. 1H-NMR (400 MHz, acetone-d6) δ (ppm) = 1.74 (tt, J = 7.5, 6.2 Hz, 2H), 2.67 (t, J = 7.6 Hz, 2H), 2.73 (s, 1H), 3.46 (t, J = 5.8 Hz, 2H), 6.79–6.81 (m, 2H), 7.06 (dd, J = 8.2, 1.6 Hz, 1H), 7.38 (s, 1H), 7.47–7.65 (m, 4H), 8.54 (s, 1H). 13C-NMR (100 MHz, acetone-d6) δ (ppm) = 31.9, 34.9, 60.8, 115.9, 117.8, 121.8, 122.7, 125.1, 125.9, 127.6 (2C), 136.3, 139.0, 141.5, 144.4, 157.9. HR-MS analyst [M + H]+ m/z (predicted) = 285.0944, m/z (measured) = 285.0934, difference = 3.56 ppm.
2.1.47. (E)-4-(5-(Prop-1-en-1-yl)benzo[b]thiophen-2-yl)phenol (25b)
Prepared according to the general procedure E. mp 212–213 °C. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 1.74 (dd, J = 6.6, 1.6 Hz, 3H), 6.21 (dq, J = 15.7, 6.6 Hz, 1H), 6.38 (dd, J = 15.8, 1.5 Hz, 1H), 6.79–6.81 (m, 2H), 7.23 (dd, J = 8.4, 1.7 Hz, 1H), 7.37 (s, 1H), 7.47–7.49 (m, 2H), 7.56 (d, J = 1.2 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H), 8.54 (s, 1H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 17.8, 115.9 (2C), 117.9, 120.6, 122.0, 124.8, 125.7, 127.6 (2C), 127.6, 131.1, 134.8, 137.3, 141.6, 144.8, 158.0. HR-MS analyst [M + H]+ m/z (predicted) = 267.0838, m/z (measured) = 267.0830, difference = 3.22 ppm.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






















