The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders


Department of Clinical and Molecular Medicine, “La Sapienza” University, 00189 Rome, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2020, 9(8), 1812; https://doi.org/10.3390/cells9081812
Submission received: 30 June 2020
/
Revised: 24 July 2020
/
Accepted: 28 July 2020
/
Published: 30 July 2020
(This article belongs to the Special Issue Roles of Inflammasomes in Aging and Age-Related Diseases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Diabetes mellitus (DM) ranks seventh as a cause of death worldwide. Chronic complications, including cardiovascular, renal, and eye disease, as well as DM-associated non-alcoholic fatty liver disease (NAFLD) account for most of the morbidity and premature mortality in DM. Despite continuous improvements in the management of late complications of DM, significant gaps remain. Therefore, searching for additional strategies to prevent these serious DM-related conditions is of the utmost importance. DM is characterized by a state of low-grade chronic inflammation, which is critical in the progression of complications. Recent clinical trials indicate that targeting the prototypic pro-inflammatory cytokine interleukin-1β (IL-1 β) improves the outcomes of cardiovascular disease, which is the first cause of death in DM patients. Together with IL-18, IL-1β is processed and secreted by the inflammasomes, a class of multiprotein complexes that coordinate inflammatory responses. Several DM-related metabolic factors, including reactive oxygen species, glyco/lipoxidation end products, and cholesterol crystals, have been involved in the pathogenesis of diabetic kidney disease, and diabetic retinopathy, and in the promoting effect of DM on the onset and progression of atherosclerosis and NAFLD. These metabolic factors are also well-established danger signals capable of regulating inflammasome activity. In addition to presenting the current state of knowledge, this review discusses how the mechanistic understanding of inflammasome regulation by metabolic danger signals may hopefully lead to novel therapeutic strategies targeting inflammation for a more effective treatment of diabetic complications.
1. Introduction
Diabetes mellitus (DM) is a chronic metabolic disease characterized by hyperglycemia resulting from a defect in insulin secretion, insulin sensitivity, or both. Genetic and environmental factors—the latter associated with lifestyle such as unhealthy diet, physical inactivity/sedentariness, overweight/obesity, and alcohol and tobacco consumption—are well-recognized contributors to the increasing incidence of DM and its complications [1]. DM is a major cause of cardiovascular disease (CVD), such as heart attack and stroke, lower limb amputation, and microvascular disease leading to retinopathy, nephropathy, and neuropathy [2]. Understanding the mechanisms of DM-induced organ damage is critical to prevent and delay long-term complications and to reduce the health and economic burden of DM.
According to the World Health Organization, the prevalence of DM is steadily increasing, particularly in low- and middle-income countries where the percentage of deaths attributable to DM that occurs prior to age 70 is higher than in high-income countries [2]. Globally, an estimated 422 million adults were living with this metabolic disease in 2014 and 1.6 million died as a result of their DM in 2016, with DM ranking the seventh cause of death [2]. Considering that the prevalence of DM is predicted to rise to 642 million by 2040 [3] and that the principal cause of DM mortality is CVD [2,3], improving strategies to prevent long-term complications of DM is more relevant than ever.
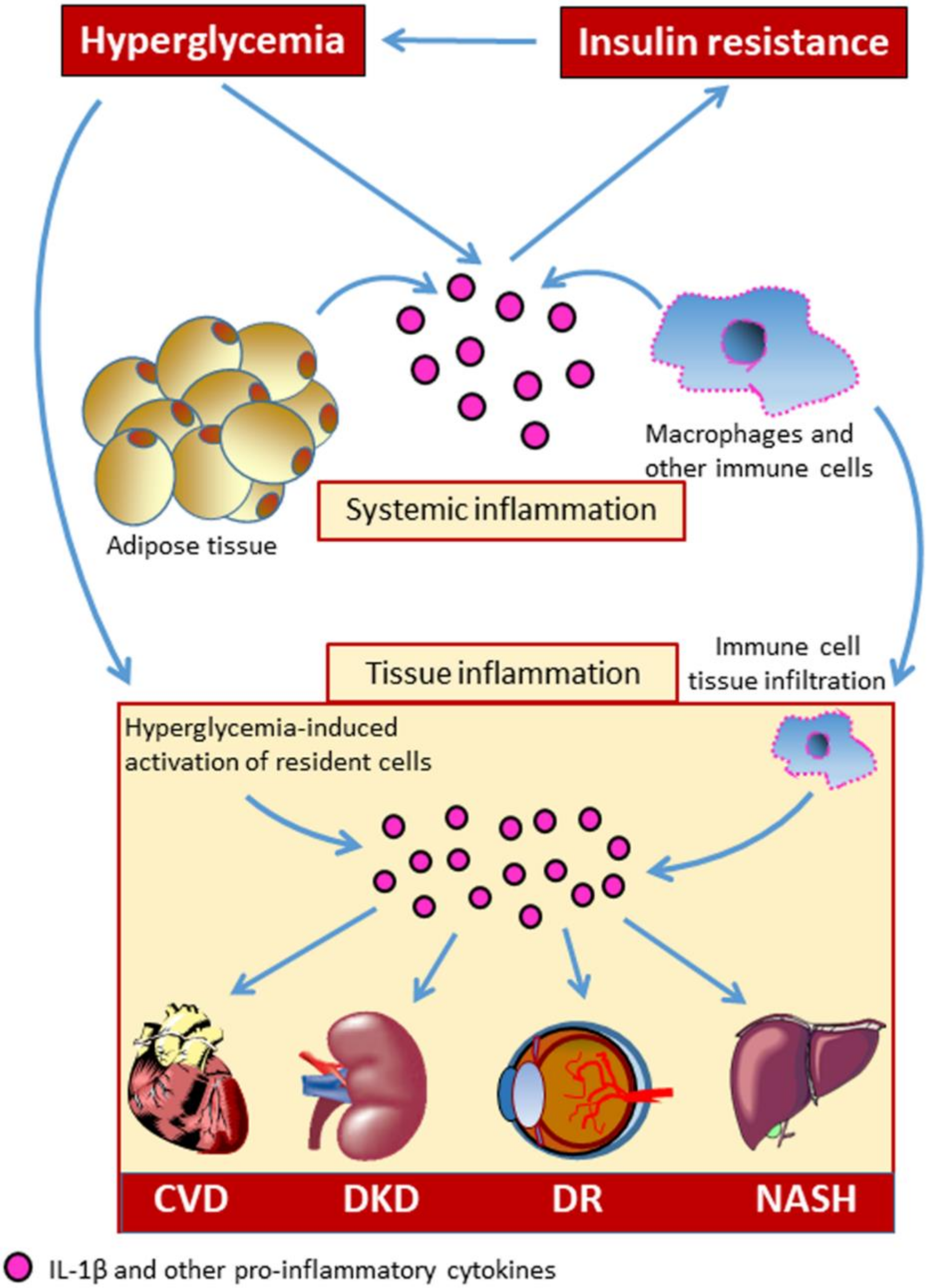
Systemic, sterile, low-grade chronic inflammation has long been recognized as a central biological mainstay of DM, and growing evidence suggests that inflammation plays an important role in DM-related complications (Figure 1) [4,5]. Over the recent past, great interest has developed regarding the involvement of the immune system in the development of chronic complications of DM and related metabolic disorders. The aberrant immune cell activation is thought to be one of the contributing mechanisms to the development of DM-related CVD [6,7].
Among the complex network of pro-inflammatory cytokines that are related to chronic metabolic diseases, interleukin (IL)-1β has been recognized to be an important player in initiating and sustaining inflammation-induced organ dysfunction in DM [8,9,10]. Therefore, the identification of factors capable of modulating the secretion of bioactive IL-1β would have therapeutic implications. In this effort, a growing number of studies on the pathogenesis of diabetic complications have focused on the involvement of multi-protein scaffolding complexes, termed inflammasomes, that coordinate the inflammatory response. These components of the innate immune system are signaling platforms that, once assembled, lead to caspase-1 activation and the processing of pro-IL-1β and pro-IL-18 to their mature bioactive forms [11,12], as well as to gasdermin D-mediated pyroptosis [13], an inflammatory form of programmed cell death. Classically, inflammasomes consist of an upstream sensor protein of the NOD-like receptor (NLR) family, the adaptor protein Apoptosis-associated speck-like protein containing a CARD (ASC), and the downstream effector cysteine protease procaspase-1. Several NLR proteins (NLRPs) capable to initiate the formation of an inflammasome were reported, including NLRP1, 2, 3, 6, and 7, NLRC4, and the hematopoietic expression, interferon-inducible nature, and nuclear localization (HIN) domain-containing family members absent in melanoma 2 (AIM2) and interferon γ-inducible protein 16 (IFI16) [12,14].
In this review, we outline the mechanisms of inflammasome activation and regulation in DM and related metabolic diseases and summarize the state of the art of our understanding of the involvement of inflammasome in the pathogenesis of chronic complications of these disorders. Along with this, we also discuss how the mechanistic understanding of inflammasome regulation by metabolic danger signals may hopefully lead to novel therapeutic strategies targeting the inflammasome for a more effective treatment of diabetic complications.
2. Activation and Regulation of the Inflammasome in Diabetes Mellitus and Related Metabolic Disorders
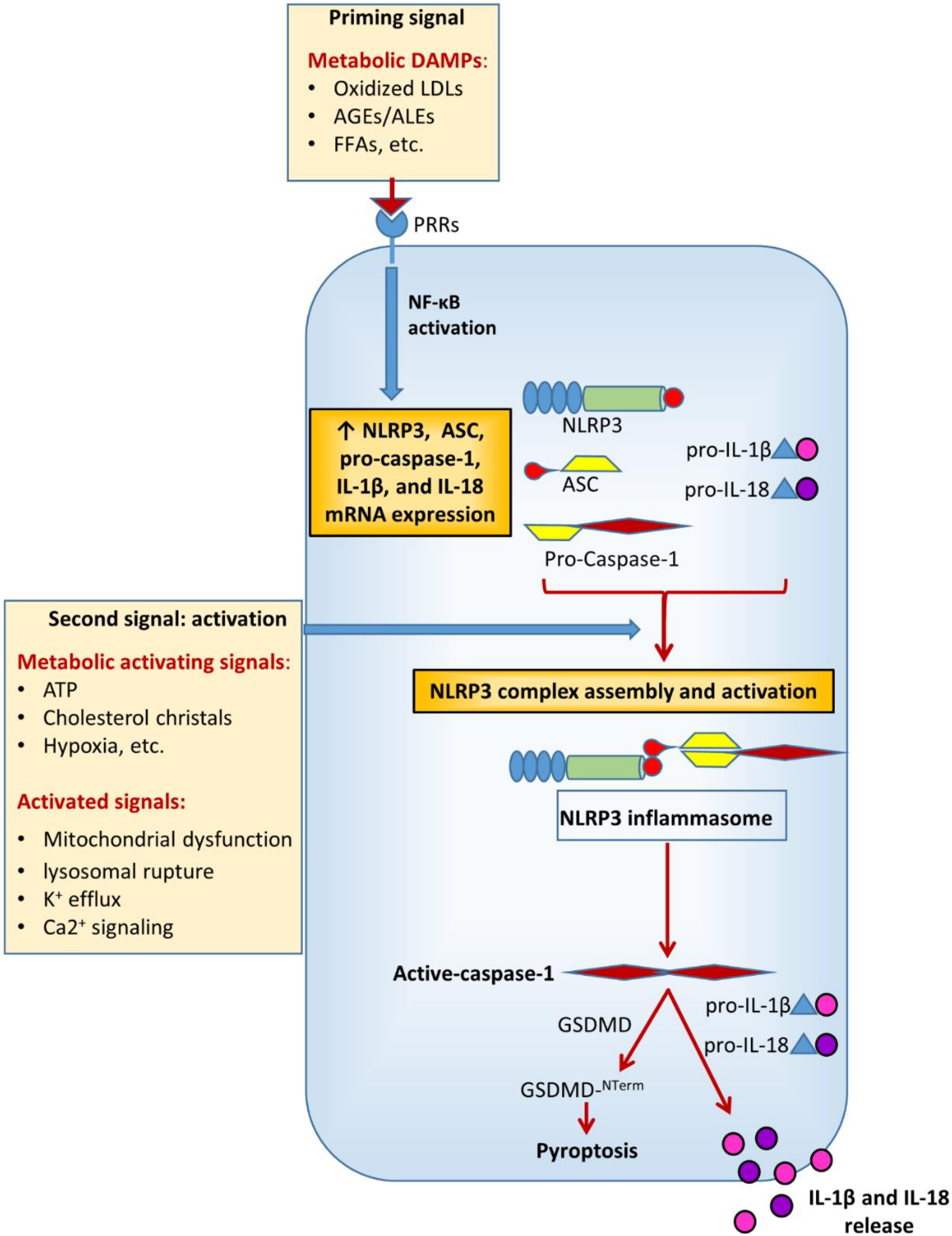
In metabolic research, particularly on DM and its chronic complications, much of the attention has been focused on NACHT, LRR, and PYD domains-containing protein 3 (NLRP3), also known as cryopyrin. This is the most characterized and investigated member of the NLR family. NLRs cooperate with toll-like receptors (TLRs) in the innate immune response to pathogens. Functionally, they are pattern recognition receptors (PRRs) that can be activated in response to a wide range of microbial and non-microbial (i.e., sterile) insults [15]. Like TLRs, NLRs are found in cells of the myeloid lineage (i.e., macrophages, granulocytes lymphocytes, etc.) and also in non-immune cells (e.g., endothelial cells) [15,16]. The current view is that NLRP3 activation in macrophages is a two-step process [17] requiring a first signal provided by microbial or endogenous molecules, termed pathogen- (PAMPS) and damage- (DAMPs) associated molecular patterns, respectively. The first signal induces a priming event which triggers NLRP3 and pro-IL-1β expression through activation of nuclear factor (NF)-κB [18,19], though more recent evidence demonstrates that priming also regulates NLRP3 activation at the post-transcriptional level [20,21]. The second signal (activation) is provided by a number of factors, including pore-forming toxins, adenosine triphosphate (ATP), viral RNA, or endogenous crystals and particulate matter [17]. The disparate chemical and structural nature of the NLRP3-activating stimuli makes their direct interaction with NLRP3 unlikely and rather suggests a common cellular signal in response to these NLRP3 activators. However, to date, there is no agreement on the identity of the signal involved in NLRP3 activation and it is not clear whether different activating stimuli can induce different signals. In fact, several cellular and molecular events have been suggested to initiate the assembly and activation of NLRP3, including mitochondrial dysfunction, lysosomal rupture, reactive oxygen species (ROS), K+ efflux, and Ca2+ signaling [17].
Multiple DM-related metabolic factors may function as NLRP3-inducing stimuli [7,22] (Figure 2). The increased production of ROS [23,24,25,26] and advanced glycation end-products (AGEs) [26,27,28], enhanced lipotoxicity due to high free fatty acids (FFAs) and cholesterol levels [24,29,30,31], defective autophagy and unfolded protein response [23,32], as well as uric acid [33] and extracellular ATP levels [34,35] have been directly involved in the development of diabetic complications and are widely recognized as potent metabolic danger signals able to regulate inflammasome activity [7,24,29,30,35,36,37,38,39].
Depending on the metabolic alteration (i.e., insulin resistance, hyperglycemia, gout, dyslipidemia, etc.) and the tissue (i.e., vascular wall, kidney, liver, etc.) considered, a plethora of metabolic danger signals have been identified so far. Given that different DAMPs have been reported to induce different control mechanisms of inflammasome assembly and activation, including mitochondrial dysfunction, endoplasmic reticulum stress, and lysosomal rupture, [40], the specific DAMP-induced mechanisms will be discussed in the paragraphs regarding each chronic complication of DM.
3. Diabetic Cardiovascular Disease
DM is a prime risk factor for CVD, conferring a two- to three-fold greater risk of major adverse cardiovascular events (MACEs), such as heart attack and stroke [2,3]. Together with hypertension, accelerated atherosclerosis and the increased prevalence of chronic kidney disease are the major contributors to DM-associated CVD [32,41]. The classical therapeutic approach for diabetic atherosclerosis and associated MACEs includes strict glycemic, lipid, and blood pressure control [42,43], though intensive blood glucose regimen is effective only if initiated early in the course of the disease [44]. Recent evidence from cardiovascular outcome trials has indicated that two new classes of anti-hyperglycemic agents, i.e., the Glucagon-like peptide (GLP)-1 receptor agonists and the sodium glucose cotransporter (SGLT) 2 inhibitors, are cardioprotective for diabetic patients beyond their glucose-lowering effect [45]. However, despite improvements in CVD preventive strategies, there is a need for novel therapies targeting the biological bases of the -accelerating effect of DM on atherogenesis. These may include blocking inflammation, which is a major mechanism of injury in vascular dysfunction, particularly in DM subjects [8,46]. Consistently, the recent reports from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) indicate that IL-1β inhibition with a monoclonal antibody (canakinumab) is effective in preventing MACEs among subjects with and without DM. This clinical outcome was associated with the inhibition of inflammation, as attested by the large reduction in circulating high-sensitivity C-reactive protein (hsCRP) and IL-6 levels [8,47,48]. Interestingly, the beneficial effects of IL-1β antagonism on atherosclerosis occurred despite no effect on low-density lipoprotein cholesterol levels and incident DM, indicating that MACE reduction was not dependent on the improvement of dyslipidemia or prevention of DM onset during the study [8,47,48]. These clinical findings indicate that the benefits of IL-1β antagonism on atherosclerosis are solely due to the inhibition of inflammation, and are in agreement with previous experimental studies demonstrating that IL-1β deficiency and IL-1 receptor antagonism were successful in decreasing the severity of lesions in murine models of atherosclerosis [49,50]. Similar results were also obtained in IL-18 deficient mice [51]. By contrast, a nonspecific approach with the generic anti-inflammatory agent methotrexate did not reduce IL-1β nor lowered MACEs in the recent Cardiovascular Inflammation Reduction Trial (CIRT) [52]. Altogether, these findings support a role for inflammasome activation in the atherosclerotic process, as IL-1β and IL-18 are processed to their mature and bioactive forms by this critical component of the innate immune system.
In a pioneering study, Lee et al. laid the foundation for investigating the role of NLRP3 in DM-associated CVD [7]. Increased expression of the inflammasome components NLRP3 and ASC was found in monocytes from newly identified, untreated type 2 DM subjects, along with increased basal and induced inflammasome activation when exposed to DAMP signals. Consistently, drug-naïve type 2 DM patients had higher IL-1β and IL-18 serum levels compared with healthy subjects, and in vitro knockdown of NLRP3 in monocytes of DM patients prevented the ability of metabolic DAMPs to induce IL-1β and IL-18 secretion [7]. In the same direction were the results of a preclinical study on a rat model of type 2 DM showing that excessive activation of Nlrp3 and pyroptosis were associated with cardiac structural and functional pathological changes, which were reverted by Nlrp3 silencing [53]. In cooperation with TLR2, NLRP3 inflammasome activation in cardiac macrophages was also involved in DM-induced, potentially life-threatening arrhythmias, which could be successfully treated by either NLRP3 inflammasome inhibition or IL-1 receptor antagonism [54]. Preliminary genetic data have also suggested an association between polymorphisms in inflammasome coding genes and increased risk for diabetic macrovascular complications, especially myocardial infarction [55].
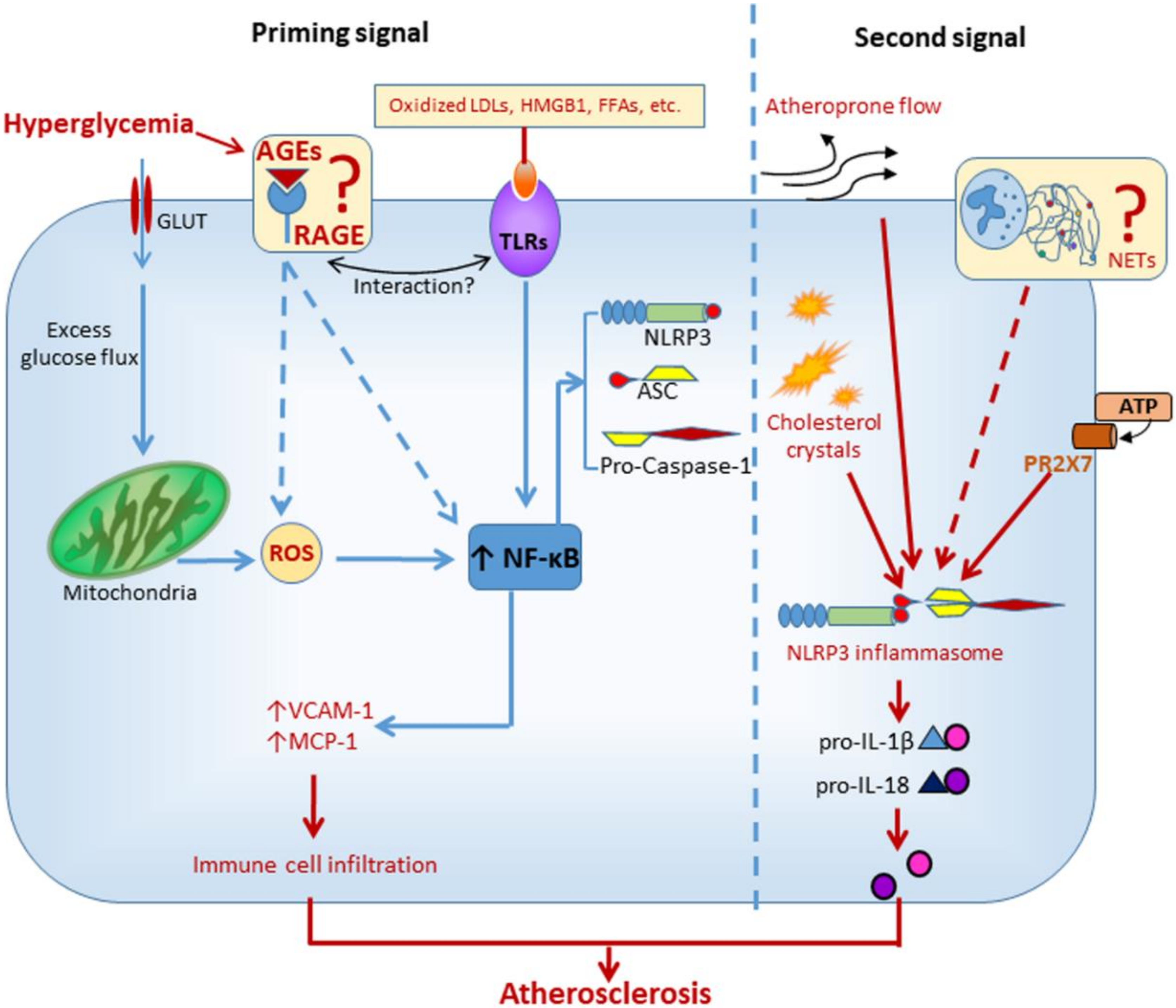
Mechanistic insights into inflammasome activation in diabetic macrovascular complications come from experimental studies in animal and cellular models of atherosclerosis. NLRP3 was involved in hyperglycemia-induced endothelial inflammation, both in vitro and in vivo. In human umbilical vein endothelial cells and in atherosclerotic plaques of diabetic mice, the overexpression of adhesion molecules induced by high glucose was suppressed by both NLRP3 knockdown and IL-1 receptor antagonism [56]. In addition, carotid atherosclerosis was positively associated with plasma levels of IL-1β in DM subjects [56]. Similarly, another study found that high glucose induced the assembly and activation of NLRP3 inflammasome in rat endothelial cells, an effect that could be reverted by blocking NADPH oxidases 4-dependent ROS generation [57]. In addition to hyperglycemia-induced ROS overproduction, oxidized LDLs (oxLDLs) [58] and high mobility group box Protein 1 [59], two well-known agonist ligands of the receptor of AGEs (RAGE) [60,61], have been involved in the activation of NLRP3 that accompanies the atherosclerotic process. Although AGEs have been definitively involved in diabetic atherosclerosis [27,62], it is currently unknown whether the AGE/RAGE axis contributes to the atherogenic process through the activation of NLRP3 (Figure 3).
Besides glucose toxicity, disrupted lipid metabolism in diabetic vessels might also be involved in inflammasome regulation. Increased aortic levels of sterol regulatory element binding protein (SREBP)-1 were associated with elevated expression of NLRP3 inflammasome components in a porcine model of atherosclerosis and DM [63]. Intense immunostaining for this lipogenic transcription factor was observed in macrophages and endothelial cells of early lesions (i.e., fatty streaks) as well as in the fibrous cap and cholesterol crystals of advanced atherosclerotic lesions [63]. Interestingly, similar findings were observed in aortic biopsy specimens from humans with atherosclerosis and DM [63]. It is now a widely accepted notion that cholesterol and its by-products, such as oxLDLs and cholesterol crystals, are metabolic stress factors playing a causal role in vascular inflammation and atherosclerosis. Among the metabolic danger signals, cholesterol crystals are now regarded as the main trigger for NLRP3 activation in atherosclerosis [64]. Besides inducing lysosomal damage, it is currently under debate whether cholesterol crystals would induce NLRP3 activation through inciting the formation of neutrophil extracellular traps (NETs) [65]. NETs are web-like structures released by neutrophils and composed of DNA, myeloperoxidase, proteases, etc. that were previously implicated in atherothrombosis [66]. However, the mechanism linking NETs to NLRP3 activation has not yet been delineated and warrants future investigation (Figure 3).
Some studies also suggest that NLRP3 activity may be modulated by mechanical forces [16,64], including hemodynamic stress [16], that can modify endothelial cell response patterns. Oscillatory shear stress was identified as a novel regulator of endothelial inflammasome activation for atherogenesis through forkhead box P transcription factor 1 (Foxp1) downregulation via repressing Kruppel-like factor 2 expression in endothelium [16]. Consistently, Foxp1 expression was significantly reduced in human and mouse coronary atherosclerotic endothelium [16]. Moreover, mitochondrial DNA was recently shown to contribute to DM-associated endothelial dysfunction and vascular inflammation via NLRP3 activation through mechanisms that involve Ca2+ influx and ROS generation [67].
A final note concerns the possible role of the anti-inflammatory effects of the new anti-diabetic medications SGLT2 inhibitors in providing their cardiovascular benefit in type 2 DM patients [68]. The recent finding that SGLT2 inhibitors attenuate NLRP3 inflammasome activation [69] might help to explain the cardioprotective effects of these anti-diabetic drugs. Therefore, this is an area that deserves further investigation.
4. Diabetic Kidney Disease
Diabetic kidney disease (DKD) is the leading cause of chronic kidney disease and end-stage kidney disease (ESKD) in most developed countries [70,71]. Approximately 30% of patients with type 1 DM and approximately 40% of patients with type 2 DM eventually develop this microvascular complication [71,72]. DKD patients, even in the early disease stages, carry a significant financial burden to the national health systems due to both direct (e.g., laboratory tests, medications, dialysis, etc.) and indirect (e.g., increased CVD morbidity) costs [73]. In the classical view of the natural history of DKD, five stages are recognized according to changes in urinary albumin excretion rate and glomerular filtration rate (GFR): hyperfiltration (stage 1), normoalbuminuria or intermittent episodes of microalbuminuria (silent nephropathy, stage 2), persistent microalbuminuria (incipient nephropathy, stage 3), macroalbuminuria and progressive GFR reduction (overt nephropathy, stage 4), and, finally, ESKD (stage 5) [74]. Recently, the traditional view that albuminuria invariably precedes and promotes GFR loss has been challenged by increasing evidence suggesting that both initiation and progression of GFR decline may occur also independently of albuminuria or even in the absence of it [75,76]. At least in part, such changes in the clinical course of DKD are likely a consequence of the introduction into clinical practice of blockers of the renin-angiotensin system (RAS), the increasing use of which has not translated into a lower prevalence of reduced GFR and DKD, and has only marginally decreased the incidence of ESKD [76]. Moreover, it is currently unclear if the heterogeneity in the clinical presentation and course of DKD reflects different anatomic entities and pathogenic mechanisms.
The current management scheme for DKD includes strict glycemic control, anti-hypertensive drugs, lipid-lowering treatment, obesity reduction, and dietary and lifestyle adjustments. DKD has been traditionally considered as the result of the interaction between hemodynamic factors, such as hyperfiltration, hypertension, and persistent local RAS activation, and metabolic factors, such as hyperglycemia and dyslipidemia. Recent evidence also indicates an important role for immune and inflammatory mechanisms, which have been implicated in the development of DKD and its progression to ESRD [77,78]. However, the inflammatory mediators, their sources (i.e., systemic or renal), and the molecular mechanisms underlying the relationship between chronic inflammation and DKD are largely unknown. Using an aptamer-based proteomic approach to examine 194 circulating inflammatory proteins in subjects with type 1 and type 2 DM, a circulating kidney risk inflammatory signature (KRIS) composed of 17 inflammatory proteins has been recently identified [79]. In addition to tumor necrosis factor (TNF) receptors 1 and 2, which were previously identified as robust predictors of renal function decline in type 1 and type 2 DM [80], 15 new circulating inflammatory proteins, including other four members of the TNF-receptor superfamily, were found to be strongly associated with renal outcomes in people with DM. Of note, all these inflammatory proteins had a systemic, non-renal source. Therefore, the signature comprising KRIS proteins highlights the importance of systemic factors as relevant pathogenic factors in DKD.
One plausible non-renal source of these inflammatory proteins are leucocytes. However, renal monocyte infiltration is observed in the course of both human and experimental DKD [81,82], and the inhibition of macrophage/monocyte recruitment into the kidney has been shown to be protective in experimental DKD models [78]. Therefore, targeted anti-inflammatory therapy has been suggested for both prevention and treatment of DKD [83].
Activated macrophages are known to secrete large amounts of cytotoxic products, including both pro-inflammatory and pro-fibrotic cytokines that may contribute to renal cells dysfunction and tissue injury. Among pro-inflammatory cytokines, serum and urinary levels of IL-18 and IL-1β have been reported to be higher in patients with DM [84,85]. While IL-1β secretion is largely restricted to infiltrating monocytes, IL-18 is also expressed by kidney cells [86,87], particularly upon kidney injury [86]. Accordingly, IL-18 has been proposed as a useful urinary biomarker of kidney injury [87]. However, essential elements of the NLRP3 inflammasome were also found to be expressed in podocytes [88,89], renal endothelial cells [88,90] and tubular epithelial cells [91,92,93,94,95]. Consistently, pro-IL-1β processing and IL-1β release from renal parenchymal cells has been reported [9,88]. Moreover, ongoing renal tissue injury exposes renal cells to numerous inflammasome activators, such as uric acid, ATP, uromodulin, histones, oxalate or cystine crystals, and matrix degradation products [39,87,96,97,98,99]. Altogether, these findings suggest that inflammasomes may contribute to the pathogenesis of kidney damage through the production of pro-inflammatory cytokines by both infiltrating immune cells and non-immune, resident renal cells [80].
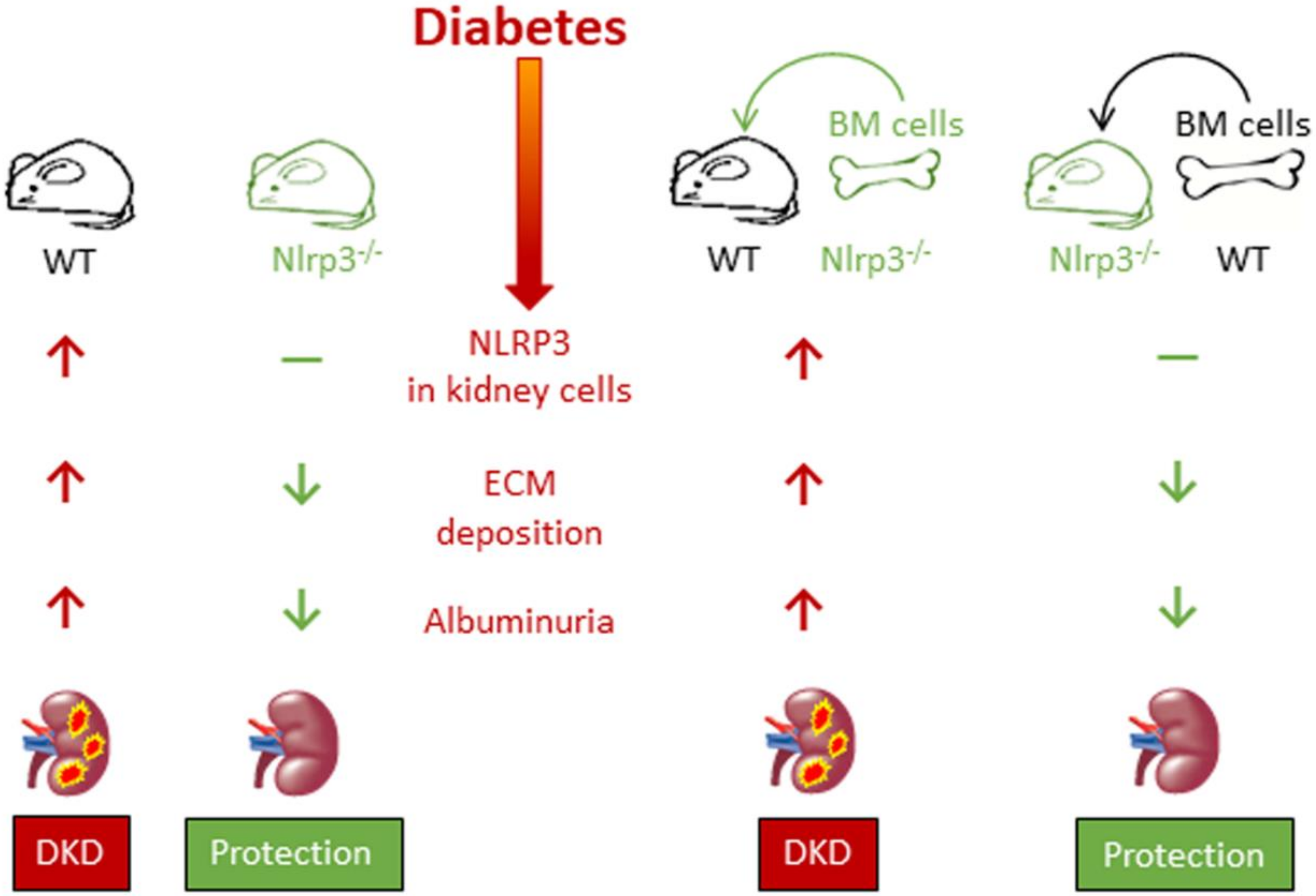
Significant evidence exists supporting the importance of caspase-1-dependent NLRP3 inflammasome activation in the pathogenesis of DKD. Circulating IL-1β and IL-18 levels, glomerular expression of IL-1β and IL-18 and inflammasome markers (NLRP3, ASC, etc.) were found to be increased in db/db mice, an experimental model of type 2 DM. Importantly, all these increases preceded structural and functional abnormalities characteristic of diabetic glomerulopathy, such as extracellular matrix accumulation and albuminuria, suggesting that NLRP3 activation triggers the onset of DKD. Importantly, all these changes were significantly reduced by NLRP3 or caspase-1 deficiency, and by pharmacological inhibition of inflammasome or caspase-1 activation [9,88]. Moreover, the transplantation of Nlrp3- or caspase-1-deficient bone marrow (BM) cells failed to ameliorate albuminuria and mesangial expansion in wild type diabetic mice, whereas Nlrp3-/- diabetic mice maintained protection from DKD despite transplantation of wild type BM cells, indicating that the Nlrp3 inflammasome in renal resident cells, particularly podocytes and glomerular endothelial cells, is the main contributor to the pathogenesis of DKD [9] (Figure 4). Finally, renal inflammasome activation was also observed in DM patients, in whom glomerular NLRP3 expression was found to be increased compared with non-diabetic subjects. In addition, serum IL1β levels were found to be higher in diabetic than in non-diabetic subjects and, among DM individuals, in albuminuric than in non-albuminuric patients [9].
A number of experimental studies have subsequently provided additional mechanistic information about the involvement of the NLRP3 inflammasome in DKD. Among the mechanisms implicated, glyco- and lipotoxicity were shown to promote inflammasome activation in the diabetic kidney through the mitochondrial ROS/Thioredoxin Interacting Protein (TXNIP) pathway [100,101,102]. In addition, the long intergenic noncoding RNA-Gm4419 was found to activate the NF-κB pathway/NLRP3 inflammasome signaling in glomerular mesangial cells exposed to high glucose conditions [103]. Recently, there has been an increasing interest in the ncRNA field and, specifically, in the role of ncRNAs in chronic complications of diabetes and related metabolic disorders. However, this topic has been reviewed recently elsewhere [104,105,106]. Mitochondrial ROS [9], uric acid [96], and ATP signaling through the purinergic receptor 2X7 (PR2X7) [34,107] have also been involved in the activation of the NLRP3 inflammasome in the diabetic kidney. Finally, D-ribose, the level of which is increased in DM, was demonstrated to induce NLRP3 inflammasome assembly and activation in podocytes via AGEs/RAGE signaling pathway [108].
Among current anti-diabetic drugs, dipeptidyl peptidase (DPP) 4 inhibitors [109], SGLT2 inhibitors [109,110], and thiazolidinediones [111] have been reported to have pleiotropic anti-inflammatory activity by reducing NLRP3 activation in DKD experimental models, independently of their glucose lowering effect. These are preliminary results obtained in preliminary experimental studies. Nonetheless, they support the need for more strictly controlled studies aimed at searching for new agents specifically targeting the NLRP3 inflammasome as potential therapeutic tools for DKD. Currently, there are only a few studies on this topic, investigating the anti-inflammasome activity of minocycline [112], curcumin [113], IL-22 [114], IL6-receptor blockade [115], and thrombomodulin gene therapy [116], with suggestive but not entirely conclusive results. To complicate matters, a protective role of the NLRP1 inflammasome in DKD was recently suggested [117]. It follows that there remains a need for more determined steps in the direction of understanding the clinical relevance of inflammasome signaling in DKD.
5. Diabetic Retinopathy
Diabetic retinopathy (DR) is a common complication of type 1 and type 2 DM, and a major cause of visual impairment and blindness among working-age and elderly people [118]. Nearly 35% of people with DM have DR and the prevalence increases with age and disease duration [118,119]. DR is a multifactorial disease with a complex interaction of microvascular, genetic, neurodegenerative, immunological, and inflammatory-related events. DR can be subdivided into two stages: the earlier stage of non-proliferative DR (non-PDR) and the advanced stage of PDR. Non-PDR lesions include microaneurysms, retinal hemorrhages, changes in vascular caliber and capillary nonperfusion [120], while PDR is characterized by pathological preretinal neovascularization coupled with fibrosis at the vitreoretinal interface [121].
By producing pro-inflammatory and pro-angiogenic cytokines, macrophages are actively involved in an aberrant ocular healing response. Persistent tissue inflammation leads to chronic tissue damage and repair resulting in severe fibrosis (gliosis) and blood-retinal barrier breakdown, with consequent extravasation of fluid into the neural retina [122,123,124]. A leakage of fluid and circulating lipoproteins from retinal vessels results in the formation of hard exudates and macular edema, a severe complication of DR that can occur both in non-PDR and PDR, and nowadays represents the leading cause of blindness in patients with DM [120,121].
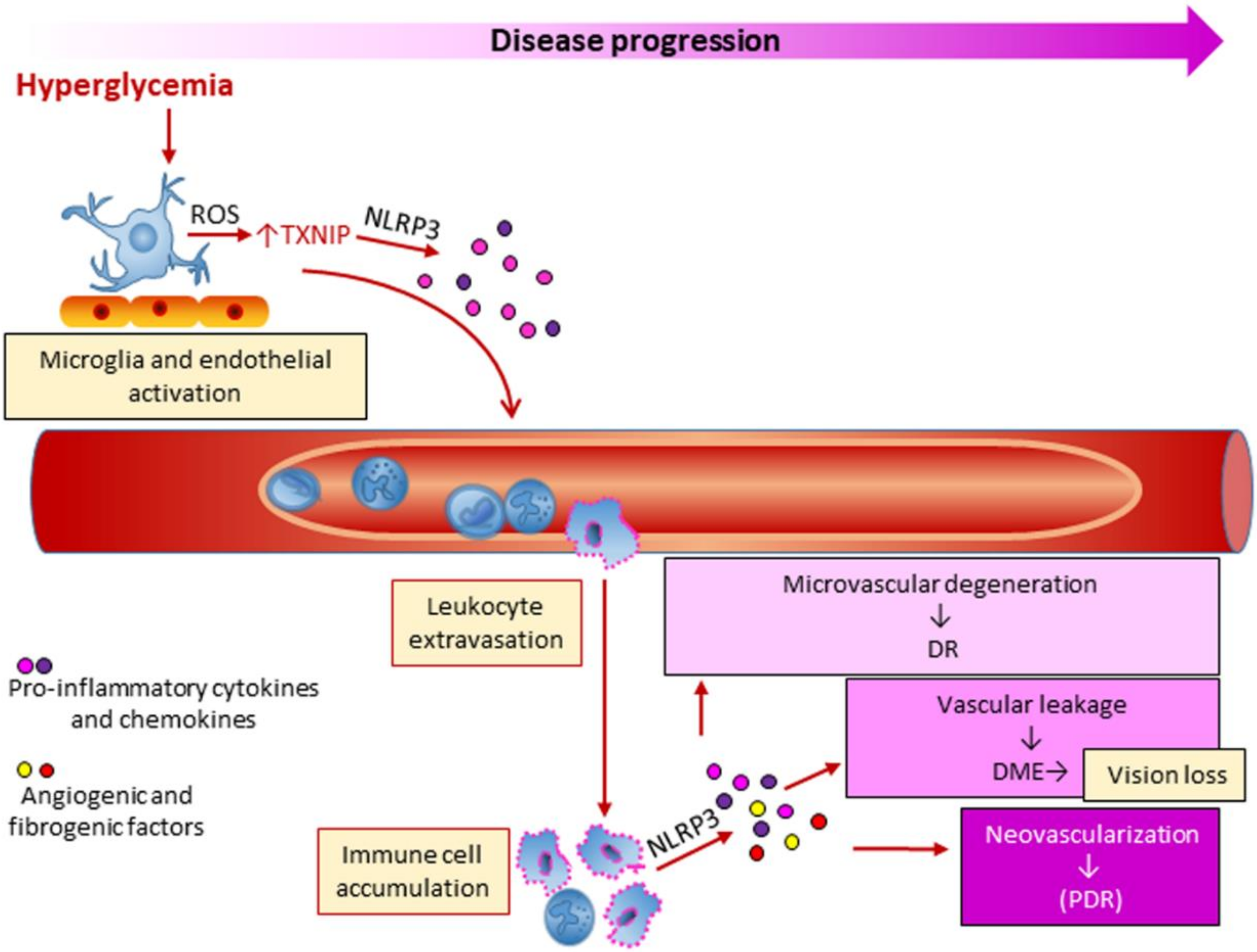
Many factors may influence DR, including hyperglycemia, dyslipidemia and hypertension, but the underlying biochemical and molecular mechanisms are still poorly defined. Seminal large-scale clinical trials have provided evidence that hyperglycemia is closely linked to DR, as intensive glycemic control delayed the onset and slowed the progression of this microvascular complication in both type 1 [42] and type 2 [43] DM. The relationship between hyperglycemia and chronic low-grade inflammation is believed to play a role also in DR. However, it is not completely clear how inflammation is initiated in DR and whether it is the initial instigator or an adjunct player in the pathogenic process. Microglia and endothelial cell activation may play a role in early vascular and neural inflammation in DR [125,126,127]. In the initial stages of DR, pro-inflammatory molecules produced by both microglia and endothelial cells may in fact be responsible for retinal leukostasis and leukocyte extravasation (Figure 5). This view is supported by the finding that chronic hyperglycemia induced the upregulation of the oxidative sensor and NLRP3 modulator TXNIP, with consequent activation of inflammatory signaling pathways in both glial and microvascular endothelial cells [126,128]. As DR progresses, multiple immune cell types, including neutrophils and monocytes, accumulate in the retina, leading the local concentration of inflammatory molecules to rise [121,125]. Consistently, increased intravitreal levels of proinflammatory cytokines including IL-6, TNF-α, Monocyte Chemoattractant Protein 1, hsCRP, and the inflammasome-related cytokines IL-1β and IL-18, have been detected in DR patients [129,130,131,132]. This adds to the growing body of evidence that inflammatory pathways, and possibly inflammasome activation, play a crucial role in DR development.
In the last decade, and particularly over the last four years, experimental studies have provided proof of the involvement of various members of the inflammasome family in DR. A study performed in the high-fat fed model of obesity-related type 2 DM demonstrated an association between inflammasome activation, attested by NLRP3 upregulation and increased levels of cleaved IL-1β and caspase-1, and retinal dysfunction. What is more, NLRP3 activation and electroretinographic defects preceded the appearance of microvascular disease, providing the rational for targeting the NLRP3 inflammasome to treat DR [133]. These findings are consistent with data from humans, where functional deficits are clinically detectable before microvascular structural lesions [133,134]. Another study that analyzed the retinas from db/db mice and human diabetic donors to investigate the role of microglia polarization dynamics in DR found that the modulation of this process towards a M2 phenotype at the early stages of DR might have therapeutic relevance [135]. Consistent with a pathogenic role of the NLRP3 inflammasome in human DR, treatment of human retinal endothelial cells with MCC950, a potent and specific inhibitor of the NLRP3 inflammasome, inhibited high-glucose-induced cell dysfunction, including apoptosis [127]. Moreover, elevated NLRP3, caspase 1 and IL-1β expression in proliferative membranes [127], and high levels of IL-1 β and IL-18 in vitreous humor [136] were observed in PDR compared with non-PDR patients. Consistent with a role of NLRP3 inflammasome in the pathological neovascularization of the retina, increased mRNA and protein levels of inflammasome components were also detected in the retina of Akimba mice, a double transgenic model displaying characteristic features of the advanced stages of DR (i.e., PDR) [137]. Finally, the expression of NLRP1, another member of the NLR family, was recently found to be increased in streptozotocin (STZ)-induced diabetic mice with features of DR [138], but not in the Akimba mouse model of PDR [137]. The finding that DR was attenuated in diabetic Nlrp1-deficient mice support the view of a contributing role of this NLR in DR [138].
Multiple non-mutually exclusive mechanisms of inflammasome activation in DR have been proposed. Hyperglycemia-induced ROS production may stimulate NLRP3 inflammasome activity through different pathways, including thioredoxin (TRX1)-TXNIP dissociation and accumulation of AGEs and advanced lipoxidation endproducts (ALEs) [139]. ROS-dependent TRX1-TXNIP dissociation was found to stimulate TXNIP-NLRP3 interaction and release of IL-1β in retinal Müller glia under chronic hyperglycemia [126,128]. AGE/ALEs were reported to induce NLRP3 mRNA expression and production of IL-1β and IL-18 in retinal pigment epithelial cells (RPE) cells [140]. Moreover, ROS-dependent TXNIP–NLRP3 association was also observed in macrophages treated with monosodium urate [141], a purine metabolite whose contributing role in DM-induced retinal inflammation was recently demonstrated [142]. Finally, ATP signaling through PR2X7) was shown to contribute to both the hyperglycemia-induced release of IL-1β from retinal pericytes and pericyte loss [143], leading to vascular leakage and macular edema.
Current therapies focus on advanced disease to prevent progression of PDR and vision loss [120]. Standard treatment options for DR include laser photocoagulation, intravitreal injections of anti-VEGF and steroid agents, and vitreoretinal surgery. The only interventions aimed at preventing DR are tight glucose and blood pressure control and much remains to be done in this direction. Most of the known inhibitors of inflammasomes are yet to be tested in humans or animal models of DR. As for DKD, the tetracycline antibiotic minocycline showed potential for treatment benefit in rodent DR, mainly by downregulating the ROS/TXNIP/NLRP3 inflammasome pathway [126]. Similar results were obtained in diabetic rats treated with 1,25-dihydroxyvitamin D [144]. Recent experimental evidence identified Prostaglandin E2 (PGE2) and its E-prostanoid receptor (EP2R) as critical regulators of inflammation and retinal endothelial cell damage in DR [145]. In STZ-induced diabetic rats, the intravitreal injection of PGE2 or butaprost, a PGE2/EP2R agonist, accelerated retinal vascular leakage, leukostasis, and endothelial cell apoptosis. These alterations were attenuated in rats that were pre-treated with AH6809, an EP2R antagonist. In addition, pretreatment of human retinal endothelial cells with AH6809 significantly inhibited PGE2- and butaprost-induced activation of caspase 1, NLRP3, and ASC [145], suggesting that both PGE2/EP2R signaling pathway and the inflammasome may be considered as potential target for DR prevention and treatment.
Finally, therapeutic agents commonly used for metabolic disorders may also be taken into consideration for their off-label beneficial effect on DR. An intriguing candidate protective factor for DR is the triglyceride-lowering drug fenofibrate, a peroxisome proliferator-activated receptor-α (PPARα) agonist. Fenofibrate has been demonstrated to delay progression and improve multiple abnormalities in experimental DR, including inflammation and vascular leakage [146,147]. What is more, the benefit was independent of the lipid-lowering activity of this drug [148]. Two large clinical trials showed the efficacy of this PPARα agonist in slowing the progression of DR and reducing retinal laser requirement, [149,150]. Interestingly, recent experimental evidence has been provided that fenofibrate ameliorates DR by modulating nuclear factor erythroid-2-related factor 2 signaling and NLRP3 inflammasome activation [151].
6. Diabetic Neuropathy
Nerve injury is a common complication of DM that leads to chronic pain, numbness, weakness and substantial loss of quality of life. Good glycemic control can prevent or retard the development of diabetic neuropathy, but more than half of all patients with diabetes eventually develop this complication [152]. Currently, there is no approved treatment to prevent or treat diabetic neuropathy, and only symptomatic pain therapies of variable efficacy are available. Though traditionally counted among microvascular complications, abnormalities in neuronal cells likely provide a critical contribution to the development of diabetic neuropathy [32]. Unfortunately, the study of the molecular mechanisms and, specifically, pathogenic mechanisms involving the inflammasome has lagged somewhat behind other complications of diabetes. To date, a handful of articles on the involvement of NLRP3 in some pathophysiological changes and clinical manifestations of diabetic neuropathy are found in the literature [153,154,155,156,157]. Though suggesting a contribution of the TXNIP [153,154], PR2X4 [155], and HMGB1/TLR4-NLRP3 signaling pathways [156], these studies do not provide conclusive evidence on the role of NLRP3 in diabetic neuropathy and need to be confirmed in larger controlled studies.
7. Diabetes-Associated Non-Alcoholic Fatty Liver Disease
Nonalcoholic fatty liver disease (NAFLD) is one of the most common forms of chronic liver disease, affecting one fourth of the population worldwide [158]. Prevalence estimates even increase among people with obesity and type 2 DM, reaching nearly 75% of the individuals affected by DM [158,159]. NAFLD is defined as the presence of ≥5–10% of hepatocytes containing lipid droplets after the exclusion of other causes of fat accumulation, such as excessive alcohol intake, viral infections, and use of steatogenic medications. NAFLD encompasses a wide spectrum of liver pathology, from the simple accumulation of fat (hepatic steatosis) to nonalcoholic steatohepatitis (NASH), cirrhosis, and liver cancer [160]. Hepatic steatosis is a reversible condition characterized by hepatic fat accumulation with no evidence of inflammation and hepatocellular injury [158,159,160]. It represents the hepatic manifestation of the metabolic syndrome [161], a condition closely associated with insulin resistance [162]. By favoring FFA mobilization from adipose tissue and increased hepatic FFA uptake and synthesis, insulin resistance favors build-up of extra fat in the liver [163]. Depending on the presence of other factors that act synergistically with insulin resistance, including nutritional and genetic factors, a minority of affected patients develops NASH [164]. Besides steatosis, NASH includes the presence of inflammation with hepatocyte injury, which is often, but not always, associated with fibrosis. Over time, chronic cycles of injury and inflammation may lead to fibrosis progression that can eventually develop into cirrhosis with its clinical consequences [160].
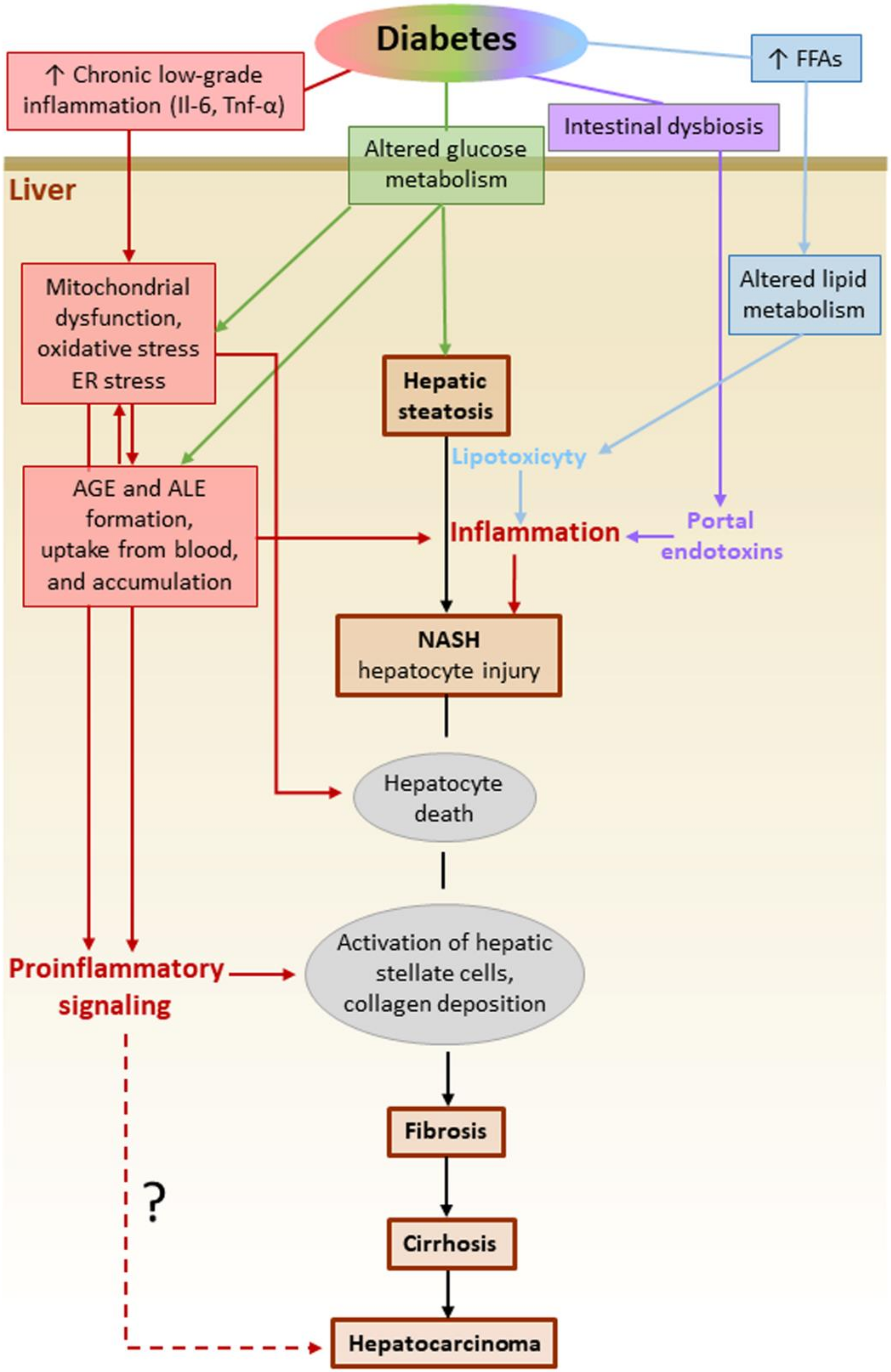
Diabetic patients are at increased risk not only of developing NAFLD [159], but also of progressing to end-stage liver disease. It has been estimated that DM increases the risk of NAFLD progression to NASH by two- to three-fold [165]. In turn, NAFLD confers a higher risk of CVD and malignancies in DM subjects [159]. In this scenario, the understanding of the molecular mechanisms underlying the accelerating effect of DM on the initiation and progression of NAFLD is of extreme importance. Regardless of the presence of DM, both local and systemic stimuli concur in activating and sustaining an inflammatory response in the liver. In the context of the dysregulated glucose metabolism associated with insulin resistance, several DM-related factors have now been recognized to drive progression to NASH and more advanced stages of NAFLD (Figure 6) [166]. These factors include systemic low-grade chronic inflammation, excessive circulating levels and liver uptake of FFAs, intrahepatic de novo lipid synthesis, systemic and liver glucose metabolism alterations, oxidative stress, and the consequent accumulation of ALEs. These are lipotoxic compounds that cause hepatocellular damage and apoptosis, endoplasmic reticulum stress, liver inflammation and fibrosis [167,168]. In addition to intrahepatic formation, circulating ALEs and their glyco-equivalents AGEs, the levels of which are increased in DM [169,170], might accumulate in the liver also because of increased hepatic uptake [171]. Besides the glycotoxic and lipotoxic effects exerted by AGEs and ALEs in the liver, other extrahepatic factors may favor hepatic inflammation in DM patients with NAFLD. Among them, the inflammatory mediators released by dysfunctional adipose tissue, such as IL-6 and TNFα [172], and endotoxins derived from gut microbiota [173,174] have been shown to activate hepatic Kupffer cells to produce IL-1β, TNF-α, and IL-6, thus exacerbating liver inflammation, hepatocyte injury, and fibrosis [175]. The search for a link between glyco/lipotoxicity and NASH has received much attention in recent years. In this context, the finding that the expression of NLRP3 and inflammasome-associated cytokines (pro-IL-1β and pro-IL-18) is higher in patients with NASH compared to those with simple steatosis [176] has spurred research on this topic.
Studies performed in Nlrp3 knockout mouse models or using NLRP3 inhibitors actually suggest that the activation of the inflammasome is important in NAFLD progression to NASH and fibrosis development [176]. Several NAFLD-associated factors have been shown to be effective stimuli for inflammasome activation, including FFAs [177], cholesterol crystals [178], increased ROS production by dysfunctional mitochondria [176,178], and AGE/ALE-mediated toxicity [169,179,180]. In addition, by providing the priming signal, membrane-bound receptors such as the PRRs TLR9 [181] and TLR4 [178] and PR2X7 [107] have been demonstrated to be critical for NLRP3 activation, IL-1β production, and NASH development. As in the kidney and the eye, both BM-derived and resident liver cells, including hepatocytes and hepatic stellate and sinusoidal cells, seem to contribute to AIM2 and NLRP3 inflammasome activation in NAFLD progression. The activation of these two inflammasomes in dietary NASH was found to be mediated by the myeloid differentiation primary response gene 88 and, in the case of AIM2, supported by TLR9 binding activity [182].
In the specific case of NAFLD associated with DM, there is experimental evidence suggesting that ROS-mediated mechanisms play a major role in the activation of NLRP3 inflammasome. In fact, quercetin and allopurinol, two compounds with proven antioxidant properties [183,184], were shown to be effective in reducing both liver inflammation and lipid accumulation under hyperglycemic conditions [185]. Treatment with these compounds significantly reduced liver inflammation via inhibiting the ROS/TXNIP/NLRP3 pathway, and positively regulated lipid liver metabolism by preventing the upregulation of PPARα and the down-regulation of SREBP1c and 2, fatty acid synthase, and liver X receptor α. Accordingly, targeting the ROS-TXNIP-NLRP3 pathway has been proposed as a therapeutic opportunity for preventing the progression of NAFLD to NASH in DM [185]. However, these experimental data should be confirmed by further studies with larger sample size and under genetically and environmentally controlled conditions, as there is conflicting evidence on the role of inflammasomes in NAFLD. In particular, the crosstalk between the gut and the liver should be explored in depth [168], especially in DM. In fact, some evidence indicates that genetic deficiency of the inflammasomes NLRP3 and NLRP6 promotes NAFLD/NASH progression by inducing intestinal microbiota dysbiosis and liver accumulation of abnormal bacterial products via portal circulation [186]. In addition, NLRP6 and NLRP3 ablation has been associated with a worsening of multiple aspects of the metabolic syndrome, including obesity, via modulation of gut microbiota. Accordingly, a protective role in NAFLD has been proposed for NLRP3 and NLRP6. [186].
There is no specific treatment yet for NAFLD irrespective of the presence or absence of DM. One of the possible strategies is to mitigate the metabolic instigators of NASH. Consistently, current therapies targeted at insulin resistance show some benefit in treating NAFLD [187,188]. Insulin-sensitizing anti-diabetic medications, such as metformin and thiazolidinediones, have been proven to be effective in treating NAFLD in DM patients. Another class of anti-diabetic agents, the GLP-1 receptor agonists, was recently demonstrated to improve all the features of NAFLD, including steatosis, inflammation, and fibrosis [189]. Besides its insulinotropic action, additional metabolic effects, including insulin sensitization properties, likely contribute to GLP-1 anti-NASH activity [190]. However, these medications mainly target organs than the liver, especially muscle and adipose tissue. The intrahepatic drug targets include lipid and glucose homeostasis regulators, mitochondrial and antioxidative stress targets in hepatocytes, inflammatory signals in hepatocytes, and pathways related to stellate cell activation and fibrogenesis [158]. The drugs for these targets are currently tested in phase 2 or 3 clinical trials and were recently thoroughly reviewed by Friedman et al. [158]. At the time of writing, there are no clinical trials specifically designed to evaluate the effectiveness and safety of targeting the inflammasomes in NASH. It must be concluded that much experimental work still needs to be done to clarify the role of inflammasomes in NAFLD before moving into clinical studies.
8. Conclusions and Perspectives
Inflammation plays a pivotal role in the development of chronic complications of DM, both microvascular and macrovascular, and also in DM-related metabolic disorders such as NAFLD. The clinical and experimental data discussed in this review highlight the role of systemic and local chronic low-grade inflammation as a key etiological process leading to chronic complications of DM. Accordingly, the benefit of directly targeting inflammation in CVD, especially when associated with DM, has been recently confirmed in randomized controlled trials [8,49,50]. Moreover, the benefit provided by current anti-diabetic medications via pleiotropic effects on inflammation further support the concept that modulating the inflammatory process in DM may be a viable option for the prevention and treatment of chronic complications.
Among the anti-diabetic drugs showing a beneficial off-target effect on inflammation, DPP-4 inhibitors, GLP-1 receptor agonists, SGLT2 inhibitors, and thiazolidinediones have been demonstrated to attenuate NLRP3 activation in experimental models of DM, independently of their glucose lowering effect. This observation would suggest the utility of specific inhibitors of inflammasomes as potential additional treatment for DM-related complications. However, current evidence on the potential benefits of such agents for chronic complications of DM and of other metabolic diseases, is limited by several factors, including conflicting findings and gaps that still exist in literature. Among the available therapeutics that target the inflammasomes, only the NLRP3 inhibitor MCC950 has been tested with encouraging results in preclinical studies of DKD [191], DR [127], and NAFLD [192]. The same compound also showed some benefit in atherosclerosis [193], but in the absence of DM. Finally, in this field, current scientific efforts are also directed at characterizing the effects of natural compounds as potential therapeutic strategies for inflammasome-related metabolic disorders. This topic has not been discussed in this review. For recent developments in the research on phytochemicals as regulators of NLRP3 activity in experimental models of DM, please refer to the recent review of Pellegrini et al. [194].
In summary, more strictly controlled experimental studies aimed at searching for and testing new agents specifically targeting the inflammasomes in diabetic complications should be performed before moving forward to clinical testing in humans.
Author Contributions
S.M., C.I. and G.P. drafted the article. M.V. critically revised the article for important intellectual content. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants of “La Sapienza” University of Rome, Italy, to S.M. and G.P. and of the Ministry of University and Research of Italy, PRIN 20178YTNWC, to G.P. The sponsors had no role in the design and conduct of the studies; collection, management and interpretation of the data; or preparation, review and approval of the manuscript.
Conflicts of Interest
The Authors declare no conflicts of interest.
Abbreviations
| DM | diabetes mellitus |
| CVD | cardiovascular disease |
| IL | interleukin |
| NLR | NOD-like receptor |
| ADC | Apoptosis-associated speck-like protein containing a CARD |
| NLRPs | NLR proteins |
| HIN | interferon-inducible nature, and nuclear localization |
| AIM2 | absent in melanoma 2 |
| IFI16 | interferon γ-inducible protein 16 |
| NLRP3 | NACHT, LRR and PYD domains-containing protein 3 |
| TLRs | toll-like receptors |
| PRRs | pattern recognition receptors |
| PAMPS | pathogen- associated molecular patterns |
| DAMPs | damage-associated molecular patterns |
| NF-κB | nuclear factor-κB |
| ATP | adenosine triphosphate |
| ROS | reactive oxygen species |
| AGEs | advanced glycation end-products |
| FFAs | free fatty acids |
| MACEs | major adverse cardiovascular events |
| CANTOS | Canakinumab Anti-Inflammatory Thrombosis Outcomes Study |
| GLP | Glucagon-like peptide |
| SGLT | sodium glucose cotransporter |
| hsCRP | high-sensitivity C-reactive protein |
| CIRT | Cardiovascular Inflammation Reduction Trial |
| OxLDLs | oxidized LDLs |
| RAGE | receptor of AGEs |
| SREBP | sterol regulatory element binding protein |
| NETs | neutrophil extracellular traps |
| Foxp1 | forkhead box P transcription factor 1 |
| DKD | diabetic kidney disease |
| ESKD | end-stage kidney disease |
| GFR | glomerular filtration rate |
| RAS | renin-angiotensin system |
| KRIS | kidney risk inflammatory signature |
| TNF | tumor necrosis factor |
| BM | bone marrow |
| TXNIP | Thioredoxin Interacting Protein |
| PR2X7 | purinergic receptor 2X7 |
| DPP | dipeptidyl peptidase |
| DR | diabetic retinopathy |
| PDR | proliferative DR |
| STZ | streptozotocin |
| TRX1 | thioredoxin |
| ALEs | advanced lipoxidation endproducts |
| PGE2 | Prostaglandin E2 |
| EP2R | E-prostanoid receptor |
| PPARα | peroxisome proliferator-activated receptor-α |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
References
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Media Centre. Diabetes. Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 30 October 2018).
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, R.M.; Donath, M.Y.; Leroith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39, S244–S252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttle, K.R. Linking Metabolism and Immunology: Diabetic Nephropathy Is an Inflammatory Disease. J. Am. Soc. Nephrol. 2005, 16, 1537–1538. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Glaser, N.; Griffen, S.; Wang-Polagruto, J.; Miguelino, E.; Jialal, I. Increased Monocytic Activity and Biomarkers of Inflammation in Patients With Type 1 Diabetes. Diabetes 2006, 55, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 Inflammasome Activation in Patients With Type 2 Diabetes. Diabetes 2012, 62, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Nair, V.; Komorowsky, C.V.; Weil, E.J.; Yee, B.; Hodgin, J.; Harder, J.L.; Godfrey, B.; Ju, W.; Boustany-Kari, C.M.; Schwarz, M.; et al. A molecular morphometric approach to diabetic kidney disease can link structure to function and outcome. Kidney Int. 2018, 93, 439–449. [Google Scholar] [CrossRef]
- Christgen, S.; Kanneganti, T.-D. Inflammasomes and the fine line between defense and disease. Curr. Opin. Immunol. 2020, 62, 39–44. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Zhuang, T.; Liu, J.; Chen, X.; Zhang, L.; Pi, J.; Sun, H.; Li, L.; Bauer, R.C.; Wang, H.; Yu, Z.; et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ. Res. 2019, 125, 590–605. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.G.; Horvath, G.L.; Stutz, A.; Alnemri, E.S.; Macdonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Núñez, G. Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J. Immunol. 2009, 183, 792–796. [Google Scholar] [CrossRef]
- Schroder, K.; Sagulenko, V.; Zamoshnikova, A.; Richards, A.A.; Cridland, J.A.; Irvine, K.; Stacey, K.J.; Sweet, M.J. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012, 217, 1325–1329. [Google Scholar] [CrossRef] [Green Version]
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional Priming and Deubiquitination Regulate NLRP3 Inflammasome Activation. J. Biol. Chem. 2012, 287, 36617–36622. [Google Scholar] [CrossRef] [Green Version]
- Mirza, R.E.; Fang, M.M.; Weinheimer-Haus, E.M.; Ennis, W.J.; Koh, T.J. Sustained Inflammasome Activity in Macrophages Impairs Wound Healing in Type 2 Diabetic Humans and Mice. Diabetes 2014, 63, 1103–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgos-Morón, E.; Abad-Jiménez, Z.; De Marañon, A.M.; Iannantuoni, F.; López, E.-; López-Domènech, S.; Salom, C.; Jover, A.; Llabata, V.; Torres, I.R.; et al. Relationship Between Oxidative Stress, ER Stress, and Inflammation in Type 2 Diabetes: The Battle Continues. J. Clin. Med. 2019, 8, 1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.S.; Brownlee, M. Molecular and Cellular Mechanisms of Cardiovascular Disorders in Diabetes. Circ. Res. 2016, 118, 1808–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menini, S.; Poirier, H.; Shapiro, J.S.; Kim, R.J.; Lazar, M.A.; Amadio, L.; Oddi, G.; Ricci, C.; Pesce, C.; Pugliese, F.; et al. Deletion of p66Shc Longevity Gene Protects Against Experimental Diabetic Glomerulopathy by Preventing Diabetes-Induced Oxidative Stress. Diabetes 2006, 55, 1642–1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Menini, S.; Iacobini, C.; Ricci, C.; Fantauzzi, C.B.; Pugliese, G. Protection from diabetes-induced atherosclerosis and renal disease by d-carnosine-octylester: Effects of early vs late inhibition of advanced glycation end-products in Apoe-null mice. Diabetologia 2014, 58, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N. Advanced glycation endproducts—role in pathology of diabetic complications. Diabetes Res. Clin. Pr. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Low Wang, C.C.; Hess, C.N.; Hiatt, W.R.; Goldfine, A.B. Clinical Update: Cardiovascular Disease in Diabetes Mellitus: Atherosclerotic Cardiovascular Disease and Heart Failure in Type 2 Diabetes Mellitus—Mechanisms, Management, and Clinical Considerations. Circulation 2016, 133, 2459–2502. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, D.I.; Maahs, D.M.; Hovind, P.; Nakagawa, T. Uric Acid as a Mediator of Diabetic Nephropathy. Semin. Nephrol. 2011, 31, 459–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzies, R.I.; Booth, J.W.; Mullins, J.J.; Bailey, M.A.; Tam, F.W.; Norman, J.T.; Unwin, R.J. Hyperglycemia-induced Renal P2X7 Receptor Activation Enhances Diabetes-related Injury. EBioMedicine 2017, 19, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solini, A.; Menini, S.; Rossi, C.; Ricci, C.; Santini, E.; Fantauzzi, C.B.; Iacobini, C.; Pugliese, G. The purinergic 2X7receptor participates in renal inflammation and injury induced by high-fat diet: Possible role of NLRP3 inflammasome activation. J. Pathol. 2013, 231, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Iacobini, C.; Menini, S.; Fantauzzi, C.B.; Pesce, C.; Giaccari, A.; Salomone, E.; Lapolla, A.; Orioli, M.; Aldini, G.; Pugliese, G. FL-926-16, a novel bioavailable carnosinase-resistant carnosine derivative, prevents onset and stops progression of diabetic nephropathy indb/dbmice. Br. J. Pharmacol. 2017, 175, 53–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.; Hwang, I.; Han, S.H.; Shin, J.-S.; Shin, O.S.; Yu, J.-W. Advanced glycation end products impair NLRP3 inflammasome–mediated innate immune responses in macrophages. J. Biol. Chem. 2017, 292, 20437–20448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pejnovic, N.; Pantic, J.; Jovanovic, I.; Radosavljevic, G.; Milovanovic, M.; Nikolić, I.; Zdravkovic, N.; Djukic, A.; Arsenijević, N.N.; Lukic, M.L. Galectin-3 Deficiency Accelerates High-Fat Diet–Induced Obesity and Amplifies Inflammation in Adipose Tissue and Pancreatic Islets. Diabetes 2013, 62, 1932–1944. [Google Scholar] [CrossRef] [Green Version]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Kanneganti, T.-D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712. [Google Scholar] [CrossRef]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Chronic Kidney Disease and Coronary Artery Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef]
- American Diabetes Association Addendum. 10. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2020. Diabetes Care 2020, 43, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; E Grobbee, D.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Hear. J. 2019, 41, 255–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Prato, S. Megatrials in type 2 diabetes. From excitement to frustration? Diabetol. 2009, 52, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Furtado, R.H.M.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; et al. Comparison of the effects of glucagon-like peptide receptor agonists and sodium-glucose cotransporter 2 inhibitors for prevention of major adverse cardiovascular and renal outcomes in type 2 diabetes mellitus: Systematic review and meta-analysis of cardiovascular outcomes trials. Circulation 2019, 139, 2022–2031. [Google Scholar]
- Kanter, J.E.; Averill, M.M.; Leboeuf, R.C.; Bornfeldt, K.E. Diabetes-accelerated atherosclerosis and inflammation. Circ. Res. 2008, 103, e116–e117. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Everett, B.M.; Libby, P.; Thuren, T.; Glynn, R.J.; Kastelein, J.; Koenig, W.; Genest, J.; Lorenzatti, A.; et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: A secondary analysis from the CANTOS randomised controlled trial. Lancet 2018, 391, 319–328. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. New Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wad, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Rader, D. IL-1 and atherosclerosis: A murine twist to an evolving human story. J. Clin. Investig. 2011, 122, 27–30. [Google Scholar] [CrossRef]
- Elhage, R.; Jawien, J.; Rudling, M.; Ljunggren, H.-G.; Takeda, K.; Akira, S.; Bayard, F.; Hansson, G.K. Reduced atherosclerosis in interleukin-18 deficient apolipoprotein E-knockout mice. Cardiovasc. Res. 2003, 59, 234–240. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. New Engl. J. Med. 2018, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 Gene Silencing Ameliorates Diabetic Cardiomyopathy in a Type 2 Diabetes Rat Model. PLOS ONE 2014, 9, e104771. [Google Scholar] [CrossRef] [PubMed]
- Monnerat, G.; Alarcón, M.L.; Vasconcellos, L.R.; Hochman-Mendez, C.; Brasil, G.V.; Bassani, R.A.; Casis, O.; Malan, D.; Travassos, L.H.; Sepúlveda, M.; et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016, 7, 13344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klen, J.; Goričar, K.; Janež, A.; Dolžan, V. NLRP3 Inflammasome Polymorphism and Macrovascular Complications in Type 2 Diabetes Patients. J. Diabetes Res. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Z.; Fan, Y.; Liu, X.; Xue, J.; Han, Z.; Zhu, C.; Wang, X. NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis. Diabetes, Metab. Syndr. Obesity Targets Ther. 2019, 12, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.X.; Ling, S.K.; Hu, M.Y.; Ma, Y.; Li, Y.; Huang, P.L. Protective Effects of Acarbose Against Vascular Endothelial Dysfunction through Inhibiting Nox4/NLRP3 Inflammasome Pathway in Diabetic Rats. Free Radic. Biol. Med. 2019, 145, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Ko, Y.S.; Park, S.W.; Kim, H.J.; Par, S.W. P2Y2R activation by ATP induces oxLDL-mediated inflammasome activation through modulation of mitochondrial damage in human endothelial cells. Free. Radic. Biol. Med. 2019, 136, 109–117. [Google Scholar] [CrossRef]
- Wang, R.; Wu, W.; Huang, S.; Li, Z.; Liu, R.; Shan, Z.; Zhang, C.; Li, W.; Wang, S. Activation of NLRP3 Inflammasome Promotes Foam Cell Formation in Vascular Smooth Muscle Cells and Atherogenesis Via HMGB1. J. Am. Hear. Assoc. 2018, 7, 008596. [Google Scholar] [CrossRef] [Green Version]
- Chellan, B.; Reardon, C.A.; Getz, G.S.; Bowman, M.A.H. Enzymatically Modified Low-Density Lipoprotein Promotes Foam Cell Formation in Smooth Muscle Cells via Macropinocytosis and Enhances Receptor-Mediated Uptake of Oxidized Low-Density Lipoprotein. Arter. Thromb. Vasc. Biol. 2016, 36, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Saremi, A.; Howell, S.; Schwenke, D.C.; Bahn, G.; Beisswenger, P.J.; Reaven, P.D. Advanced Glycation End Products, Oxidation Products, and the Extent of Atherosclerosis During the VA Diabetes Trial and Follow-up Study. Diabetes Care 2017, 40, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Xu, S.; Jiang, B.; Cohen, R.A.; Zang, M. Activation of Sterol Regulatory Element Binding Protein and NLRP3 Inflammasome in Atherosclerotic Lesion Development in Diabetic Pigs. PLOS ONE 2013, 8, e67532. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Fu, J. Novel Insights Into the NLRP3 Inflammasome in Atherosclerosis. J. Am. Hear. Assoc. 2019, 8, e012219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Avondt, K.; Maegdefessel, L.; Soehnlein, O. Therapeutic Targeting of Neutrophil Extracellular Traps in Atherogenic Inflammation. Thromb. Haemost. 2019, 119, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Pereira, C.A.; Carlos, D.; Ferreira, N.S.; Silva, J.F.; Zanotto, C.Z.; Zamboni, D.S.; Garcia, V.D.; Ventura, D.F.; Silva, J.S.; Tostes, R.C. Mitochondrial DNA Promotes NLRP3 Inflammasome Activation and Contributes to Endothelial Dysfunction and Inflammation in Type 1 Diabetes. Front. Physiol. 2020, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- A Zelniker, T.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Furtado, R.H.M.; et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: A systematic review and meta-analysis of cardiovascular outcome trials. Lancet 2019, 393, 31–39. [Google Scholar] [CrossRef]
- Ye, Y.; Bajaj, M.; Yang, H.C.; Perez-Polo, J.R.; Birnbaum, Y. SGLT-2 Inhibition with Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice with Type 2 Diabetes. Further Augmentation of the Effects with Saxagliptin, a DPP4 Inhibitor. Cardiovasc. Drugs Ther. 2017, 31, 119–132. [Google Scholar] [CrossRef]
- Afkarian, M.; Zelnick, L.R.; Hall, Y.N.; Heagerty, P.J.; Tuttle, K.; Weiss, N.S.; De Boer, I.H. Clinical Manifestations of Kidney Disease Among US Adults With Diabetes, 1988–2014. JAMA 2016, 316, 602–610. [Google Scholar] [CrossRef]
- Saran, R.; Li, Y.; Robinson, B.; Ayanian, J.; Balkrishnan, R.; Bragg-Gresham, J.; Chen, J.T.; Cope, E.; Gipson, D.; He, K.; et al. US Renal Data System 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am. J. Kidney Dis. 2015, 66, S1–S305. [Google Scholar] [CrossRef] [Green Version]
- Reutens, A.T. Epidemiology of Diabetic Kidney Disease. Med. Clin. North Am. 2013, 97, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, A.A.; Segel, J.E.; Zhuo, X.; Hoerger, T.J.; Imai, K.; Williams, D. Medical Costs of CKD in the Medicare Population. J. Am. Soc. Nephrol. 2013, 24, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E.; Christensen, C.K.; Vittinghus, E. The Stages in Diabetic Renal Disease: With Emphasis on the Stage of Incipient Diabetic Nephropathy. Diabetes 1983, 32, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G. Updating the natural history of diabetic nephropathy. Acta Diabetol. 2014, 51, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, G.; Italian Diabetes Society and the Italian Society of Nephrology; Penno, G.; Natali, A.; Barutta, F.; Di Paolo, S.; Reboldi, G.; Gesualdo, L.; De Nicola, L. Diabetic kidney disease: New clinical and therapeutic issues. Joint position statement of the Italian Diabetes Society and the Italian Society of Nephrology on “The natural history of diabetic kidney disease and treatment of hyperglycemia in patients with type 2 diabetes and impaired renal function”. J. Nephrol. 2019, 33, 9–35. [Google Scholar] [CrossRef] [Green Version]
- Goldfine, A.B.; Shoelson, S.E. Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk. J. Clin. Investig. 2017, 127, 83–93. [Google Scholar] [CrossRef]
- Sakai, N.; Wada, T. Revisiting inflammation in diabetic nephropathy: The role of the Nlrp3 inflammasome in glomerular resident cells. Kidney Int. 2015, 87, 12–14. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Pavkov, M.E.; Skupien, J.; Smiles, A.; Dom, Z.I.M.; Wilson, J.M.; Park, J.; Nair, V.; Schlafly, A.; Saulnier, P.-J.; et al. A signature of circulating inflammatory proteins and development of end-stage renal disease in diabetes. Nat. Med. 2019, 25, 805–813. [Google Scholar] [CrossRef]
- Niewczas, M.A.; Gohda, T.; Skupien, J.; Smiles, A.M.; Walker, W.H.; Rosetti, F.; Cullere, X.; Eckfeldt, J.H.; Doria, A.; Mayadas, T.N.; et al. Circulating TNF Receptors 1 and 2 Predict ESRD in Type 2 Diabetes. J. Am. Soc. Nephrol. 2012, 23, 507–515. [Google Scholar] [CrossRef]
- Furuta, T.; Saito, T.; Ootaka, T.; Soma, J.; Obara, K.; Abe, K.; Yoshinaga, K. The Role of Macrophages in Diabetic Glomerulosclerosis. Am. J. Kidney Dis. 1993, 21, 480–485. [Google Scholar] [CrossRef]
- Viedt, C.; Orth, S. Monocyte chemoattractant protein-1 (MCP-1) in the kidney: Does it more than simply attract monocytes? Nephrol. Dial. Transplant. 2002, 17, 2043–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Gomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Zheng, B.; Cleary, C.M.; Ruiz-Ortega, M.; Ortiz, A.; Fernandez-Fernandez, B. Targeting inflammation in diabetic kidney disease: Early clinical trials. Expert Opin. Investig. Drugs 2016, 25, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Alexandraki, K.I.; Piperi, C.; Kalofoutis, C.; Singh, J.; Alaveras, A.; Kalofoutis, A. Inflammatory Process in Type 2 Diabetes: The Role of Cytokines. Ann. New York Acad. Sci. 2006, 1084, 89–117. [Google Scholar] [CrossRef]
- Nakamura, A.; Shikata, K.; Hiramatsu, M.; Nakatou, T.; Kitamura, T.; Wada, J.; Itoshima, T.; Makino, H. Serum Interleukin-18 Levels Are Associated With Nephropathy and Atherosclerosis in Japanese Patients With Type 2 Diabetes. Diabetes Care 2005, 28, 2890–2895. [Google Scholar] [CrossRef] [Green Version]
- Stokman, G.; Kers, J.; Ünsal, Y.; Hoelbeek, J.; Claessen, N.; De Boer, O.J.; Netea, M.G.; Hilbrands, L.; Bemelman, F.J.; Berge, I.J.T.; et al. Predominant Tubular Interleukin-18 Expression in Polyomavirus-Associated Nephropathy. Transplant. 2016, 100, e88–e95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Yuan, J.; Zhao, Y.; Zha, Y. Urine interleukin-18 in prediction of acute kidney injury: A systemic review and meta-analysis. J. Nephrol. 2014, 28, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Kohli, S.; Nazir, S.; Ghosh, S.; Ranjan, S.; Wang, H.; Madhusudhan, T.; et al. Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. J. Am. Soc. Nephrol. 2016, 27, 2270–2275. [Google Scholar] [CrossRef] [Green Version]
- Abais, J.M.; Xia, M.; Li, G.; Chen, Y.; Conley, S.M.; Gehr, T.W.B.; Boini, K.M.; Li, P.-L. Nod-like Receptor Protein 3 (NLRP3) Inflammasome Activation and Podocyte Injury via Thioredoxin-Interacting Protein (TXNIP) during Hyperhomocysteinemia. J. Biol. Chem. 2014, 289, 27159–27168. [Google Scholar] [CrossRef] [Green Version]
- Martin-Rodriguez, S.; Caballo, C.; Gutierrez, G.; Vera, M.; Cruzado, J.M.; Cases, A.; Escolar, G.; Diaz-Ricart, M. TLR4 and NALP3 inflammasome in the development of endothelial dysfunction in uraemia. Eur. J. Clin. Investig. 2015, 45, 160–169. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-Independent NLRP3 Augments TGF-β Signaling in Kidney Epithelium. J. Immunol. 2012, 190, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Wen, Y.; Tang, T.-T.; Lv, L.-L.; Tang, R.-N.; Liu, H.; Ma, K.-L.; Crowley, S.D.; Liu, B.-C. Megalin/Cubulin-Lysosome-mediated Albumin Reabsorption Is Involved in the Tubular Cell Activation of NLRP3 Inflammasome and Tubulointerstitial Inflammation. J. Biol. Chem. 2015, 290, 18018–18028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Mueller, J.L.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; Correia, J.D.S.; Ulevitch, R.J.; Hoffman, H.M.; McKay, D.B. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 2010, 185, 6277–6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Xu, M.; Ding, L.-H.; Lv, L.-L.; Liu, H.; Ma, K.-L.; Zhang, A.-H.; Crowley, S.D.; Liu, B. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int. J. Biochem. Cell Biol. 2014, 57, 7–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Lee, S.-H.; Kim, Y.-G.; Kim, S.-Y.; Seo, J.-W.; Choi, Y.-W.; Kim, D.-J.; Jeong, K.-H.; Lee, T.; Ihm, C.-G.; et al. Hyperuricemia-induced NLRP3 activation of macrophages contributes to the progression of diabetic nephropathy. Am. J. Physiol. Physiol. 2015, 308, F993–F1003. [Google Scholar] [CrossRef]
- Darisipudi, M.N.; Thomasova, D.; Mulay, S.R.; Brech, R.; Noessner, E.; Liapis, H.; Anders, H.-J. Uromodulin Triggers IL-1β–Dependent Innate Immunity via the NLRP3 Inflammasome. J. Am. Soc. Nephrol. 2012, 23, 1783–1789. [Google Scholar] [CrossRef] [Green Version]
- Allam, R.; Darisipudi, M.N.; Tschopp, J.; Anders, H.-J. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur. J. Immunol. 2013, 43, 3336–3342. [Google Scholar] [CrossRef]
- Prencipe, G.; Caiello, I.; Cherqui, S.; Whisenant, T.; Petrini, S.; Emma, F.; De Benedetti, F. Inflammasome Activation by Cystine Crystals: Implications for the Pathogenesis of Cystinosis. J. Am. Soc. Nephrol. 2014, 25, 1163–1169. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Gu, J.; Gou, F.; Huang, W.; Gao, C.; Chen, G.; Long, Y.; Zhou, X.; Yang, M.; Liu, S.; et al. High Glucose and Lipopolysaccharide Prime NLRP3 Inflammasome via ROS/TXNIP Pathway in Mesangial Cells. J. Diabetes Res. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Liu, S.; Wang, H.; Dou, H. Role of the thioredoxin interacting protein in diabetic nephropathy and the mechanism of regulating NOD-like receptor protein 3 inflammatory corpuscle. Int. J. Mol. Med. 2019, 43, 2440–2450. [Google Scholar] [CrossRef]
- Han, Y.; Xu, X.; Tang, C.; Gao, P.; Chen, X.; Xiong, X.; Yang, M.; Yang, S.; Zhu, X.; Yuan, S.; et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018, 16, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Peng, R.; Zhang, L.-Y.; Sun, Y.; Peng, H.-M.; Liu, H.-D.; Yu, L.-J.; Li, A.-L.; Zhang, Y.-J.; Jiang, W.-H.; et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis. 2017, 8, e2583. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.; Natarajan, R. Long Noncoding RNAs in Diabetes and Diabetic Complications. Antioxidants Redox Signal. 2018, 29, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Leti, F.; Distefano, J.K. Long Noncoding RNAs as Diagnostic and Therapeutic Targets in Type 2 Diabetes and Related Complications. Genes 2017, 8, 207. [Google Scholar] [CrossRef] [Green Version]
- Raut, S.K.; Khullar, M. The Big Entity of New RNA World: Long Non-Coding RNAs in Microvascular Complications of Diabetes. Front. Endocrinol. 2018, 9, 300. [Google Scholar] [CrossRef]
- Fantauzzi, C.B.; Menini, S.; Iacobini, C.; Rossi, C.; Santini, E.; Solini, A.; Pugliese, G. Deficiency of the Purinergic Receptor 2X7 Attenuates Nonalcoholic Steatohepatitis Induced by High-Fat Diet: Possible Role of the NLRP3 Inflammasome. Oxidative Med. Cell. Longev. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Hong, J.; Li, G.; Zhang, Q.; Ritter, J.; Li, W.; Li, P.-L. D-Ribose Induces Podocyte NLRP3 Inflammasome Activation and Glomerular Injury via AGEs/RAGE Pathway. Front. Cell Dev. Biol. 2019, 7, 259. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Bajaj, M.; Qian, J.; Ye, Y. Dipeptidyl peptidase-4 inhibition by Saxagliptin prevents inflammation and renal injury by targeting the Nlrp3/ASC inflammasome. BMJ Open Diabetes Res. Care 2016, 4, e000227. [Google Scholar] [CrossRef]
- Birnbaum, Y.; Bajaj, M.; Yang, H.C.; Ye, Y. Combined SGLT2 and DPP4 Inhibition Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Nephropathy in Mice with Type 2 Diabetes. Cardiovasc. Drugs Ther. 2018, 32, 135–145. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, B.; Wang, L.; Yang, M.; Xia, Z.; Wei, W.; Zhang, F.; Yuan, X. Pioglitazone ameliorates glomerular NLRP3 inflammasome activation in apolipoprotein E knockout mice with diabetes mellitus. PLOS ONE 2017, 12, e0181248. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Bock, F.; Al-Dabet, M.M.; Gadi, I.; Nazir, S.; Wang, H.; Kohli, S.; Ranjan, S.; Mertens, P.R.; Nawroth, P.P.; et al. Stabilization of endogenous Nrf2 by minocycline protects against Nlrp3-inflammasome induced diabetic nephropathy. Sci. Rep. 2016, 6, 34228. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Yin, N.; Liu, W.; Cui, X.; Chen, S.; Wang, E. Curcumin Ameliorates Diabetic Nephropathy by Suppressing NLRP3 Inflammasome Signaling. BioMed Res. Int. 2017, 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Fan, J.; Zhang, X.; Luan, J.; Bian, Q.; Ding, T.; Wang, Y.; Wang, Z.; Song, P.; et al. Interleukin-22 ameliorated renal injury and fibrosis in diabetic nephropathy through inhibition of NLRP3 inflammasome activation. Cell Death Dis. 2017, 8, e2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Liu, X.; Yin, J.; Wu, H.; Cai, X.; Wang, N.; Qian, Y.; Wang, F. IL-6 receptor blockade ameliorates diabetic nephropathy via inhibiting inflammasome in mice. Metabolism 2018, 83, 18–24. [Google Scholar] [CrossRef]
- Yang, S.-M.; Ka, S.-M.; Wu, H.-L.; Yeh, Y.-C.; Kuo, C.-H.; Hua, K.-F.; Shi, G.-Y.; Hung, Y.-J.; Hsiao, F.-C.; Yang, S.-S.; et al. Thrombomodulin domain 1 ameliorates diabetic nephropathy in mice via anti-NF-κB/NLRP3 inflammasome-mediated inflammation, enhancement of NRF2 antioxidant activity and inhibition of apoptosis. Diabetologia 2013, 57, 424–434. [Google Scholar] [CrossRef]
- Soares, J.L.; Fernandes, F.P.; Patente, T.A.; Monteiro, M.B.; Parisi, M.C.; Giannella-Neto, D.; Correa-Giannella, M.L.; Pontillo, A. Gain-of-function variants in NLRP1 protect against the development of diabetic kidney disease: NLRP1 inflammasome role in metabolic stress sensing? Clin. Immunol. 2018, 187, 46–49. [Google Scholar] [CrossRef]
- Wong, T.Y.; Cheung, C.M.G.; Larsen, M.; Sharma, S.; Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Prim. 2016, 2, 16013. [Google Scholar] [CrossRef]
- Richter, C.U. Management of diabetic retinopathy. In Primary Care Medicine; Gorrol, A.H., May, L.A., Mulley, A.G., Jr., Eds.; J.P. Lippincott Company: London, UK, 1987; pp. 857–858. [Google Scholar]
- Duh, E.J.; Sun, J.K.; Stitt, A.W. Diabetic retinopathy: Current understanding, mechanisms, and treatment strategies. JCI Insight 2017, 2, 93751. [Google Scholar] [CrossRef]
- Stitt, A.W.; Curtis, T.M.; Chen, M.; Medina, R.J.; McKay, G.J.; Jenkins, A.J.; Gardiner, T.A.; Lyons, T.J.; Hammes, H.-P.; Simó, R.; et al. The progress in understanding and treatment of diabetic retinopathy. Prog. Retin. Eye Res. 2016, 51, 156–186. [Google Scholar] [CrossRef]
- Schröder, S.; Palinski, W.; Schmid-Schönbein, G.W. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am. J. Pathol. 1991, 139, 81–100. [Google Scholar]
- Friedlander, M. Fibrosis and diseases of the eye. J. Clin. Investig. 2007, 117, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Loukovaara, S.; Nurkkala, H.; Tamene, F.; Gucciardo, E.; Liu, X.; Repo, P.; Lehti, K.; Varjosalo, M. Quantitative Proteomics Analysis of Vitreous Humor from Diabetic Retinopathy Patients. J. Proteome Res. 2015, 14, 5131–5143. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.-Y.; Green, W.R.; Tso, M.O.M. Microglial Activation in Human Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Zhao, M.; Zhao, S.; Lu, Q.; Ni, L.; Zou, C.; Lu, L.; Xu, X.; Guan, H.; Zheng, Z.; et al. Activation of the TXNIP/NLRP3 inflammasome pathway contributes to inflammation in diabetic retinopathy: A novel inhibitory effect of minocycline. Inflamm. Res. 2016, 66, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lv, X.; Hu, Z.; Ye, X.; Zheng, X.; Ding, Y.; Xie, P.; Liu, Q. Protection of Mcc950 against high-glucose-induced human retinal endothelial cell dysfunction. Cell Death Dis. 2017, 8, e2941. [Google Scholar] [CrossRef] [PubMed]
- Devi, T.S.; Lee, I.; Hüttemann, M.; Kumar, A.; Nantwi, K.D.; Singh, L.P. TXNIP Links Innate Host Defense Mechanisms to Oxidative Stress and Inflammation in Retinal Muller Glia under Chronic Hyperglycemia: Implications for Diabetic Retinopathy. Exp. Diabetes Res. 2012, 2012, 1–19. [Google Scholar] [CrossRef]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [Green Version]
- Boss, J.D.; Singh, P.K.; Pandya, H.K.; Tosi, J.; Kim, C.; Tewari, A.; Juzych, M.S.; Abrams, G.W.; Kumar, A. Assessment of Neurotrophins and Inflammatory Mediators in Vitreous of Patients With Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2017, 58, 5594–5603. [Google Scholar] [CrossRef]
- Wu, H.; Hwang, D.-K.; Song, X.; Tao, Y. Association between Aqueous Cytokines and Diabetic Retinopathy Stage. J. Ophthalmol. 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Loukovaara, S.; Piippo, N.; Kinnunen, K.; Hytti, M.; Kaarniranta, K.; Kauppinen, A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. 2017, 95, 803–808. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, R.; Bligard, G.W.; Zhang, S.; Yin, L.; Lukasiewicz, P.; Semenkovich, C.F. Functional Deficits Precede Structural Lesions in Mice With High-Fat Diet–Induced Diabetic Retinopathy. Diabetes 2016, 65, 1072–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresnick, G.H.; Korth, K.; Groo, A.; Palta, M. Electroretinographic Oscillatory Potentials Predict Progression of Diabetic Retinopathy. Arch. Ophthalmol. 1984, 102, 1307–1311. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Alcalde-Estevez, E.; García-Ramírez, M.; Cazzoni, D.; De La Villa, P.; Sánchez-Fernández, E.M.; Mellet, C.O.; García-Fernández, J.M.; Hernández, C.; Simó, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db / db mice. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, X.; Liao, N.; Mi, L.; Peng, Y.; Liu, B.; Zhang, S.; Wen, F. Enhanced Expression of NLRP3 Inflammasome-Related Inflammation in Diabetic Retinopathy. Investig. Opthalmology Vis. Sci. 2018, 59, 978–985. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Lim, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, S.B.B.; Wong, T.Y.; Luu, C.D.; Agrawal, R.; Ghosh, A.; Mortellaro, A.; et al. The NLRP3 Inflammasome May Contribute to Pathologic Neovascularization in the Advanced Stages of Diabetic Retinopathy. Sci. Rep. 2018, 8, 2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Liu, C.; Wan, X.-S.; Li, S.-W. NLRP1 deficiency attenuates diabetic retinopathy (DR) in mice through suppressing inflammation response. Biochem. Biophys. Res. Commun. 2018, 501, 351–357. [Google Scholar] [CrossRef]
- Akhtar-Schäfer, I.; Wang, L.; Krohne, T.U.; Xu, H.; Langmann, T. Modulation of three key innate immune pathways for the most common retinal degenerative diseases. EMBO Mol. Med. 2018, 10, e8259. [Google Scholar] [CrossRef]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells—Implications for age-related macular degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2009, 11, 136–140. [Google Scholar] [CrossRef]
- Thounaojam, M.C.; Montemari, A.; Powell, F.L.; Malla, P.; Gutsaeva, D.R.; Bachettoni, A.; Ripandelli, G.; Repossi, A.; Tawfik, A.; Martin, P.M.; et al. Monosodium Urate Contributes to Retinal Inflammation and Progression of Diabetic Retinopathy. Diabetes 2019, 68, 1014–1025. [Google Scholar] [CrossRef] [Green Version]
- Platania, C.B.M.; Giurdanella, G.; Di Paola, L.; Leggio, G.M.; Drago, F.; Salomone, S.; Bucolo, C. P2X7 receptor antagonism: Implications in diabetic retinopathy. Biochem. Pharmacol. 2017, 138, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Lu, Q.; Chen, W.; Li, J.; Li, C.; Zheng, Z. Vitamin D3 Protects against Diabetic Retinopathy by Inhibiting High-Glucose-Induced Activation of the ROS/TXNIP/NLRP3 Inflammasome Pathway. J. Diabetes Res. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wang, Y.; Xie, T.; Zhan, P.; Zou, J.; Nie, X.; Shao, J.; Zhuang, M.; Tan, C.; Tan, J.; et al. Prostaglandin E2/EP2 receptor signalling pathway promotes diabetic retinopathy in a rat model of diabetes. Diabetologia 2019, 62, 335–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Hu, Y.; Lin, M.; Jenkins, A.J.; Keech, A.C.; Mott, R.; Lyons, T.J.; Ma, J.-X. Therapeutic Effects of PPARα Agonists on Diabetic Retinopathy in Type 1 Diabetes Models. Diabetes 2012, 62, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Chen, Y.; Ding, L.; He, X.; Takahashi, Y.; Gao, Y.; Shen, W.; Cheng, R.; Chen, Q.; Qi, X.; et al. Pathogenic role of diabetes-induced PPAR- down-regulation in microvascular dysfunction. Proc. Natl. Acad. Sci. 2013, 110, 15401–15406. [Google Scholar] [CrossRef] [Green Version]
- Noonan, J.E.; Jenkins, A.J.; Ma, J.-X.; Keech, A.C.; Wang, J.J.; Lamoureux, E.L. An Update on the Molecular Actions of Fenofibrate and Its Clinical Effects on Diabetic Retinopathy and Other Microvascular End Points in Patients With Diabetes. Diabetes 2013, 62, 3968–3975. [Google Scholar] [CrossRef] [Green Version]
- Keech, A.; Mitchell, P.; Summanen, P.; O’Day, J.; Davis, T.; Moffitt, M.; Taskinen, M.-R.; Simes, R.; Tse, D.; Williamson, E.; et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): A randomised controlled trial. Lancet 2007, 370, 1687–1697. [Google Scholar] [CrossRef]
- The ACCORD Study Group and ACCORD Eye Study Group; ACCORD Study Group; ACCORD Eye Study Group; Chew, E.Y.; Ambrosius, W.T.; Davis, M.D.; Danis, R.P.; Gangaputra, S.; Greven, C.M.; Hubbard, L.; et al. Effects of Medical Therapies on Retinopathy Progression in Type 2 Diabetes. New Engl. J. Med. 2010, 363, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, F.; Zhang, X.; Cheng, R.; Ma, J.-X.; Yi, J.; Li, J. Fenofibrate ameliorates diabetic retinopathy by modulating Nrf2 signaling and NLRP3 inflammasome activation. Mol. Cell. Biochem. 2017, 445, 105–115. [Google Scholar] [CrossRef]
- Vincent, A.M.; Callaghan, B.C.; Smith, A.L.; Feldman, E. Diabetic neuropathy: Cellular mechanisms as therapeutic targets. Nat. Rev. Neurol. 2011, 7, 573–583. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, C.; Yan, B.; Shi, X.; Shi, Y.; Qu, L.; Liang, X. Jinmaitong Ameliorates Diabetic Peripheral Neuropathy Through Suppressing TXNIP/NLRP3 Inflammasome Activation In The Streptozotocin-Induced Diabetic Rat Model. Diabetes, Metab. Syndr. Obesity Targets Ther. 2019, 12, 2145–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Lin, X.; Guan, M.; Zeng, Y.; Liu, Y. Verapamil Attenuated Prediabetic Neuropathy in High-Fat Diet-Fed Mice through Inhibiting TXNIP-Mediated Apoptosis and Inflammation. Oxidative Med. Cell. Longev. 2019, 2019, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Huang, Y.; Zhou, F.; Zhao, B.; Zhongyuan, X. Dexmedetomidine attenuates P2X4 and NLRP3 expression in the spine of rats with diabetic neuropathic pain. Acta Cir. Bras. 2019, 34. [Google Scholar] [CrossRef] [Green Version]
- Thakur, V.; Sadanandan, J.; Chattopadhyay, M. High-Mobility Group Box 1 Protein Signaling in Painful Diabetic Neuropathy. Int. J. Mol. Sci. 2020, 21, 881. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Dai, J.; Li, L.; Chen, H.; Chai, Y. NLRP3 Inflammasome Expression and Signaling in Human Diabetic Wounds and in High Glucose Induced Macrophages. J. Diabetes Res. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Noureddin, M.; Rinella, M.E. Nonalcoholic Fatty liver disease, diabetes, obesity, and hepatocellular carcinoma. Clin. Liver Dis. 2015, 19, 361–379. [Google Scholar] [CrossRef]
- Puri, P.; Sanyal, A.J. Nonalcoholic fatty liver disease: Definitions, risk factors, and workup. Clin. Liver Dis. 2012, 1, 99–103. [Google Scholar] [CrossRef]
- Howard, W. Diagnosis and Management of the Metabolic Syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Yearb. Endocrinol. 2006, 2006, 113–114. [Google Scholar] [CrossRef]
- Bradbury, M.W.; Berk, P.D. Lipid metabolism in hepatic steatosis. Clin. Liver Dis. 2004, 8, 639–671. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High Prevalence of Nonalcoholic Fatty Liver Disease in Patients With Type 2 Diabetes Mellitus and Normal Plasma Aminotransferase Levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.-F.; Bian, H.; Gao, X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front. Pharmacol. 2019, 10, 877. [Google Scholar] [CrossRef] [Green Version]
- Muraki, Y.; Makita, Y.; Yamasaki, M.; Amano, Y.; Matsuo, T. Elevation of liver endoplasmic reticulum stress in a modified choline-deficient l -amino acid-defined diet-fed non-alcoholic steatohepatitis mouse model. Biochem. Biophys. Res. Commun. 2017, 486, 632–638. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Stitt, A.W.; Gardiner, T.A.; Anderson, N.L.; Canning, P.; Frizzell, N.; Duffy, N.; Boyle, C.; Januszewski, A.S.; Chachich, M.; Baynes, J.W.; et al. The AGE Inhibitor Pyridoxamine Inhibits Development of Retinopathy in Experimental Diabetes. Diabetes 2002, 51, 2826–2832. [Google Scholar] [CrossRef] [Green Version]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free. Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Iacobini, C.; Menini, S.; Ricci, C.; Fantauzzi, C.B.; Scipioni, A.; Salvi, L.; Cordone, S.; Delucchi, F.; Serino, M.; Federici, M.; et al. Galectin-3 ablation protects mice from diet-induced NASH: A major scavenging role for galectin-3 in liver. J. Hepatol. 2011, 54, 975–983. [Google Scholar] [CrossRef]
- Di Maira, G.; Pastore, M.; Marra, F. Liver fibrosis in the context of nonalcoholic steatohepatitis: The role of adipokines. Minerva Gastroenterol. e Dietol. 2017, 64, 39–50. [Google Scholar]
- Carnevale, R.; Pastori, D.; Nocella, C.; Cammisotto, V.; Baratta, F.; Del Ben, M.; Angelico, F.; Sciarretta, S.; Bartimoccia, S.; Novo, M.; et al. Low-grade endotoxemia, gut permeability and platelet activation in patients with impaired fasting glucose. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Y.; An, W.; Song, J.; Zhang, Y.; Zhao, X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J. Clin. Investig. 2018, 129, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Kakisaka, K.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Gores, G.J. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am. J. Physiol. Liver Physiol. 2011, 302, G77–G84. [Google Scholar] [CrossRef] [Green Version]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef]
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; A Brenner, D.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Pillai, A.; Ganz, M.; Lippai, R.; Petrášek, J.; Park, J.-K.; Kodys, K.; Dolganiuc, A.; Kurt-Jones, E.A.; Szabo, S.B.G. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 2014, 34, 1402–1413. [Google Scholar] [CrossRef] [Green Version]
- Hertog, M.G.; Hollman, P.C. Potential health effects of the dietary flavonol quercetin. Eur. J. Clin. Nutr. 1996, 50, 63–71. [Google Scholar] [PubMed]
- Spahr, L.; Amann, P.; Kern, I.; Golaz, O.; Frossard, J.-L.; Hadengue, A.; Bresson-Hadni, S. Allopurinol, oxidative stress and intestinal permeability in patients with cirrhosis: An open-label pilot study. Liver Int. 2007, 27, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, C.; Ding, X.-Q.; Pan, Y.; Gu, T.-T.; Wang, M.-X.; Liu, Y.-L.; Wang, F.-M.; Wang, S.-J.; Kong, L.-D. Quercetin and allopurinol reduce liver thioredoxin-interacting protein to alleviate inflammation and lipid accumulation in diabetic rats. Br. J. Pharmacol. 2013, 169, 1352–1371. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.-C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Musso, G.; Cassader, M.; Rosina, F.; Gambino, R. Impact of current treatments on liver disease, glucose metabolism and cardiovascular risk in non-alcoholic fatty liver disease (NAFLD): A systematic review and meta-analysis of randomised trials. Diabetol. 2012, 55, 885–904. [Google Scholar] [CrossRef]
- Gao, X.; Fan, J.-G. Diagnosis and management of non-alcoholic fatty liver disease and related metabolic disorders: Consensus statement from the Study Group of Liver and Metabolism, Chinese Society of Endocrinology. J. Diabetes 2013, 5, 406–415. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; A Aldersley, M.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Jinnouchi, H.; Sugiyama, S.; Yoshida, A.; Hieshima, K.; Kurinami, N.; Suzuki, T.; Miyamoto, F.; Kajiwara, K.; Matsui, K.; Jinnouchi, T. Liraglutide, a Glucagon-Like Peptide-1 Analog, Increased Insulin Sensitivity Assessed by Hyperinsulinemic-Euglycemic Clamp Examination in Patients with Uncontrolled Type 2 Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, X.; Li, L.; Ma, T.; Shi, M.; Yang, Y.; Fan, Q.-L. A small molecule inhibitor MCC950 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3 inflammasome activation. Diabetes, Metab. Syndr. Obesity Targets Ther. 2019, 12, 1297–1309. [Google Scholar] [CrossRef] [Green Version]
- Mridha, A.; Wree, A.; Robertson, A.A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, F.; Wang, Y.; Gisterå, A.; Roy, J.; Paulsson-Berne, G.; Hedin, U.; Lerman, A.; Hansson, G.K.; Herrmann, J.; et al. Inflammasome-Driven Interleukin-1α and Interleukin-1β Production in Atherosclerotic Plaques Relates to Hyperlipidemia and Plaque Complexity. JACC Basic Transl. Sci. 2019, 4, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as Novel Therapeutic Strategies for NLRP3 Inflammasome-Related Neurological, Metabolic, and Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Systemic and local tissue inflammation in diabetic complications. CVD = cardiovascular disease; DKD = diabetic kidney disease; DR = diabetic retinopathy; NASH = non-alcoholic steatohepatitis. IL-1β = interleukin-1β.
Figure 1.
Systemic and local tissue inflammation in diabetic complications. CVD = cardiovascular disease; DKD = diabetic kidney disease; DR = diabetic retinopathy; NASH = non-alcoholic steatohepatitis. IL-1β = interleukin-1β.

Figure 2.
Diabetes mellitus-related metabolic factors acting as NLRP3-priming and activating signals. LDLs = low-density lipoproteins; AGEs = advanced glycation endproducts; ALEs = advanced lipoxidation endproducts; FFAs = free fatty acids; ATP = adenosine triphosphate; ASC = apoptosis-associated speck-like protein containing a CARD; IL-1β = interleukin-1β; IL-18 = interleukin-18; GSDMD = gasdermin D; GSDMD-N = GSDMD N-terimanl domain.
Figure 2.
Diabetes mellitus-related metabolic factors acting as NLRP3-priming and activating signals. LDLs = low-density lipoproteins; AGEs = advanced glycation endproducts; ALEs = advanced lipoxidation endproducts; FFAs = free fatty acids; ATP = adenosine triphosphate; ASC = apoptosis-associated speck-like protein containing a CARD; IL-1β = interleukin-1β; IL-18 = interleukin-18; GSDMD = gasdermin D; GSDMD-N = GSDMD N-terimanl domain.

Figure 3.
Diabetes mellitus-related factors possibly involved in the activation of NLRP3 and acceleration of the atherogenic process. GLUT = glucose transporter; AGEs = advanced glycation endproducts; RAGE = Receptor for AGEs; TLRs = toll-like receptors; LDLs = loe density lipoproteins; HMGB1 = high mobility group box 1; NETs = neutrophil extracellular traps; ROS = radical oxygen species; NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells; ASC = apoptosis-associated speck-like protein containing a CARD; P2RX7 = P2X purinoreceptor 7; VCAM-1 = vascular cell adhesion molecule 1; MCP-1 = monocyte chemoattractant protein 1; IL-1β = interleukin-1β; IL-18 = interleukin-18.
Figure 3.
Diabetes mellitus-related factors possibly involved in the activation of NLRP3 and acceleration of the atherogenic process. GLUT = glucose transporter; AGEs = advanced glycation endproducts; RAGE = Receptor for AGEs; TLRs = toll-like receptors; LDLs = loe density lipoproteins; HMGB1 = high mobility group box 1; NETs = neutrophil extracellular traps; ROS = radical oxygen species; NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells; ASC = apoptosis-associated speck-like protein containing a CARD; P2RX7 = P2X purinoreceptor 7; VCAM-1 = vascular cell adhesion molecule 1; MCP-1 = monocyte chemoattractant protein 1; IL-1β = interleukin-1β; IL-18 = interleukin-18.

Figure 4.
Nlrp3 inflammasome activation in renal resident cells contributes to the pathogenesis of diabetic kidney disease. WT = wild type; NLRP3−/− = NOD-like receptor pyrin domain-containing-3 knocked down; BM = bone marrow; ECM = extracellular matrix; DKD = diabetic kidney disease.
Figure 4.
Nlrp3 inflammasome activation in renal resident cells contributes to the pathogenesis of diabetic kidney disease. WT = wild type; NLRP3−/− = NOD-like receptor pyrin domain-containing-3 knocked down; BM = bone marrow; ECM = extracellular matrix; DKD = diabetic kidney disease.

Figure 5.
Role of inflammation and inflammasome NLRP3 activation in the early and advanced stages of diabetic retinopathy. ROS = radical oxygen species; TXNIP = thioredoxin interacting protein; DR = diabetic retinopathy; DME = diabetic macular edema; PDR = proliferative DR.
Figure 5.
Role of inflammation and inflammasome NLRP3 activation in the early and advanced stages of diabetic retinopathy. ROS = radical oxygen species; TXNIP = thioredoxin interacting protein; DR = diabetic retinopathy; DME = diabetic macular edema; PDR = proliferative DR.

Figure 6.
Molecular mechanisms underlying the accelerating effect of diabetes mellitus on the initiation and progression of NAFLD. The dashed line indicates a hypothesis under investigation. IL-6 = interleukin-6; TNF-α = Tumor necrosis factor-α; FFAs = free fatty acids; ER = endoplasmic reticulum; NASH = non-alcoholic steatohepatitis; AGE = advanced glycation endproducts; ALE = advanced lipoxidation endproducts.
Figure 6.
Molecular mechanisms underlying the accelerating effect of diabetes mellitus on the initiation and progression of NAFLD. The dashed line indicates a hypothesis under investigation. IL-6 = interleukin-6; TNF-α = Tumor necrosis factor-α; FFAs = free fatty acids; ER = endoplasmic reticulum; NASH = non-alcoholic steatohepatitis; AGE = advanced glycation endproducts; ALE = advanced lipoxidation endproducts.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Menini, S.; Iacobini, C.; Vitale, M.; Pugliese, G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells 2020, 9, 1812. https://doi.org/10.3390/cells9081812
AMA Style
Menini S, Iacobini C, Vitale M, Pugliese G. The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders. Cells. 2020; 9(8):1812. https://doi.org/10.3390/cells9081812
Chicago/Turabian StyleMenini, Stefano, Carla Iacobini, Martina Vitale, and Giuseppe Pugliese. 2020. "The Inflammasome in Chronic Complications of Diabetes and Related Metabolic Disorders" Cells 9, no. 8: 1812. https://doi.org/10.3390/cells9081812
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.