
Abstract
Climatic oscillations of the Quaternary rapidly compelled plant species to shift their geographical range. How alpine plant species responded to climate change, however, remains elusive and remnants of the cold-adapted flora that currently strive in restricted ranges as small, isolated populations have been particularly overlooked. To address the evolutionary history of such a ‘glacial relict’, we here sampled and genotyped all known native populations of a narrow endemic species from the northwestern Alps, Papaver occidentale, as well as closely related taxa with double digest restriction-site Associated DNA (ddRAD) sequencing. Spatial patterns of genetic variation across populations coupled with insights from climatic niche modelling through time address underpinings of the long-term persistence of the species in face of climate changes. Evidence from population genetics and ecological modelling indicates that P. occidentale likely persisted through the last glacial maximum outside of the Western Prealps and that a major lineage recolonized the area from lower elevation, external regions. Differentiated lineages at the Northern margins of the species distribution range highlight highly divergent and geographically restricted populations that include considerable share of private markers and may indicate local glacial survival in isolated conditions. Our data thus imply that processes having shaped intraspecific spatial genetic structure within the Alps can be complex and lead to mosaic of populations with a mixed-history of local survival and immigration. A better understanding of spatio-temporal aspects of range contraction–expansion is crucial to shed light on processes underlying the evolution of remnant populations of such endemic species and set conservation priorities considering current climate changes.
Similar content being viewed by others

Introduction
Climatic oscillations of the Quaternary rapidly compelled species to shift their geographical range (Hewitt 2004), especially in the alpine regions that were heavily glaciated during cold periods such as the last glacial maximum (LGM) some 22,000 years ago (Schonswetter et al. 2005). How plant species react to climate change, however, remains poorly understood and the European Alps offer an adequately heterogeneous model landscape to address the impact of range expansion–contraction on the evolution of alpine plants (Ozenda 2009). The European Alps are among main regional centers of biodiversity and their flora contains around 10% of endemic species (Aeschimann et al. 2011). How arctic-alpine plants shifted their distribution ranges in between cold-phases of the LGM and current times is largely unknown, as remain consequences for their diversification (Kadereit 2017; Schönswetter and Schneeweiss 2019).
Glacial history of alpine plants in the European Alps
The current distribution of cold-adapted species across the European Alps is commonly interpreted as the result of a tabula rasa scenario (reviewed in Stehlik 2000). This hypothesis typically postulates extinction of all populations within the heavily glaciated Alps during cold phases and hence species persistence in nearby lowlands or secondary massifs at the periphery of ice sheets. Extra-alpine refugia were accordingly postulated as main sources of current populations for several alpine species and generally supported by clines of genetic diversity left by recolonization towards the center of the Alps (Stehlik et al. 2002a; Taberlet et al. 2012; Thiel-Egenter et al. 2011).
The so-called nunatak hypothesis, contrastingly, postulates in situ survival (or resistance) of populations throughout climatic cold phases in ice-free mountain tops (nunataks) within otherwise highly glaciated alpine areas. Endemic alpine species being particularly abundant at both extremities of the Alps, those regions (i.e., south-western and north-eastern Alps) were early postulated as major refugia with relatively stable climates, where alpine plants persisted throughout cycles of range contraction–expansion (Harrison and Noss 2017; Smyčka et al. 2017; Tribsch 2004). Consistent with long-term persistence of cold-adapted plant populations in nunataks across the Alps (i.e., so-called, cryptic refugia), specific mountain ranges in the central Alps host narrow endemic species such as Phyteuma humile (Schneeweiss et al. 2013) or represent hotspots of genetically differentiated populations in several widespread species such as Eritrichium nanum (Stehlik et al. 2002b) or Senecio halleri (Bettin et al. 2007).
Despite continuous efforts to disentangle intricate patterns of genetic variation within species in the light of the tabula rasa paradigm, the contribution of different LGM refugia to the current flora of the Alps remains elusive. Accordingly, the extent to which different cold-adapted species from alpine habitats recolonized the Alps from specific external low-elevation populations or from in situ refugia in the peripheral vs the central Alps remains a largely open question (Schönswetter and Schneeweiss 2019). More generally, the spatial arrangement of refugia likely influences gene-flow during range contraction–expansion and how it interacts with the evolutionary diversification of plants from different elevations is still poorly understood (Kadereit 2017). In contrast to several widespread species that have been investigated at the scale of the entire Alps (e.g., Taberlet et al., 2012), species that currently occur as small, isolated populations (i.e., so-called glacial relicts) have been neglected and deserve further attention (Jiménez-Alfaro et al. 2016).
Endemic species of the Western Prealps
The Western Prealps (i.e., the area covering North-Western calcareous periphery of central Alps, from Haute-Savoie in France to Lake of Thun in Switzerland) form a relatively small but well-defined geological and biogeographical unit (Gilomen 1941; Caron 1973). They are part of the external calcareous Alps extending from Isère in France to Lower Austria (Berthouzoz et al. 2013), and represent the intersection between the major LGM refugia at both extremities of the Alps (Gerber et al. 2010; Parisod 2008; Thiel-Egenter et al. 2011). Currently dominated by subalpine vegetation, those calcareous mountains show several alpine species with fragmented distribution on mountain tops (Gerber et al. 2010). Postulated as favorable for species from calcareous bedrocks during the LGM, candidate refugia remain to be precisely identified (Paun et al. 2008; Schonswetter et al. 2005). The area certainly played a major role as early source of postglacial recolonizers towards the central Alps (Parisod 2008), and several species such as Biscutella laevigata or Eryngium alpinum present genetically diversified and differentiated populations across the Western Prealps (Naciri and Gaudeul 2007; Parisod and Besnard 2007). The area appears as a major hybrid zone for lineages of species such as Arabis alpina (Rogivue et al. 2018; Thiel-Egenter et al. 2011) and, consistent with the hypothesis of secondary contact among lineages, the narrow endemic of the Western Prealps Arenaria bernensis was demonstrated to be of allopolyploid origin (Berthouzoz et al. 2013). It calls for additional case studies on processes at the origin of endemic species in the Western Prealps to shed light on their role as a museum vs cradle of biodiversity (Gerber et al. 2010).
The genus Papaver includes a hundred species in several sections among which the monophyletic sect. Meconella presents exclusively arctic-alpine and high-mountain species (Carolan et al. 2006). It includes Papaver radicatum, which is among most northerly-growing plants with records up to 83° North, and the alpine poppy complex (Papaver alpinum s.l.) that includes over 13 outcrossing species distributed in small isolated regions across the European Alps and some other major central- and southern European mountain ranges (Markgraf 1958). Notably, white-flowered and narrow-lobed alpine poppies growing on calcareous bedrock were circumscribed as a distinctive group (Kadereit 1990) and taxa nested within the white-flowered clade (i.e., P. ernesti-mayeri, P. occidentale, P. sendtneri, and P. tatricum) were traditionally treated as separate species or subspecies of P. alpinum (e.g., Jalas and Suominen 1991). Among them, Papaver occidentale (Markgr.) H.E. Hess, Landolt & Hirzel has early been recognized as an endemic species with a distribution range matching the boundaries of the Western Prealps (Hess et al. 1977). Molecular markers hardly corroborated the traditionally delimited taxa based on highly polymorphic morphological characters, and internal division within P. alpinum s.l. remain debatable (Schönswetter et al. 2009). As populations of P. occidentale form a well supported genetic group testifying its history as a separate lineage (Schönswetter et al. 2009) and are on priority lists regarding action plans for protection (BAFU 2011), this genetically and geographically-delimited conservation unit deserves further investigation.
Several aspects of the biology and ecology of P. occidentale remain poorly investigated besides very few local studies and reports (Bétrisey 2014; Bornand and Hoffer-Massard 2003; Delarze 2012). The taxon appears to be currently restricted to north facing calcareous screes of the alpine zone (Thlaspion rotundifolii). Although such habitat is usually not disturbed by human activities, this stenothermic plant requires high-elevation, cold screes on calcareous and such habitats are becoming rare among mid-elevation summits of the Western Prealps. Vulnerable to future climate changes, the majority of population present between 50 and 800 individuals, and largest ones include up to 10,000 individuals. As all members of the sect. Meconella, P. occidentale is perennial with leaves in basal rosettes, but presents no means of vegetative propagation (Kadereit 1990). Individual plants are outcrossing and often start to flower 1 year after germination, with an average generation time estimated at ca. 8 years supporting high seed production (Nordal et al. 1997; B. Clémant, personal observations). The present study aims at describing the distribution of genetic variation among extant populations of the endemic P. occidentale to understand how such a taxon persisted in face of past climate changes. More specifically, it characterizes a comprehensive sample of P. occidentale, as well as closely related outgroups selected based on earlier literature, to delineate its native distribution and address the phylogeography of remnant populations using genome-wide double digest Restriction-site Associated DNA (ddRAD) sequencing. Such historical insights on the genetic structure within this endemic taxon are used to set conservation priorities.
Materials and methods
Sampling of plant material
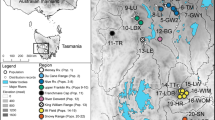
Papaver occidentale was early treated as a species distinct from other white-flowered taxa of the P. alpinum complex such as P. sendtneri (Hess et al. 1977), whereas Kadereit (1990) suggested its affinity to poppies of the Tatra Mountains in the Carpathians (P. tatricum). To account for taxonomic uncertainty, all known populations of the P. alpinum complex showing four white petals across the Western Prealps were considered in this study (incl. P. sendtneri) and we further included samples from P. tatricum. Current populations were comprehensively sampled across the whole Western Prealps (Fig. 1) by revisiting all localities reported in Info Flora (www.infoflora.ch), the literature (Markgraf 1958) and unpublished reports (Delarze 2012; Bétrisey 2014).

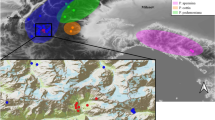
Habitat suitability and distribution of Papaver occidentale at the last glacial maximum (a) and at present time (b). The current distribution of the different taxa of the P. alpinum complex across the European Alps is illustrated, with the range of known populations of P. occidentale shown as black dots across the Western Prealps. Maximum extent of the ice sheet is shown in light grey
In each field population, 1 m-radius plots of similar density (considering subpopulations including at least five individuals) were surveyed and georeferenced using a GPS (Satmap Active 10). One or two plots were sampled in populations of less than 25 individuals, whereas three to five plots were sampled in larger populations as to maximizing the distance between plots and encompass the local distribution while collecting in similarly dense subpopulations. Leaf material was non-invasively sampled from one individual per plot for a total of 112 native samples from 19 populations nested in six regions (Massif des Bornes, Grammont, Gummfluh, Vanil Noir, Dent de Savigny and Spillgerten; Table 1). Four individuals from two small non-native populations from the Jura Mountains were additionally here included to confirm their introduction from the Massif des Bornes (Delarze 2012). In addition, we included outgroup samples of close relatives from the North of the Alps (Schönswetter et al. 2009) as twelve individuals of P. sendtneri from the Northern Alps (Pilatus and surroundings) and eight individuals of P. tatricum from the Tatra Mountains, for a total of 136 samples (Supplementary Information Table S1).
Climatic niche modelling and potential distribution through time
Species distribution modelling based on all known occurrences of P. occidentale was performed following Guisan et al. (2017) to generate coarse predictions supporting interpretations. A general additive model was developed, calibrated and validated at the locale scale, in the area currently occupied by P. occidentale (see Supplementary Text 1). We identified potential distribution at large scale (present and LGM) based on the same response curves. As pertinent variables such as the distribution of calcareous screes developed in Fragnière et al. (2020) are available at neither the large scale nor across time, we here described the potential distribution of P. occidentale during LGM using temperature and precipitation from Worldclim data (Hijmans et al. 2005; CCSM4 model at 2.5 arc-minutes resolution), after exclusion of the surface covered by ice following Ehlers et al. (2011).
Preparation of ddRAD sequencing libraries
DNA was extracted from silica-dried leaves of all 136 individual samples using DNeasy Plant Mini Kit (QIAGEN) according to the manufacturer’s protocol. Extracted DNA was checked for integrity on agarose electrophoresis and quantified using Nanodrop before genotyping by the ddRAD sequencing approach.
Libraries for ddRAD sequencing were prepared following (Peterson et al. 2012) with slight modifications. In brief, 250 nanograms of genomic DNA from each of the 136 individual sample was digested with 0.5 µl EcoRI-HF and 1 µl MseI (New England Biolabs) in 3 µl NEB Smartcut Buffer for 50 min at 37 °C. After inactivation at 65 °C for 20 min and cleaning with 1 × AMPure XP beads (Beckman Coulter), adapters were ligated in a 30 µl reaction volume using 2 µl P2-biotin adapters, 2 µl P1_X adapters, 3 µl 10 × T4 DNA ligase buffer and 1 µl T4 DNA ligase. The P1_X EcoR1 adapters included 48 barcodes (5 bp long), whereas the P2 MseI adapters included a Biotin tag and four degenerated bases to identify PCR duplicates on one strand as well as the index and the flow cell annealing region on the other strand. Samples were then pooled and DNA fragments of targeted length of 550 bp were size-selected using 0.6 × followed by 0.12 × undiluted AMPure XP beads. Biotin-tagged fragments were further selected with Dynabeads M-270 Streptavidin (Invitrogen). For each MseI index, remaining ddRAD fragments were PCR-amplified for ten cycles and cleaned using 1 × AMPure XP beads. The concentration and distribution of fragment sizes of each library as well as their final pool were checked using the Bioanalyzer with the High Sensitivity DNA kit (Agilent). The pooled library supplemented with 20% PhiX (Illumina) for quality control was sequenced as 100-bp single-end reads on one lane of Illumina HiSeq3000 lane at the Next Generation Sequencing Platform of the University of Bern.
Reference assembly and SNP calling
Raw sequences were quality controlled with fastqc (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and processed with the ddocent pipeline (www.ddocent.com) following Grünig et al. (2020). After demultiplexing and trimming with the process_radtag component of stacks v.1.26 (Catchen et al. 2013), reads above 80% similarity were clustered using cd-hit to generate the reference catalogue of loci against which individual reads were mapped using bwa (Li and Durbin 2009). Such an approach based on de novo reference fits best practice (LaCava et al. 2020) and was here priviledged to alternative reference-based approaches using the distantly related P. somniferum and its recent history of genomic duplication (Guo et al. 2018).
Multi-allelic SNPs were called with freebayes v1.1 (https://github.com/ekg/freebayes) and further filtered with vcftools (Danecek et al. 2011). Only variants with a minimum depth of 3, a mean depth of 10, a minor allele count of 3 and a minor allele frequency of 0.05, were retained. SNPs with more than 20% missing data per population and coverage larger than 100 were further excluded. After decomposition of multi-allelic SNPs and removal of indels, only bi-allelic loci with less than 10% missing data among individuals were kept. Genome-wide Tajima’s D was estimated using PopGenome (Pfeifer et al. 2014). SNPs in (global) Hardy–Weinberg equilibrium were haplotyped using rad_haplotyper v.1.1.5 (Willis et al. 2017) to generate a dataset of independent and polymorphic ddRAD loci.
To further exclude variants possibly under selection, those ddRAD loci encompassing coding regions (i.e., within an annotated gene) were identified using the recent P. somniferum reference genome (Guo et al. 2018) using the Burrows-Wheeler aligner. Although not all coding regions are under selection and specific non-coding sequences may be non-neutral, such a non-genic dataset should include a majority of nearly-neutral loci for historical inferrences (Nielsen 2005).
Genetic structure and isolation by distance
Genotypes of all individuals were visualized with a Neighbornet network based on the proportion of different sites between all individuals (uncorrectedP) using splitstree4 v4.14.8 (Huson and Bryant 2006). The number of private alleles and allelic richness using rarefaction, as well as observed and expected heterozygosity and FIS within populations and regions, were estimated from ddRAD-derived SNPs using the R-package diversity (Keenan et al. 2013). Genetic diversity was further partitioned among populations nested within regions using analyses of molecular variance in arlequin ver.3.5 (Excoffier and Lischer 2010) with 1000 permutations.
Model-based clustering of individuals based on multilocus allele frequencies inferred the genetic structure among samples using structure v2.3.4 with admixture and correlated allele frequencies, but without a priori information (Pritchard et al. 2000). Analyses included 10 replicated runs for each number of clusters (K) between K = 1 and K = 9 with 50,000 iterations of burn-in followed by 1,000,000 iterations. The optimal K was inferred following the ΔK method based on change in Ln probabilities L(K) (Evanno et al. 2005) using structure Harvester (https://omictools.com/structure-harvester-tool). Outcomes of replicates were integrated using clumpp (Jakobsson and Rosenberg 2007) and, after permutation of matrices across replicated analyses with the same K, the mean matrix output was visualized using distruct (Rosenberg 2003). These analyses were performed for all 136 samples as well as for the 112 native P. occidentale samples separately, based on all loci as well as non-genic loci only. Given the large number of loci and the limited influence of cluster size on individual membership estimates, resulting patterns are reliable (Rosenberg et al. 2005).
Genetic isolation by geographical distance expected under restricted dispersal of neutral genes was assessed by Mantel tests. Pairwise genetic distances estimated by Rousset’s â (Rousset 2000) in spagedi v1.3 (Hardy and Vekemans 2002) were associated to Euclidean spatial distances between individuals using the mantel function in the R-package vegan (https://cran.r-project.org/web/packages/vegan/index.html), with significance evaluated by 999 permutations. Analyses were first carried out for all the 112 native P. occidentale individuals and, then, within each genetic cluster out of structure analyses.
Results
Potential distribution of Papaver occidentale during the Last Glacial Maximum
Coarse niche modelling based on the current distribution of native populations of P. occidentale confirmed that current areas with suitable humidity and temperature mostly occur at high elevation across the Western Prealps. In contrast, during the LGM, this area was mostly unavailable due to covering by ice sheet, while climatically suitable space was distributed across large areas outside of the Alps. In particular, large potential LGM refugia appeared close to the South, the North and the West edge of the current distribution of P. occidentale (Fig. 1).
Genotyping of ddRAD loci in Papaver occidentale
The 136 samples analyzed here using the ddRAD sequencing approach included 112 native and four introduced individuals of P. occidentale as well as 20 outgroup individuals from P. sendtneri and P. tatricum. Around 272 × 106 sequencing reads passed our quality control and were mapped at 76.8% on our de novo reference catalogue made of 66,940 contigs. Stringent filtering of 338,463 raw SNPs yielded 5912 biallelic SNPs (Supplementary Information Table S2) that revealed a large excess of frequent variants consistent with a positive Tajima’s D among all 136 individuals (D = 1.78) or among individuals within each region (Table 1).
SNPs were haplotyped into 3070 independent ddRAD loci in all 136 individuals. Loci identified in coding sequences highlighted 701 independent loci that may be non-neutral and that were removed to yield a non-genic dataset with 136 genotypes at 2369 ddRAD loci. Datasets resulted in similar patterns of genetic variation (Supplementary Information Figure S1) and analyses based on all 3070 ddRAD loci are presented here.
Genetic structure
Neighbor-Net network based on proportions of SNPs among all 136 individuals clustered genotypes according to taxonomy and geography of samples, with outgroups from P. sendtneri and P. tatricum forming a group consistently differentiated from samples of P. occidentale (Fig. 2).

Neigbour-Net network based on proportions of SNPs among all 136 studied samples from the Papaver alpinum complex analyzed here. Outgroups from P. sendtneri and P. tatricum form a group consistently differentiated from native samples of P. occidentale whose samples cluster according to their region of origin
Based on multilocus allele frequencies, STRUCTURE clustered all samples from the North-East of the study area with the close geographically outgroup samples of P. sendtneri. Within P. occidentale, genotypes from populations of the Jura Mountains clustered with samples from the Massif des Bornes, as expected based on the documented introduction of those populations. After their exclusion, 112 samples of P. occidentale were considered representative of native populations in our dataset and all presented distinct genotypes distributed among the six regions (Table 1).
Hierarchical AMOVA among the 112 native samples showed that individuals harbour most of the genetic variation, with only 5.1% (FSC) and 12.1% (FCT) of the total variance segregating among populations and regions, respectively (Supplementary Information Table S3). Further congruent with such genetic homogeneity within P. occidentale, individual inbreeding coefficients within populations (mean FIS = − 0.21) and regions (FIT = 0.001) were non-significantly different from zero, as could be expected for a representative of Papaveraceae with an efficient self-incompatibility system. Pairwise FST supported significant genetic structure between most of the 19 native populations of P. occidentale, with higher differentiation among populations from different regions (Supplementary Information Table S4). Although genetic diversity appeared evenly distributed among regions, allelic richness as well as observed heterozygosity were significantly greater in large populations of the Massif des Bornes, Vanil Noir and Spillgerten than in other regions, where the species is locally less abundant (Table 1). Accordingly, 85.4% of the private alleles segregating within the species were represented among those three regions that also host largest populations.
Clustering of genotypes among all 136 samples using structure showed a mean Ln probabilities L(K) increasing from K = 1 to K = 9 (Fig. S1) and identified optimal numbers of genetic clusters at K = 2, K = 3 and K = 5. Clustering based on the full dataset of SNPs vs SNPs in non-genic loci detected largely similar hierarchical genetic clusters across K (Fig. S1), supporting patterns representative of neutral genetic structure within P. occidentale. clumpp and distruct outputs confirmed individual assignment among clusters across replicated structure runs and, from K = 2, populations of Spillgerten formed a genetically differentiated cluster (Fig. 3). Outgroups and individuals from Vanil Noir were detected as genetically distinct from K = 3 and, at higher K, further genetic differentiation was detected among taxa and regions. Similar analyses focused on the 112 native samples of P. occidentale confirmed this hierarchical structure with optimal K at 2 and 3, and supported the Spillgerten population as being most divergent lineage (Fig. S1).

Genetic structure of Papaver occidentale based on 3070 ddRAD loci. Structure barplots show the hierarchical partitioning of genetic variation from K = 2 to K = 5 clusters among native samples of the species (112 samples; Table 1) as well as introduced populations in Jura and outgroups (P. sendtneri and P. tatricum; 136 samples). The distribution of native populations among genetic clusters from the Western Prealps shows a widespread Southern lineage of P. occidentale that is genetically close to outgroups and extends up to the North of the species range. Differentiated gene pools are restricted to specific regions such as Spielgerten and Vanil noir in the North of the species range
Isolation by distance among all native individuals of P. occidentale showed a significant correlation of 0.81 (p = 0.001). Similar analyses within each genetic cluster highlighted significant isolation by distance within the Massif des Bornes (0.54, p = 0.001), Spillgerten (0.47, p = 0.001) and Vanil Noir (0.66, p = 0.001) as well as within the remaining regions (Gummfluh and Dent de Savigny: 0.87, p = 0.001).
Discussion
Pattern of genetic variation within Papaver occidentale
The fine-scale genetic composition of the narrow endemic Papaver occidentale is here investigated for the first time. Individuals genotyped with ddRAD sequencing congruently clustered into coherent groups using distance-based or model-based inferences in structure, and highlighted P. occidentale as consistently differentiated from other white-flowered taxa of the P. alpinum complex included in this analysis. Despite overall genetic similarity between P. occidentale, P. sendtneri and P. tatricum, samples were unambiguously identified as P. occidentale or P. sendtneri based on morphology and multilocus genotypes, clarifying the distribution of native populations of P. occidentale considered as a conservation unit.
Native populations from the southern part of the distribution range of P. occidentale were assigned to a well defined genetic cluster that is substantially differentiated from northern populations growing in the Vanil Noir or Spillgerten regions. As the species presented overall genetic isolation by distance across its range, detected clusters may reflect inflated coincidence with geographic regions along such a genetic cline (Serre and Pääbo 2004). Such artefactual clustering is, however, unlikely to explain the high genetic differentiation of specific populations such as here observed in Spillgerten. As the assignment of individuals into geographical clusters chiefly results from discontinuity in genetic distances reflecting geographic barriers to gene flow (Rosenberg et al. 2005), the restricted genetic clusters detected here at Vanil Noir and Spillgerten appear largely isolated from other populations of P. occidentale that form a genetic cluster highlighting a widespread Southern lineage related to outgroups and showing large expansion across the Western Prealps. Despite their restricted spatial extent across specific mountains, these Northern populations appear not only differentiated but also highly diversified, harbouring exceptionally high levels of allelic richness and private variants suggestive of long-term accumulation of genetic diversity. Within the widespread southern lineage, comparable variation was only apparent among southern-most populations of the Massif des Bornes. Such a pattern with highest allelic richness at both the northern and the southern margins, whereas the geographic centre of the species distribution range is less diverse, appears mostly consistent with long-isolated lineages.
Potential source(s) of postglacial (re)colonization
As the current distribution area of P. occidentale in the Western Prealps was largely covered by the massive LGM Alpine ice sheet, populations likely survived across some of the low-elevation regions highlighted by niche modelling as putatively climatically suitable during the cold period. In northeastern and southeastern Alps, individual plants and even large populations of P. alpinum s.l. were observed in gravel river beds and road sides at low altitudes (Schönswetter et al. 2009). Therefore, it is not unlikely that ancestral populations of P. occidentale were frequent in calcareous-rich glacier forelands and vast alluvial terraces that surrounded heavily-glaciated parts of central Alps during the LGM. They then followed the retracting glaciers and survived only in few isolated and small areas (Kadereit 1990). The excess of frequent variants at genome-wide SNPs is indicative of population contraction across the species range and, therefore, consistent with scenarios involving range shift of large LGM populations towards the currently restricted distribution of genetically differentiated lineages across the Western Prealps.
Descriptive insights on current genetic variation at ddRAD loci within P. occidentale directly assess neither the location of LGM refugia nor whether recolonization occurred from one or multiple regions. Southern and northern lineages appearing genetically divergent from other white-flowered taxa, with samples from the Spielgerten population standing out at the lower level of hierarchical clustering (i.e., from K = 2), support relatively ancient isolation. Without reliable fossil calibration available, it, however, remains challenging to firmly distinguish between patterns of genetic differentiation shaped in independent LGM refugia or through early allopatry at the onset of postglacial recolonization (e.g., following long-distance dispersal). Patterns of spatial genetic variation were, therefore, privileged to identify potential refugia and postglacial recolonization pathways used by P. occidentale towards its current range in the Western Prealps.
South-western Alps have regularly been discussed as a large refugium, where alpine plants survived throughout the LGM (Schönswetter et al. 2005). Although the P. alpinum complex is represented by P. aurantiacum there (Aeschimann et al. 2011), a long-term persistence of P. occidentale at the edge of the Alps can hardly be rejected and may appear consistent with the highly diverse populations currently observed at the southern margin of its distribution range (Hampe and Petit 2005). The considerable areas in the north and the west of the current range of P. occidentale that were climatically and edaphically suitable during the LGM could indeed have contributed to all or specific lineages of the Western Prealps (Parisod 2008). Large areas in the north of the Alps were suggested to have sheltered favorable conditions for alpine plants during the LGM (Schmitt 2009) and harbour relict populations of alpine plants on extrazonal rocky habitats (Reisch et al. 2003; Vogler and Reisch 2013). Such a scenario would suggest a mostly postglacial differentiation of taxa currently occurring across the northern Alps (P. occidentale and P. sendtneri) and possibly the Tatra Mountains (P. tatricum). A western refugium looks also plausible and may support not only a genetically differentiated P. occidentale but would further be consistent with a diversification of the P. alpinum complex in distinct LGM refugia (Kadereit et al. 2004). Similar scenarios were priviledged for other species such as Erinus alpinus (Stehlik et al. 2002a) and commonly postulated to explain the disjunct distribution of alpine plant species in the Massif Central, the Jura Mountains and the Western Alps (Kropf et al. 2008; Parisod 2008). Although the species may have arguably gone locally extinct before notice, the absence of native populations of P. occidentale in the Jura Mountains despite favorable conditions for reproduction must be considered discordant with the otherwise plausible hypothesis of a (north-)western refugium.
Provided that spatial patterns of genetic variation in P. occidentale hardly favor one over the multiple competing scenarios fitting the tabula rasa paradigm, it may be worth considering debated alternatives involving in situ persistence within the Alps. Being a cold-adapted species from periglacial habitats, P. occidentale has an ecological potential to have survived the LGM on micro-topographically favorable sites on mountain tops extruding from otherwise inactive ice sheets across the external calcareous Alps (Stehlik 2000). The patchy distribution of genetic clusters from P. occidentale across the Western Prealps matches scenarios involving long-term persistence of divergent lineages in nunataks closer to the current species range. The high accumulation of private alleles in northern populations of P. occidentale such as Spillgerten indeed seems coherant with in situ survival of populations across patches of rocky habitats at the northern edge of the ice sheets during the LGM. Accordingly, the current distribution of P. occidentale would result from an interplay of local survival in isolation and postglacial immigration from external refugia that yielded lineages such as the widespread genetic cluster of P. occidentale currently observed from the Massif des Bornes to the Dents de Savigny and that almost swamped former residents. Such a mixed-scenario, whereby high-mountain species adapted to periglacial habitats survived the LGM across peripheral and interior refugia is realistic (Schönswetter and Schneeweiss 2019) and may explain the distribution of several plants across the Western Prealps. Given that the extant range of P. occidentale is restricted and that no populations exist outside of formerly glaciated areas, a comparative context is not available to explicitly test alternative scenarios.
Complex demographic processes under climate changes
Provided the intricate genetic variation within alpine species such as P. occidentale, how divergence may have accumulated in isolation and then homogeneized during contraction and expansion phases remains difficult to quantify with available approaches. As the tabula rasa and the nunatak hypotheses are likely non-mutually exclusive, it remains challenging to test for all realistic demographic scenarios that could account for the spatial extent of different refugia and the timing of gene flow across heterogeneous landscapes such as the Alps. Past (cold phase) refugia may not host suitable conditions anymore and remnants of glacial survivors may expectedly grow far from their LGM source, complicating inference of spatio-temporal scenarios of range contraction–expansion from the current distribution of genome-wide variation, even when based on all known native populations such as here achieved within P. occidentale. Heuristic approaches integrating demographic and ecological modelling such as recently used by Pan et al. (2019) may help to explore and favor plausible scenarios, although still awaiting the development of high-resolution proxies to be used across space and time in the context of mountainous regions such as the Alps.
To conclude on processes underlying the distribution of rare endemic taxa such as P. occidentale and, more generally, the role of Quaternary climatic oscillations on species evolutionary diversification in the Alps (Kadereit 1990; Schönswetter et al. 2009), additional work is necessary. Although consistent with the long-term isolation of divergent gene pools, the present study cannot conclude on the nature of P. occidentale as a biological species and further comparative phylogeography of high-mountain taxa of the P. alpinum complex would be necessary to shed light on the hierarchy of spatio-temporal processes having shaped such a group of cold-adapted species across the Alps. It is, however, already clear that multiple regions within the range of P. occidentale harbor high levels of genetic diversity and private alleles, and that corresponding populations are of crucial importance for the management of biodiversity. More generally, as several glacial relicts currently find a refugium in the Alps, further work is needed to understand their evolution and offer useful guidelines regarding their conservation in face of current climate changes.
References
Aeschimann D, Rasolofo N, Theurillat JP (2011) Analyse de la flore des Alpes. 2: biodiversité et chorologie. Candollea 66:225–253
BAFU (2011) Liste der National Prioritären Arten. Arten mit nationaler Priorität für die Erhaltung und Förderung, Stand 2010. Bundesamt für Umwelt, Bern. Umwelt-Vollzug Nr. 1103
Berthouzoz M, Maendly S, Bétrisey S, Mangili S, Prunier P, Lexer C, Kozlowski G (2013) Some like it cold: distribution, ecology and phylogeny of Arenaria bernensis Favarger (Caryophyllaceae) from the western Prealps in Switzerland. Alp Bot 123:65–75. https://doi.org/10.1007/s00035-013-0116-5
Bétrisey S (2014) Adaptation du plan d’action pour Papaver occidentale. Jardin botanique de l'Université de Fribourg, Suisse
Bettin O, Cornejo C, Edwards PJ, Holderegger R (2007) Phylogeography of the high alpine plant Senecio halleri (Asteraceae) in the European Alps: in situ glacial survival with postglacial stepwise dispersal into peripheral areas. Mol Ecol 16:2517–2524. https://doi.org/10.1111/j.1365-294X.2007.03273.x
Bornand C, Hoffer-Massard F (2003) La Pierreuse ou le parfum du pavot, le 20 juillet 2002. Bull Cercle Vaud Bot 32:53–61
Carolan JC, Hook ILI, Chase MW, Kadereit JW, Hodkinson TR (2006) Phylogenetics of Papaver and related regera based on DNA sequences for ITS nuclear ribosomal DNA and plastid trnL intron and trnL-F intergenic spacers. Ann Bot 98:141–155. https://doi.org/10.1093/aob/mcl079
Caron C (1973) Survol géologique des Alpes occidentales. Bull Soc Frib Sci Nat 62:73–81
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140. https://doi.org/10.1111/mec.12354
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158. https://doi.org/10.1093/bioinformatics/btr330
Delarze R (2012) Plan d’action pour Papaver occidentale (Markgraf) H.E. Hess & Landolt (Pavot des Alpes occidentales). Coordination régionale pour la protection de la flore
Ehlers J, Gibbard PL, Hughes PD (2011) Quaternary glaciations-extent and chronology: a closer look. Elsevier
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fragnière Y, Pittet L, Clément B, Bétrisey S, Gerber E, Ronikier M, Parisod C, Kozlowski G (2020) Climate change and alpine screes: no future for glacial relict Papaver occidentale (Papaveraceae) in Western Prealps (submitted)
Gerber E, Kozlowski G, Mariéthoz A-S (2010) La flore des Préalpes. Éditions Rossolis
Gilomen H (1941) Die Flora der westschweizerischen Kalkvoralpen Sonderabdruck aus den Mitteilungen der Naturforschenden Gesellschaft in Bern. Haupt, Bern
Grünig S, Fischer M, Parisod C (2020) Recent hybrid speciation at the origin of the narrow endemic Pulmonaria helvetica. Ann Bot (in press; https://doi.org/10.1093/aob/mcaa145)
Guisan A, Thuiller W, Zimmermann NE (2017) Habitat suitability and distribution models: with applications in R. Cambridge University Press, Cambridge
Guo L, Winzer T, Yang X, Li Y, Ning Z, He Z, Teodor R, Lu Y, Bowser TA, Graham IA, Ye K (2018) The opium poppy genome and morphinan production. Science 362:343–347. https://doi.org/10.1126/science.aat4096
Hampe A, Petit RJ (2005) Conserving biodiversity under climate change: the rear edge matters. Ecol Lett 8:461–467. https://doi.org/10.1111/j.1461-0248.2005.00739.x
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetics structure at the individual or population levels. Mol Ecol Notes 2:618–620. https://doi.org/10.1046/j.1471-8286.2002.00305.x
Harrison S, Noss R (2017) Endemism hotspots are linked to stable climatic refugia. Ann Bot 119:207–214. https://doi.org/10.1093/aob/mcw248
Hess NE, Landolt E, Hirzel R (1977) Flora der Schweiz und angrenzender Gebiete. Birkhäuser Verlag, Basel
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond Biol Sci 359:183–195. https://doi.org/10.1098/rstb.2003.1388
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. https://doi.org/10.1002/joc.1276
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. https://doi.org/10.1093/molbev/msj030
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Jalas J, Suominen J (1991) Atlas Florae Europaeae, Vol. 9: from Paeoniaceae to Capparaceae. Societas Biologica Fennica Vanamo, Helsinki
Jiménez-Alfaro B, García-Calvo L, García P, Acebes JL (2016) Anticipating extinctions of glacial relict populations in mountain refugia. Biol Cons 201:243–251. https://doi.org/10.1016/j.biocon.2016.07.015
Kadereit JW (1990) Notes on the taxonomy, distribution, phylogeny and ecology of Papaver alpinum L. (Papaveraceae). Botanische Jahrbücher fur Systematik Pflanzengeschichte und Pflanzengeographie 112:79–97
Kadereit JW (2017) The role of in situ species diversification for the evolution of high vascular plant species diversity in the European Alps—a review and interpretation of phylogenetic studies of the endemic flora of the Alps. Perspect Plant Ecol Evol Syst 26:28–38. https://doi.org/10.1016/j.ppees.2017.03.002
Kadereit JW, Griebeler EM, Comes HP (2004) Quaternary diversification in European alpine plants: pattern and process. Philos Trans R Soc Lond Biol Sci 359:265–274. https://doi.org/10.1098/rstb.2003.1389
Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA (2013) diveRsity: an R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol Evol 4:782–788. https://doi.org/10.1111/2041-210X.12067
Kropf M, Comes HP, Kadereit JW (2008) Causes of the genetic architecture of south-west European high mountain disjuncts. Plant Ecol Div 1:217–228. https://doi.org/10.1080/17550870802331938
LaCava MEF, Aikens EO, Megna LC, Randolph G, Hubbard C, Buerkle CA (2020) Accuracy of assembly of DNA sequences from double‐digest libraries varies substantially among software. Mol Ecol Res 20:360–370. https://doi.org/10.1111/1755-0998.13108
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Markgraf F (1958) Eine neue Gliederung des Alpenmohene. Phyton 7:302–314
Naciri Y, Gaudeul M (2007) Phylogeography of the endangered Eryngium alpinum L. (Apiaceae) in the European Alps. Mol Ecol 16:2721–2733. https://doi.org/10.1111/j.1365-294X.2007.03269.x
Nielsen R (2005) Molecular signatures of natural selection. Annu Rev Genet 39:197–218. https://doi.org/10.1146/annurev.genet.39.073003.112420
Nordal I, Hestmark G, Solstad H (1997) Reproductive biology and demography of Papaver radicatum—a key species in Nordic plant geography. Opera Bot 132:77–87
Ozenda P (2009) On the genesis of the plant population in the Alps: new or critical aspects. C R Biol 332:1092–1103. https://doi.org/10.1016/j.crvi.2009.09.018
Pan D, Hülber K, Willner W, Schneeweiss GM (2019) An explicit test of Pleistocene survival in peripheral versus nunatak refugia in two high mountain plant species. Mol Ecol. https://doi.org/10.1111/MEC.15316
Parisod C (2008) Postglacial recolonisation of plants in the western Alps of Switzerland. Bot Helv 118:1–12. https://doi.org/10.1007/s00035-008-0825-3
Parisod C, Besnard G (2007) Glacial in situ survival in the Western Alps and polytopic autopolyploidy in Biscutella laevigata L. (Brassicaceae). Mol Ecol 16:2755–2767. https://doi.org/10.1111/j.1365-294X.2007.03315.x
Paun O, Schönswetter P, Winkler M, Consortium I, Tribsch A (2008) Historical divergence vs. contemporary gene flow: evolutionary history of the calcicole Ranunculus alpestris group (Ranunculaceae) in the European Alps and the Carpathians. Mol Ecol 17:4263–4275. https://doi.org/10.1111/j.1365-294X.2008.03908.x
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135. https://doi.org/10.1371/journal.pone.0037135
Pfeifer B, Wittelsbuerger U, Ramos-Onsins SE, Lercher MJ (2014) PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol Biol Evol 31:1929–1936. https://doi.org/10.1093/molbev/msu136
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Reisch C, Poschlod P, Wingender R (2003) Genetic variation of Saxifraga paniculata Mill. (Saxifragaceae): molecular evidence for glacial relict endemism in central Europe. Biol J Linn Soc 80:11–21. https://doi.org/10.1046/j.1095-8312.2003.00215.x
Rogivue A, Graf R, Parisod C, Holderegger R, Gugerli F (2018) The phylogeographic structure of Arabis alpina in the Alps shows consistent patterns across different types of molecular markers and geographic scales. Alp Bot 128:35–45. https://doi.org/10.1007/s00035-017-0196-8
Rosenberg NA (2003) distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Rosenberg NA, Mahajan S, Ramachandran S, Zhao C, Pritchard JK, Feldman MW (2005) Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet 1:660–671. https://doi.org/10.1371/journal.pgen.0010070
Rousset F (2000) Genetic differentiation between individuals. J Evol Biol 13:58–62. https://doi.org/10.1046/j.1420-9101.2000.00137.x
Schmitt T (2009) Biogeographical and evolutionary importance of the European high mountain systems. Front Zool 6:9. https://doi.org/10.1186/1742-9994-6-9
Schneeweiss GM, Pachschwoll C, Tribsch A, Schonswetter P, Barfuss MH, Esfeld K, Weiss-Schneeweiss H, Thiv M (2013) Molecular phylogenetic analyses identify alpine differentiation and dysploid chromosome number changes as major forces for the evolution of the European endemic Phyteuma (Campanulaceae). Mol Phylogenet Evol 69:634–652. https://doi.org/10.1016/j.ympev.2013.07.015
Schonswetter P, Stehlik I, Holderegger R, Tribsch A (2005) Molecular evidence for glacial refugia of mountain plants in the European Alps. Mol Ecol 14:3547–3555. https://doi.org/10.1111/j.1365-294X.2005.02683.x
Schönswetter P, Schneeweiss GM (2019) Is the incidence of survival in interior Pleistocene refugia (nunataks) underestimated? Phylogeography of the high mountain plant Androsace alpina (Primulaceae) in the European Alps revisited. Ecol Evol 9:4078–4086. https://doi.org/10.1002/ece3.5037
Schönswetter P, Solstad H, Escobar García P, Elven R (2009) A combined molecular and morphological approach to the taxonomically intricate European mountain plant Papaver alpinum s.l. (Papaveraceae)—Taxa or informal phylogeographical groups? Taxon 58:1326–1343. https://doi.org/10.1002/tax.584020
Serre D, Pääbo S (2004) Evidence for gradients of human genetic diversity within and among continents. Genome Res 14:1679–1685. https://doi.org/10.1101/gr.2529604
Smyčka J, Roquet C, Renaud J, Thuiller W, Zimmermann NE, Lavergne S (2017) Disentangling drivers of plant endemism and diversification in the European Alps—a phylogenetic and spatially explicit approach. Perspect Plant Ecol Evol Syst 28:19–27. https://doi.org/10.1016/j.ppees.2017.06.004
Stehlik I (2000) Nunataks and peripheral refugia for alpine plants during quaternary glaciation in the middle part of the Alps. Bot Helv 110:25–30
Stehlik I, Schneller JJ, Bachmann K (2002a) Immigration and in situ glacial survival of the low-alpine Erinus alpinus (Scrophulariaceae). Biol J Linn Soc 77:87–103. https://doi.org/10.1046/j.1095-8312.2002.00094.x
Stehlik I, Blattner FR, Holderegger R, Bachmann K (2002b) Nunatak survival of the high Alpine plant Eritrichium nanum (L.) Gaudin in the central Alps during the ice ages. Mol Ecol 11:2027–2036. https://doi.org/10.1046/j.1365-294X.2002.01595.x
Taberlet P, Zimmermann NE, Englisch T, Tribsch A, Holderegger R, Alvarez N, Niklfeld H, Coldea G, Mirek Z, Moilanen A, Ahlmer W, Marsan PA, Bona E, Bovio M, Choler P, Cieslak E, Colli L, Cristea V, Dalmas JP, Frajman B, Garraud L, Gaudeul M, Gielly L, Gutermann W, Jogan N, Kagalo AA, Korbecka G, Kupfer P, Lequette B, Letz DR, Manel S, Mansion G, Marhold K, Martini F, Negrini R, Nino F, Paun O, Pellecchia M, Perico G, Piekos-Mirkowa H, Prosser F, Puscas M, Ronikier M, Scheuerer M, Schneeweiss GM, Schonswetter P, Schratt-Ehrendorfer L, Schupfer F, Selvaggi A, Steinmann K, Thiel-Egenter C, van Loo M, Winkler M, Wohlgemuth T, Wraber T, Gugerli F, IntraBioDiv C, Vellend M (2012) Genetic diversity in widespread species is not congruent with species richness in alpine plant communities. Ecol Lett 15:1439–1448. https://doi.org/10.1111/ele.12004
Thiel-Egenter C, Alvarez N, Holderegger R, Tribsch A, Englisch T, Wohlgemuth T, Colli L, Gaudeul M, Gielly L, Jogan N, Linder HP, Negrini R, Niklfeld H, Pellecchia M, Rioux D, Schönswetter P, Taberlet P, van Loo M, Winkler M, Gugerli F (2011) Break zones in the distributions of alleles and species in alpine plants. J Biogeogr 38:772–782. https://doi.org/10.1111/j.1365-2699.2010.02441.x
Tribsch A (2004) Areas of endemism of vascular plants in the Eastern Alps in relation to Pleistocene glaciation. J Biogeogr 31:747–760. https://doi.org/10.1111/j.1365-2699.2004.01065.x
Willis SC, Hollenbeck CM, Puritz JB, Gold JR, Portnoy DS (2017) Haplotyping RAD loci: an efficient method to filter paralogs and account for physical linkage. Mol Ecol Resour 17:955–965. https://doi.org/10.1111/1755-0998.12647
Acknowledgements
Open access funding provided by University of Bern. We would like to specially thank P. Wandeler, D. Jordan, D. Giovannini, B. Wolf and D. Bernátová for support, and F. Cheda, B. Holzer, O. Guex and F. Ciardo for collection permits. MR was supported by statutory funds of the W. Szafer Institute of Botany, Polish Academy of Sciences.
Funding
Natural History Museum of Fribourg.
Author information
Authors and Affiliations
Contributions
LP planned and performed field sampling for the main taxon of the study, generated and analyzed data, and drafted the paper; SB, BC, EG planned and performed field work; YF performed niche modelling; SG participated to the production and analysis of genetic data; MR planned and performed field work for outgroups, and drafted the paper, GK conceived and supervised the study, and drafted the paper; CP supervised the production and analysis of genetic data, and drafted the paper.
Corresponding author
Ethics declarations
Conflict of interest
None.
Data availability statement
Supporting data will be stored at NCBI and Dryad upon acceptance.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pittet, L., Fragnière, Y., Grünig, S. et al. Genetic structure of the endemic Papaver occidentale indicates survival and immigration in the Western Prealps. Alp Botany 130, 129–140 (2020). https://doi.org/10.1007/s00035-020-00238-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00035-020-00238-3