Conformational Equilibrium of NADPH–Cytochrome P450 Oxidoreductase Is Essential for Heme Oxygenase Reaction

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Enzymatic Assay
2.3. Analytical Ultracentrifugation
2.4. Cryo-Electron Microscopy (EM), Image Processing, and Molecular Modeling of the rCPR–Heme–rHO-1 Complex
3. Results
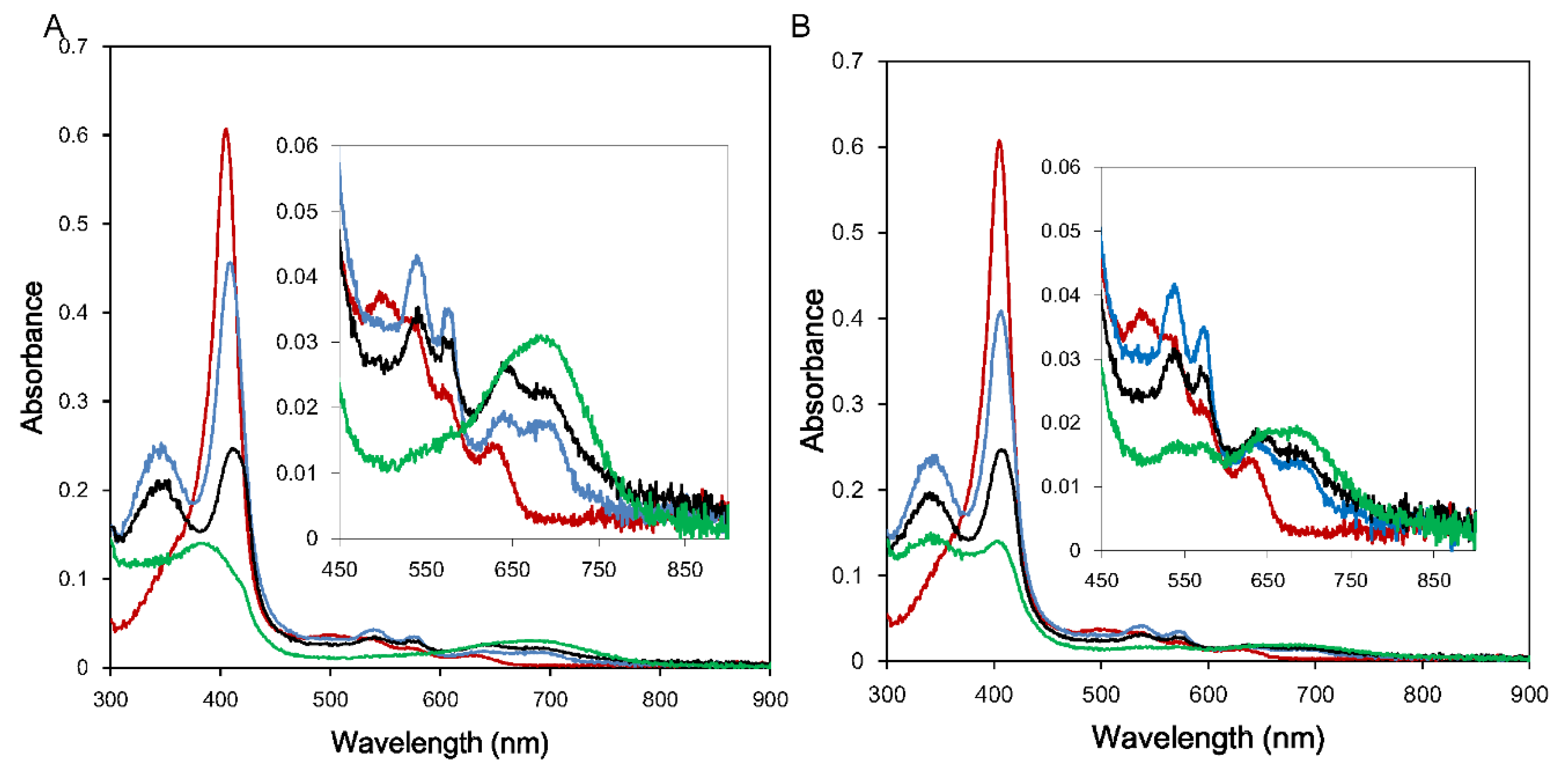
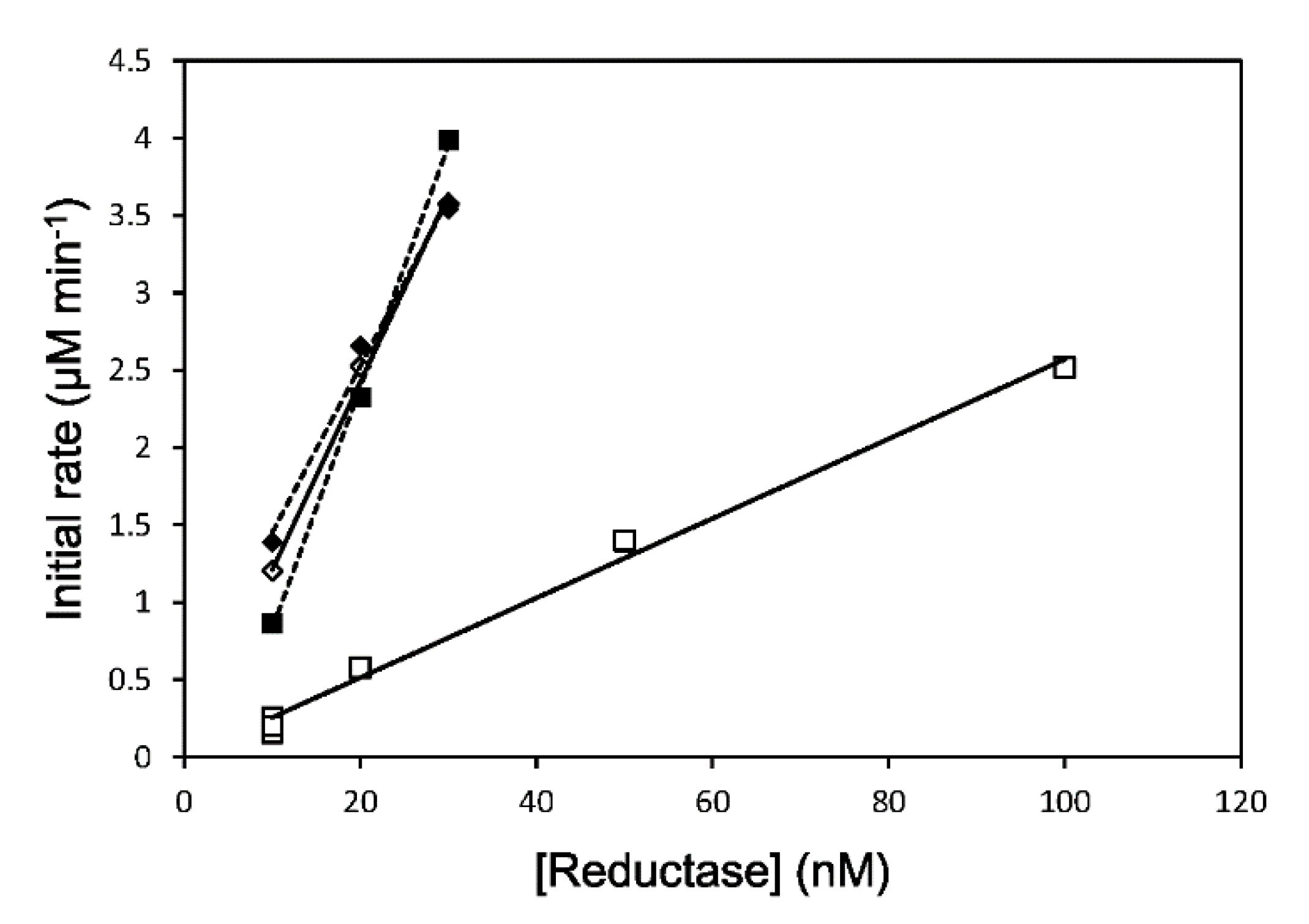
3.1. Enzymatic Assay
3.2. Reduction Kinetics
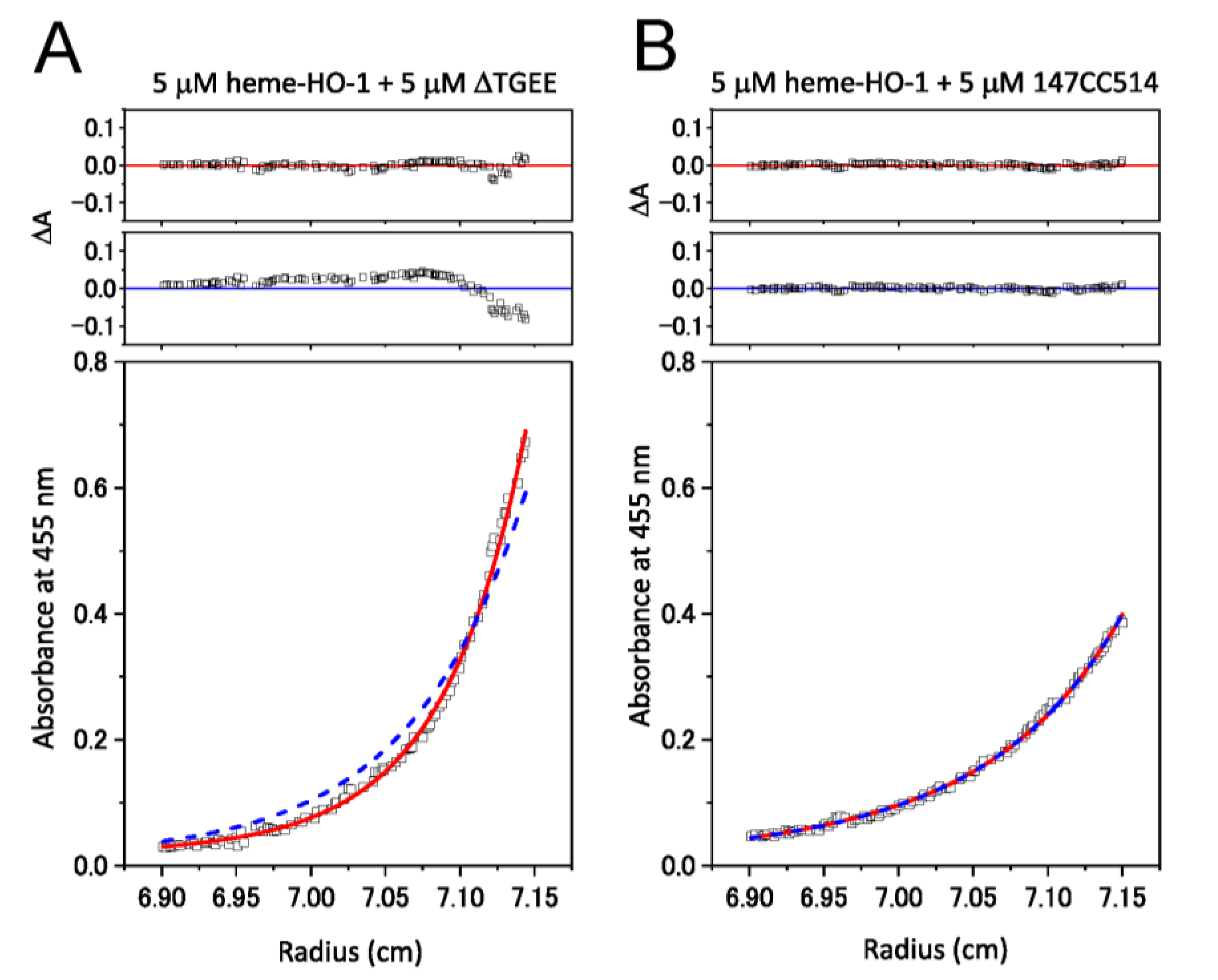
3.3. Complex Formation between CPRs and Heme–rHO-1 Evaluated by Ultracentrifugation
3.4. Cryo-EM Structure of the rCPR–Heme–rHO-1 Complex
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kikuchi, G.; Yoshida, T.; Noguchi, M. Heme oxygenase and heme degradation. Biochem. Biophys. Res. Commun. 2005, 338, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R.; Wilks, A. Heme oxygenase structure and mechanism. In Advances in Inorgranic Chemistry; Sykes, A.G., Ed.; Academic Press: San Diego, CA, USA, 2001; Volume 51, pp. 359–407. [Google Scholar]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Kajimura, M.; Nakamura, T.; Hishiki, T.; Nakanishi, T.; Yukutake, Y.; Nagahata, Y.; Ishikawa, M.; Hattori, K.; Takenouchi, T.; et al. Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide-sensitive hydrogen sulfide pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 1293–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugishima, M.; Wada, K.; Unno, M.; Fukuyama, K. Bilin-metabolizing enzymes: Site-specific reductions catalyzed by two different type of enzymes. Curr. Opin. Struct. Biol. 2019, 59, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, M.; Yoshida, T.; Kikuchi, G. Purification and properties of biliverdin reductases from pig spleen and rat liver. J. Biochem. (Tokyo) 1979, 86, 833–848. [Google Scholar] [CrossRef]
- Kutty, R.K.; Maines, M.D. Purification and characterization of biliverdin reductase from rat liver. J. Biol. Chem. 1981, 256, 3956–3962. [Google Scholar]
- Schuller, D.J.; Wilks, A.; de Ortiz Montellano, P.R.; Poulos, T.L. Crystal structure of human heme oxygenase-1. Nat. Struct. Biol. 1999, 6, 860–867. [Google Scholar]
- Sugishima, M.; Omata, Y.; Kakuta, Y.; Sakamoto, H.; Noguchi, M.; Fukuyama, K. Crystal structure of rat heme oxygenase-1 in complex with heme. FEBS Lett. 2000, 471, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Sugishima, M.; Sakamoto, H.; Kakuta, Y.; Omata, Y.; Hayashi, S.; Noguchi, M.; Fukuyama, K. Crystal structure of rat apo-heme oxygenase-1 (HO-1): Mechanism of heme binding in HO-1 inferred from structural comparison of the apo and heme complex forms. Biochemistry 2002, 41, 7293–7300. [Google Scholar] [CrossRef]
- Sugishima, M.; Sakamoto, H.; Higashimoto, Y.; Noguchi, M.; Fukuyama, K. Crystal structure of rat heme oxygenase-1 in complex with biliverdin-iron chelate: Conformational change of the distal helix during the heme cleavage reaction. J. Biol. Chem. 2003, 278, 32352–32358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugishima, M.; Sakamoto, H.; Noguchi, M.; Fukuyama, K. Crystal structures of ferrous and CO-, CN--, and NO-bound forms of rat heme oxygenase-1 (HO-1) in complex with heme: Structural implications for discrimination between CO and O2 in HO-1. Biochemistry 2003, 42, 9898–9905. [Google Scholar] [PubMed]
- Sato, H.; Sugishima, M.; Sakamoto, H.; Higashimoto, Y.; Shimokawa, C.; Fukuyama, K.; Palmer, G.; Noguchi, M. Crystal structure of rat haem oxygenase-1 in complex with ferrous verdohaem: Presence of a hydrogen-bond network on the distal side. Biochem. J. 2009, 419, 339–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugishima, M.; Moffat, K.; Noguchi, M. Discrimination between CO and O2 in heme oxygenase: Comparison of static structures and dynamic conformation changes following CO photolysis. Biochemistry 2012, 51, 8554–8562. [Google Scholar] [PubMed] [Green Version]
- Unno, M.; Matsui, T.; Chu, G.C.; Couture, M.; Yoshida, T.; Rousseau, D.L.; Olson, J.S.; Ikeda-Saito, M. Crystal structure of the dioxygen-bound heme oxygenase from Corynebacterium diphtheriae: Implications for heme oxygenase function. J. Biol. Chem. 2004, 279, 21055–21061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unno, M.; Matsui, T.; Ikeda-Saito, M. Structure and catalytic mechanism of heme oxygenase. Nat. Prod. Rep. 2007, 24, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Matsui, T.; Ikeda-Saito, M. Crystallographic studies of heme oxygenase complexed with an unstable reaction intermediate, verdoheme. J. Inorg. Biochem. 2012, 113, 102–109. [Google Scholar] [CrossRef]
- Unno, M.; Ardevol, A.; Rovira, C.; Ikeda-Saito, M. Structures of the substrate-free and product-bound forms of HmuO, a heme oxygenase from corynebacterium diphtheriae: X-ray crystallography and molecular dynamics investigation. J. Biol. Chem. 2013, 288, 34443–34458. [Google Scholar] [CrossRef] [Green Version]
- Pandey, A.V.; Fluck, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254. [Google Scholar] [CrossRef]
- Iyanagi, T.; Xia, C.; Kim, J.J. NADPH-cytochrome P450 oxidoreductase: Prototypic member of the diflavin reductase family. Arch. Biochem. Biophys. 2012, 528, 72–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Roberts, D.L.; Paschke, R.; Shea, T.M.; Masters, B.S.; Kim, J.J. Three-dimensional structure of NADPH-cytochrome P450 reductase: Prototype for FMN- and FAD-containing enzymes. Proc. Natl. Acad. Sci. USA 1997, 94, 8411–8416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamdane, D.; Xia, C.; Im, S.C.; Zhang, H.; Kim, J.J.; Waskell, L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 2009, 284, 11374–11384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugishima, M.; Sato, H.; Higashimoto, Y.; Harada, J.; Wada, K.; Fukuyama, K.; Noguchi, M. Structural basis for the electron transfer from an open form of NADPH-cytochrome P450 oxidoreductase to heme oxygenase. Proc. Natl. Acad. Sci. USA 2014, 111, 2524–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugishima, M.; Sato, H.; Wada, K.; Yamamoto, K. Crystal structure of a NADPH-cytochrome P450 oxidoreductase (CYPOR) and heme oxygenase 1 fusion protein implies a conformational change in CYPOR upon NADPH/NADP(+) binding. FEBS Lett. 2019, 593, 868–875. [Google Scholar] [CrossRef]
- Spencer, A.L.; Bagai, I.; Becker, D.F.; Zuiderweg, E.R.; Ragsdale, S.W. Protein/protein interactions in the mammalian heme degradation pathway: Heme oxygenase-2, cytochrome P450 reductase, and biliverdin reductase. J. Biol. Chem. 2014, 289, 29836–29858. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Hamdane, D.; Shen, A.L.; Choi, V.; Kasper, C.B.; Pearl, N.M.; Zhang, H.; Im, S.C.; Waskell, L.; Kim, J.J. Conformational changes of NADPH-cytochrome P450 oxidoreductase are essential for catalysis and cofactor binding. J. Biol. Chem. 2011, 286, 16246–16260. [Google Scholar] [CrossRef] [Green Version]
- Iijima, M.; Ohnuki, J.; Sato, T.; Sugishima, M.; Takano, M. Coupling of Redox and Structural States in Cytochrome P450 Reductase Studied by Molecular Dynamics Simulation. Sci. Rep. 2019, 9, 9341. [Google Scholar] [CrossRef]
- Sugishima, M.; Sakamoto, H.; Higashimoto, Y.; Omata, Y.; Hayashi, S.; Noguchi, M.; Fukuyama, K. Crystal structure of rat heme oxygenase-1 in complex with heme bound to azide. Implication for regiospecific hydroxylation of heme at the alpha-meso carbon. J. Biol. Chem. 2002, 277, 45086–45090. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Omata, Y.; Sakamoto, H.; Hara, T.; Noguchi, M. Purification and characterization of a soluble form of rat liver NADPH-cytochrome P-450 reductase highly expressed in Escherichia coli. Protein Expr. Purif. 2003, 29, 1–7. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Collaborative Computational Project No.4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Takahashi, S.; Kikuchi, G. Partial purification and reconstitution of the heme oxygenase system from pig spleen microsomes. J. Biochem. (Tokyo) 1974, 75, 1187–1191. [Google Scholar] [CrossRef]
- Sun, D.; Sato, M.; Yoshida, T.; Shimizu, H.; Miyatake, H.; Adachi, S.; Shiro, Y.; Kikuchi, A. Crystallization and preliminary X-ray diffraction analysis of a rat biliverdin reductase. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 1180–1182. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Kikuchi, G. Purification and properties of heme oxygenase from pig spleen microsomes. J. Biol. Chem. 1978, 253, 4224–4229. [Google Scholar] [PubMed]
- Lebowitz, J.; Lewis, M.S.; Schuck, P. Modern analytical ultracentrifugation in protein science: A tutorial review. Protein Sci. 2002, 11, 2067–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasunaga, T.; Wakabayashi, T. Evaluation of a 2k CCD camera with an epitaxially grown CsI scintillator for recording energy-filtered electron cryo-micrographs. J. Electron Microsc. (Tokyo) 2008, 57, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, T.; Wakabayashi, T. Extensible and object-oriented system Eos supplies a new environment for image analysis of electron micrographs of macromolecules. J. Struct. Biol. 1996, 116, 155–160. [Google Scholar] [CrossRef]
- Murakami, K.; Yasunaga, T.; Noguchi, T.Q.; Gomibuchi, Y.; Ngo, K.X.; Uyeda, T.Q.; Wakabayashi, T. Structural basis for actin assembly, activation of ATP hydrolysis, and delayed phosphate release. Cell 2010, 143, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Pudney, C.R.; Khara, B.; Johannissen, L.O.; Scrutton, N.S. Coupled motions direct electrons along human microsomal P450 Chains. PLoS Biol. 2011, 9, e1001222. [Google Scholar] [CrossRef] [Green Version]
- Frances, O.; Fatemi, F.; Pompon, D.; Guittet, E.; Sizun, C.; Perez, J.; Lescop, E.; Truan, G. A well-balanced preexisting equilibrium governs electron flux efficiency of a multidomain diflavin reductase. Biophys. J. 2015, 108, 1527–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, M.; Ellis, J.; Huang, W.C.; Lloyd Raven, E.; Roberts, G.C.; Oldham, N.J. Detection of a protein conformational equilibrium by electrospray ionisation-ion mobility-mass spectrometry. Angew. Chem. Int. Ed. Engl. 2011, 50, 8291–8294. [Google Scholar] [CrossRef]
- Huang, W.C.; Ellis, J.; Moody, P.C.; Raven, E.L.; Roberts, G.C. Redox-linked domain movements in the catalytic cycle of cytochrome p450 reductase. Structure 2013, 21, 1581–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nature 1992, 355, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Higashimoto, Y.; Sakamoto, H.; Sugishima, M.; Shimokawa, C.; Harada, J.; Palmer, G.; Noguchi, M. Reduction of oxaporphyrin ring of CO-bound alpha-verdoheme complexed with heme oxygenase-1 by NADPH-cytochrome P450 reductase. J. Inorg. Biochem. 2011, 105, 289–296. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reduction System | HO Activity | Apparent Reduction Rate Constant | ||
|---|---|---|---|---|
| (min−1) | (%) | (min−1) | (%) | |
| rCPR | 1.96 ± 0.35 | 100 | 122 ± 3.8 | 100 |
| 147CC514 | 0.229 ± 0.0037 | 11.7 ± 0.19 | 23.9 ± 5.1 | 19.6 ± 4.2 |
| DTT-treated rCPR | ND | ND | 130 ± 10.8 | 107 ± 8.9 |
| DTT-treated 147CC514 | ND | ND | 112 ± 23.6 | 91.8 ± 19 |
| IAM/DTT-treated rCPR | 1.17 ± 0.0024 | 59.7 ± 0.12 | ND | ND |
| IAM/DTT-treated 147CC514 | 1.25 ± 0.197 | 63.8 ± 10 | ND | ND |
| ΔTGEE [24] | ND | ND | 0.250 | 0.205 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugishima, M.; Taira, J.; Sagara, T.; Nakao, R.; Sato, H.; Noguchi, M.; Fukuyama, K.; Yamamoto, K.; Yasunaga, T.; Sakamoto, H. Conformational Equilibrium of NADPH–Cytochrome P450 Oxidoreductase Is Essential for Heme Oxygenase Reaction. Antioxidants 2020, 9, 673. https://doi.org/10.3390/antiox9080673
Sugishima M, Taira J, Sagara T, Nakao R, Sato H, Noguchi M, Fukuyama K, Yamamoto K, Yasunaga T, Sakamoto H. Conformational Equilibrium of NADPH–Cytochrome P450 Oxidoreductase Is Essential for Heme Oxygenase Reaction. Antioxidants. 2020; 9(8):673. https://doi.org/10.3390/antiox9080673
Chicago/Turabian StyleSugishima, Masakazu, Junichi Taira, Tatsuya Sagara, Ryota Nakao, Hideaki Sato, Masato Noguchi, Keiichi Fukuyama, Ken Yamamoto, Takuo Yasunaga, and Hiroshi Sakamoto. 2020. "Conformational Equilibrium of NADPH–Cytochrome P450 Oxidoreductase Is Essential for Heme Oxygenase Reaction" Antioxidants 9, no. 8: 673. https://doi.org/10.3390/antiox9080673