
Abstract
Pulsed electron paramagnetic resonance dipolar spectroscopy (PDS) allows to measure the distances between electron spin centers and, in favorable cases, their relative orientation. This data is frequently used in structural biology for studying biomolecular structures, following their conformational changes and localizing paramagnetic centers within them. In order to extract the inter-spin distances and the relative orientation of spin centers from the primary, time-domain PDS signals, a specialized data analysis is required. So far, the software to do such analysis was available only for isotropic S = 1/2 spin centers, such as nitroxide and trityl radicals, as well as for high-spin Gd3+ and Mn2+ ions. Here, a new data analysis program, called AnisoDipFit, was introduced for spin systems consisting of one isotropic and one anisotropic S = 1/2 spin centers. The program was successfully tested on the PDS data corresponding to the spin systems Cu2+/organic radical, low-spin Fe3+/organic radical, and high-spin Fe3+/organic radical. For all tested spin systems, AnisoDipFit allowed determining the inter-spin distance distribution with a sub-angstrom precision. In addition, the spatial orientation of the inter-spin vector with respect to the g-frame of the metal center was determined for the last two spin systems. Thus, this study expands the arsenal of the PDS data analysis programs and facilitates the PDS-based distance and angle measurements on the highly relevant class of metolloproteins.
Similar content being viewed by others
1 Introduction
Electron paramagnetic resonance spectroscopy (EPR) offers several pulsed techniques to measure nanometer-scale distances in condensed matter. These techniques include pulsed electron–electron double resonance (PELDOR or DEER) [1, 2], double quantum coherence EPR (DQC) [3], single-frequency technique for refocusing dipolar couplings (SIFTER) [4], and relaxation induced dipolar modulation enhancement (RIDME) [5, 6]. Altogether, they are often denoted as pulsed EPR dipolar spectroscopy or, shortly, PDS [7, 8]. In the last decade, PDS distance restraints were used for numerous tasks arising from structural biology, such as studying the structure of biomolecules and biomolecular complexes [9,10,11], following the conformational changes of proteins during their function [12,13,14], and localization of metal ions within biomolecules [15,16,17]. The idea behind PDS is the measurement of the dipolar coupling between electron spin centers, which has a 1/r3 dependence on the distance r between these centers. In order to be applicable, PDS requires that a biomolecule or a biomolecular complex contains at least two electron spin centers. This requirement can be fulfilled in several different ways. First, a biomolecule can contain intrinsic electron spin centers, such as Fe3+, Mn2+, Co2+, Cu2+, metal-sulfur clusters, and flavins. Second, electron spin centers can be artificially attached at the selected sites in the biomolecular surface using site-directed spin labeling (SDSL) [18]. The most common spin labels are nitroxides [19, 20], although alternative spin labels based on trityl [20, 21], Gd3+ [22,23,24], Mn2+ [25, 26], and Cu2+ [27, 28] also exist. Furthermore, intrinsic spin centers can be combined with spin labels within one biomolecule, which is especially useful if a biomolecule contains only one intrinsic spin center.
A large variety of spin centers, which can be used for PDS, is certainly an advantage. However, each of these spin centers have specific spectroscopic properties, which pose some requirements and challenges for the method. One of these challenges is related to the PDS data analysis. The aim of the PDS data analysis is the extraction of the distance distribution out of the primary PDS data, which is measured in a form of a time trace modulated by dipolar coupling frequencies. As the primary PDS data depends on the spectroscopic properties of electron spin centers and, sometimes, also on the applied PDS technique, no general procedure for such data analysis is available. Instead, several particular cases can be considered.
The most established case in PDS corresponds to S = 1/2 spin centers with isotropic or almost isotropic g-factors. This case applies to majority of organic radicals, including nitroxide or trityl spin labels. The g-factors of these radicals are fairly isotropic and only slightly deviate from the g-factor of free electron ge ≈ 2.0023. The PDS measurements on these centers are done using the PELDOR technique, if the excitation bandwidth of the microwave pulses is smaller than the spectral width of the spin system, or the DQC and SIFTER techniques, if the excitation bandwidth is comparable to the spectral width of the spin system. The time traces that are obtained from these measurements can be usually translated into distance distributions by means of the programs such as DeerAnalysis [29], LongDistances [30], and DD [31]. These programs use the assumption that that the dipolar coupling between the electron spin centers is averaged over all possible orientations of the inter-spin vector \(\overrightarrow{r}\) with respect to the applied static magnetic field \({\overrightarrow{B}}_{0}\) and, therefore, the dipolar spectrum, which is obtained from the Fourier transform of the PDS time trace, has the shape of a Pake doublet. This assumption is usually well fulfilled in frozen solution. The exception to this is the case of orientation-selective PELDOR time traces, which are obtained when the mutual orientation of spin centers is correlated and the microwave pulses of the PELDOR pulse sequence selectively excite only certain orientations of the spin centers with respect to \({\overrightarrow{B}}_{0}\). The orientation-selective PDS time traces do not yield the complete Pake doublet and, therefore, require specialized analysis. The common way to account for the orientation selectivity is to measure several PELDOR time traces with different spectral positions of pump/detection pulses and then to build the geometric model of the spin system, which provides the best fit to all acquired time traces [32,33,34,35]. The accomplishment of both steps is usually quite demanding and very time consuming.
Besides isotropic S = 1/2 spin centers, DeerAnalysis is also applicable for the analysis of PELDOR time traces acquired on the high-spin Gd3+ (S = 7/2) [36] and Mn2+ (S = 5/2) ions [25]. This, however, requires that the PELDOR experiments are done at microwave frequencies, which significantly exceed the zero-field splitting (ZFS) constant D of these spin centers. Only in this case, the tilting of the quantization axis of Gd3+ and Mn2+ away from \({\overrightarrow{B}}_{0}\) can be minimized and, thus, an additional dependence of the primary PDS data on the mutual orientation of \(\overrightarrow{r}\) with respect to the ZFS frame can be avoided [24]. In addition, DeerAnalysis should be cautiously applied for determination of short Gd3+–Gd3+ distances, for which the pseudo-secular term of the dipolar Hamiltonian can become non-negligible and contribute to the dipolar coupling frequency [24]. In addition to the PELDOR technique, the RIDME technique was recently used for PDS experiments on Gd3+ [37, 38] and Mn2+ [39,40,41]. In contrast to the PELDOR time traces, the RIDME time traces could not be analyzed by DeerAnalysis. This is because of the higher harmonics of the dipolar coupling frequency which, together with the usual dipolar coupling frequency, modulate the RIDME time traces. Thus, the extraction of the distance distributions from the RIDME data requires a data analysis, which takes these higher harmonics into account. Such data analysis was developed and implemented in the program OvertoneAnalysis [38].
The third relevant case corresponds to S = 1/2 spin centers with anisotropic g-factors. The metal ions like Cu2+, low-spin Fe3+ (ls-Fe3+), and low-spin Co2+ (ls-Co2+), as well as the iron–sulfur clusters like [2Fe-2S]+ and [4Fe-4S]+, belong to this type of spin centers. In addition, high-spin Fe3+ (hs-Fe3+) ions with large axial ZFS constants, often found in hemoproteins, can be considered as an anisotropic S = 1/2 spin center at low temperatures [42]. Due to the significant g-anisotropy and, in some cases, the additional anisotropy of the hyperfine coupling constant, the spectral width of all these spin centers largely exceeds the bandwidth of typical microwave pulses. Consequently, the PDS time traces acquired on these spin centers are usually orientation-selective. As was mentioned above, the extraction of the distance distribution from the orientation-selective PDS data is a difficult task. For anisotropic spin centers, this task is additionally complicated by the fact that the spectral widths often exceed the bandwidths of available EPR resonators. Therefore, the successful analysis of orientation-selective PELDOR time traces was reported only for Cu2+ [15, 43,44,45,46,47,48,49,50] and iron–sulfur clusters [9, 45, 51]. Importantly, the orientation selectivity can be avoided if the PDS measurements are performed between one anisotropic S = 1/2 spin center and one isotropic S = 1/2 spin center. This was demonstrated using the ultra-wideband PELDOR technique [52, 53] and, especially, RIDME technique [6, 42, 54,55,56,57,58,59,60]. In the absence of orientation selectivity, the PDS data analysis certainly becomes simpler, but it is still more complicated than the corresponding data analysis used for isotropic S = 1/2 spin centers. The reason for this is the dependence of the dipolar coupling frequency on the anisotropic g-factor of one of the spin centers, which in turn leads to the dependence of the primary PDS data on the mutual orientation of the inter-spin vector \(\overrightarrow{r}\) with respect to the g-frame of the anisotropic spin center [42, 54]. A PDS data analysis, which takes into account this dependence, was recently developed and implemented in the program AnisoDipFit (originally called DipFit) [42, 60]. However, the detailed description of this program has not been reported yet.
The aim of the present work is to provide the full description of the AnisoDipFit program and to benchmark this program for different anisotropic S = 1/2 spin centers. The description will begin with the theory, which lays the basis of the AnisoDipFit data analysis, and will be continued by an overview of the program. Next, AnisoDipFit will be used to simulate the PDS time traces of spin systems, in which the anisotropic S = 1/2 spin center has different degree of g-anisotropy. This time traces will be then analyzed by means of DeerAnalysis, which should reveal how much the DeerAnalysis-derived distance distributions deviate from the actual ones for different anisotropic spin centers. Finally, AnisoDipFit will be used for the data analysis of the experimental RIDME data reported for the spin systems Cu2+/nitroxide, ls-Fe3+/trityl, and hs-Fe3+/nitroxide. The results of this data analysis will be compared between the different spin systems, which should reveal the effect of g-anisotropy on the precision of the extracted distance and angular distributions.
2 Theory
Using an assumption that the magnetic moment \(\overrightarrow{\mu }\) of an electron spin \(\overrightarrow{S}\) is proportional to its g-tensor \(\widehat{g}\) (Chapter 9.2 in Ref. [61]),
and the point-dipole approximation, the Hamiltonian of the dipole–dipole interaction between two electron spin centers, called here spin A and spin B, can be written as
where μ0 is the vacuum permeability, βe is the Bohr magneton, r and \(\overrightarrow{n}\) are the length and the unit vector of the inter-spin vector \(\overrightarrow{r}\), \({\widehat{g}}_{A}\) and \({\widehat{g}}_{B}\) are the g-tensors of both spin centers, \({\overrightarrow{S}}_{A}\) and \({\overrightarrow{S}}_{B}\) are the spin vectors of both spin centers. The round brackets denote the scalar product of two vectors. For the case of two anisotropic spin-1/2 centers, Bedilo and Maryasov have derived a more convenient form of Eq. (2) [62]:
The expressions for all alphabetic operators can be found in the original publication [62]. If the Zeeman interaction of both electron spins is much larger than other magnetic interactions, only the secular term \(\widehat{A}\) and the pseudo-secular term \(\widehat{B}\) contribute to \({\widehat{\mathcal{H}}}_{dd}\), whereas the contribution of the terms \(\widehat{C}\)–\(\widehat{I}\) to \({\widehat{\mathcal{H}}}_{dd}\) is negligible. This case is known as the high-field case and is realized in most PDS experiments. Moreover, if the dipolar coupling between the electron spin centers is small compared to the difference between their individual resonance frequencies, the so-called secular approximation applies. Under this approximation, only the secular term \(\widehat{A}\) has a non-negligible contribution to \({\widehat{\mathcal{H}}}_{dd}\), yielding the dipolar coupling frequency
Here, h is the Planck constant, \({\overrightarrow{k}}_{A}\) and \({\overrightarrow{k}}_{B}\) determine the quantization axes of each of the spins,
where \({\overrightarrow{B}}_{0}\) is the vector of applied static magnetic field, and the symbol T denotes transposition.
If the g-anisotropy of one of the spin centers, e.g. spin A, is neglected and the g-factor of this spin center is assumed to be equal to ge, Eq. (4) can be simplified to
where gB,eff is the effective g-factor of the anisotropic spin center, and θ is the angle between \(\overrightarrow{r}\) and \({\overrightarrow{B}}_{0}\). Comparing Eq. (6) with the equation for the dipolar coupling between two isotropic spin-1/2 centers with gA = gB = ge,
reveals that both equations differ by the factor gB,eff/ge and the angular term in the square brackets. In the case of two isotropic spins, the latter term depends only on the angle θ, whereas, when one of the spins is anisotropic, it depends on the orientation of \(\overrightarrow{r}\) and \({\overrightarrow{B}}_{0}\) with respect to the g-tensor of the anisotropic spin center. The orientation of \(\overrightarrow{r}\) can be described by two spherical angles, a polar angle ξ and an azimuthal angle φ (Fig. 1). Similarly, the orientation of \({\overrightarrow{B}}_{0}\) can be given by a polar angle ξB and an azimuthal angle φB (Fig. 1). Consequently, the dipolar coupling frequency given by Eq. (6) depends on the inter-spin distance r, angles ξ, φ, ξB, and φB, and the principal g-values of the anisotropic spin center.

The geometric model of a spin system consisting of one isotropic spin center, spin A, and one anisotropic spin center, spin B. The orientation of the inter-spin vector \(\overrightarrow{r}\) with respect to the g-tensor axes of spin B is given by the spherical coordinates (r, ξ, φ). The orientation of the magnetic field \({\overrightarrow{B}}_{0}\) with respect to the g-tensor axes of spin B is given by the spherical angles (ξB, φB). The angle between \(\overrightarrow{r}\) and \({\overrightarrow{B}}_{0}\) is denoted as θ
Since the PDS measurements are usually done on disordered samples, such as frozen solutions, the dipolar coupling frequency is averaged over all possible orientations of \({\overrightarrow{B}}_{0}\) with respect to the g-tensor of the anisotropic spin center, i.e., over ξB and φB. Because the molecule, which hosts the spin pair, usually has some flexibility, the dipolar frequencies are additionally averaged over the distributions of r, ξ, and φ, namely P(r), P(ξ) and P(φ). Altogether, this provides the so-called dipolar spectrum.
Since the dipolar spectrum corresponds to the Fourier transform of the background-corrected PDS time trace, all dependencies, obtained for the dipolar spectrum, apply also for the PDS time trace. These dependencies can be expressed in a form of equation:
where V(t) is the amplitude of the background-corrected PDS signal, V(0) is the value of V(t) at t = 0, and λ is the modulation depth parameter. Note that Eq. (8) implies the absence of orientation selectivity. Taken that the principal g-values of the anisotropic spin center and the modulation depth parameter can be determined experimentally, the only variables in Eq. (8) are the distributions P(r), P(ξ), and P(φ). The extraction of these distributions from the experimentally determined V(t) is a complex, ill-posed problem. Nevertheless, a possible algorithm to solve this problem using several simplifying assumptions was proposed and implemented in the program AnisoDipFit, which is described next.
3 Program Overview
As mentioned above, the program AnisoDipFit was developed for the analysis of the PDS data, which corresponds to spin systems consisting of one isotropic and one anisotropic S = 1/2 centers. The AnisoDipFit data analysis is based on using the geometric model of the spin system. Since there is an infinite number of possible spin system geometries, some simplifying assumptions about the model have to be made. In AnisoDipFit, the following assumptions are used:
-
1.
The spin system consists of two well-localized S = 1/2 centers, denoted as spin A and spin B.
-
2.
Spin A has an isotropic or almost isotropic g-tensor, whereas spin B can also have an anisotropic g-factor.
-
3.
The reference coordinate system of the model is set to be coincident with the g-tensor axes of spin B. A vector connecting spin A with spin B is described by three spherical coordinates: a length r, a polar angle ξ, and an azimuthal angle φ (Fig. 1).
-
4.
In order to account for the conformational flexibility of the spin system, all three geometric parameters, namely r, ξ, and φ, are allowed to have either a uniform distribution or a normal distribution. In both cases, the distributions are described by two parameters, a mean value and a width. In the case of the normal distribution, the standard deviation is used as the width parameter.
-
5.
Since the PDS data does not provide enough information about potential correlations between the geometric parameters r, ξ, and φ, the geometric model is simplified by assuming the correlations to be zero.
It follows from the above that the AnisoDipFit geometric model is described by six parameters: the mean distance ‹r›, the mean angles ‹ξ› and ‹φ›, and the corresponding widths Δr, Δξ and Δφ. Depending on the g-tensor symmetry of spin B, all six parameters or only a subset of them are required to simulate the primary PDS data. If the g-tensor of spin B is rhombic, all six parameters are needed. However, if this g-tensor is axial and gB,xx = gB,yy, the parameters ‹φ› and Δφ have no effect on the PDS data. In this case, only four parameters are sufficient, whereas ‹φ› and Δφ can be set to 0. In order to account for this, AnisoDipFit allows the user to adjust the number of required parameters. In addition, it should be mentioned that the g-tensors of both spin centers possess inversion symmetry as an intrinsic property. Due to this symmetry, there are several sets of angles ‹ξ› and ‹φ›, which provide identical PDS data. For definiteness, both angles are defined in the interval of [0°, 90°], in which their values are unique [63]. By using the initial values of ‹ξ› and ‹φ›, other symmetry-related values of these angles can be readily determined as (180° − ‹ξ›, ‹φ›), (180° − ‹ξ›, ‹φ› + 180°), and (‹ξ›, ‹φ› + 180°).
In general, AnisoDipFit has two main operation modes, a simulation mode and a fitting mode. In the simulation mode, the dipolar spectrum or the PDS time trace is calculated for the above-described geometric model with the pre-defined values of ‹r›, ‹ξ›, ‹φ›, Δr, Δξ, and Δφ. In other words, the PDS data is simulated for the known distributions P(r), P(ξ), and P(φ). The calculation of dipolar spectrum is based on Eq. (6), which is averaged over random orientations of \({\overrightarrow{B}}_{0}\) (powder averaging) and, additionally, over the distributions P(r), P(ξ), and P(φ). This averaging is done by means of the Monte Carlo method using 106–107 samples. Each sample corresponds to one value of r, ξ, φ, ξB, and φB, which are randomly selected according to the respective probability distributions P(r), P(ξ), P(φ), P(ξB) = sin(ξB), and P(φB) = 1/(2π). Since the probability distribution of ξ is given not only by P(ξ) but also includes sin(ξ), the contribution of each sample to the total dipolar spectrum is weighted by sin(ξ). As follows from Eq. (6), the calculation of the dipolar coupling frequencies requires the g-factors of both spin centers. Therefore, both g-factors have to be provided among the input data of the program. The calculation of the PDS time trace is done in two steps. First, the dipolar spectrum is calculated in the same way as described above. This spectrum consist of N different values of dipolar frequency, νdd,i, and the corresponding probabilities, pi. The difference between consecutive frequency values is constant and set to 0.01 MHz. Second, the dipolar spectrum is translated into the PDS time trace using the equation:
The value of the modulation depth parameter λ can be either specified among the input data of the program or read out from the experimental PDS time trace, which can be optionally provided with the input data.
Besides the simulations described above, AnisoDipFit allows calculating the dipolar spectrum in dependence of the angular parameters ‹ξ›, ‹φ›, θ. In these calculations, all parameters of the geometric model are set to their pre-defined values and only one of the selected angles is varied in the entire interval of [0°, 90°] with a user-defined step. The rest of the calculations are done in the same way as for a single dipolar spectrum. Note that the possibility to record the angular dependencies of the dipolar spectrum can be useful if one wants to study the effect of individual angular parameters on the shape of dipolar spectrum.
The last parameter, which has to be mentioned for the AnisoDipFit simulations, is the temperature of the PDS experiment. Usually, the temperature of the PDS experiment has no effect on the PDS data. However, as it was shown recently [42], this can be violated for the RIDME data acquired at temperatures, at which the polarization of the anisotropic spin center differs between the different g-values of this spin center. For example, for hs-Fe3+ at Q-band, the RIDME data begins to depend on temperature below 3 K [42]. In this case, the dipolar coupling frequencies, which correspond to the different g-values of the anisotropic spin center, contribute to the dipolar spectrum with the following weights [42]:
where kB is the Boltzmann constant, and T is the temperature of the RIDME experiment. AnisoDipFit allows the user to include these weights into the simulations of the RIDME data. If the weights are included, the values of the temperature and the magnetic field B0 need to be specified among the input data of the program. In analogy to the angular dependencies of the dipolar spectrum, AnisoDipFit also allows to calculate the dependence of dipolar spectrum on the temperature. This data can be used to estimate the temperature ranges, in which the RIDME spectrum is temperature-dependent.
The second mode of AnisoDipFit is the fitting mode. As follows from its name, this mode allows to fit experimental PDS data using the parameters of the above-described geometric model (‹r›, ‹ξ›, ‹φ›, Δr, Δξ, and Δφ) and the temperature of the PDS experiment as fitting parameters. Depending on the symmetry of anisotropic spin center, the number of geometric parameters that are required for the fitting may vary (see above). Moreover, the temperature of the PDS experiment has to be included into the fitting only if the error of the experimentally measured temperature is above 0.1 K. Therefore, the user can choose which parameters will be included in the fitting and which ones will be set to a constant value. During the fitting, the selected fitting parameters are optimized until the simulated dipolar spectrum or PDS time trace provides the best fit to the experimental PDS spectrum or PDS time trace, respectively. The simulation of dipolar spectra and PDS time traces is done in the same way as in the simulation mode. The goodness of fit is monitored by calculating the χ2 deviation between the simulated and the experimental PDS data [64]. The calculation of χ2 requires knowledge of the standard deviation of noise in the experimental PDS data (σN), which can be either provided among the input data of the program or estimated by the program automatically using the best fit to the experimental data as a reference noise-free signal (in this case, σN is set to 1 during the fitting). The optimization of the fitting parameters is done by means of a genetic algorithm. This algorithm has been shown to be very efficient when one deals with a large number of optimization parameters and needs to find a global minimum [65,66,67,68]. The detailed description of the genetic algorithm implemented in AnisoDipFit is provided elsewhere [68]. The results of each fitting include the best fit to the experimental PDS data and the corresponding optimized values of fitting parameters. The precision of the optimized parameters is however unknown. To fill this gap, AnisoDipFit performs an error analysis. Since the parameter space is quite large (up to seven fitting parameters) and the fitting of PDS data is very time-consuming (hours), usual error analyses, such as bootstrap [64] and Bayesian analysis [69], cannot be done within a reasonable time. Therefore, the error analysis is done in a less general way. First, χ2 is recorded in dependence of ‹r› and Δr, ‹ξ› and Δξ, ‹φ and Δφ, and T. While recording each of these dependences, the remaining fitting parameters are set to their optimized values. For example, when χ2 is recorded as a function of ‹r› and Δr (Fig. 2a), the values of ‹ξ›, ‹φ›, Δξ, Δφ, and T are fixed at the corresponding optimized values (or at the user-defined values, if some of them were not optimized). Determination of parameters’ errors from the obtained dependencies relies on the assumption that the contributions to χ2 from P(r), P(ξ), P(φ), and T are uncorrelated, at least, near the global minimum. This assumption was confirmed in the previous study [60], but in general, it allows to estimate only the lower bound of parameters’ uncertainty. Prior to the error estimation, each two-dimensional dependence, e.g. χ2(‹r›, Δr), is converted into two one-dimensional dependencies that are optimized with respect to the first or second parameter of the corresponding two-dimensional dependence, e.g. χ2(‹r›) and χ2(Δr) (Fig. 2b). The obtained one-dimensional dependencies are then used to determine parameter ranges, in which the deviation of the χ2 values from the minimal χ2 is less than Δχ2. The threshold Δχ2 is build up out of two contributions. The first contribution, Δχci2, takes into account errors related to the noise in the experimental data and possible discrepancies between the actual spin system and its geometric model. Δχci2 is calculated using the user-defined confidence level, e.g. a 3σ confidence level. If one assumes that the measurement errors are distributed normally, nσ confidence level corresponds to Δχci2 = n2 [64]. Thus, for the 3σ confidence level one obtains Δχci2 = 9. The second contribution, Δχne2, takes into account the numerical error, which is mostly determined by the accuracy of the Monte Carlo integration. The value of Δχne2 is estimated by calculating χ2 for the 104 identical sets of optimized fitting parameters and, subsequently, finding the difference between the maximal and minimal values of χ2. Lastly, the determined uncertainty ranges are converted into the errors of the fitting parameters by calculating the largest deviation of each parameter from its optimized value within the corresponding uncertainty ranges.

General idea of the AnisoDipFit error analysis. a Exemplary dependence of χ2 on the fitting parameters ‹r› and Δr, χ2(‹r›, Δr). While acquiring this dependence, all other fitting parameters are set to their optimized values. The optimized values of ‹r› and Δr are depicted by a circle. b One-dimensional dependencies χ2(‹r›) and χ2(Δr), which are derived from χ2(‹r›, Δr). χ2(‹r›) is optimized with respect to Δr, and χ2(Δr) with respect to ‹r›. The χ2 threshold is depicted by the black dashed line. This threshold consists of two contributions, Δχci2, which is determined at the 3σ confidence level, and Δχne2, which takes into account the numerical error. The uncertainty ranges of ‹r› and Δr are shown as gray intervals
After discussing the function of AnisoDipFit, its technical information will be given. AnisoDipFit is a stand-alone program, which is freely available at https://github.com/dinarabdullin/AnisoDipFit. Similar to many quantum chemistry programs, AnisoDipFit can be run from the Terminal (or Command Prompt) and requires a special configuration file, which contains all input data of the program. The detailed information on how one creates such a configuration file, as well as few examples of configuration files, can be found in the program’s manual, which can be accessed using the link above. The source code of AnisoDipFit was written using the Python programming language and the libraries numpy, scipy, wx, matplotlib, and libconf. The Windows and Linux executables of the program can be downloaded from https://github.com/dinarabdullin/AnisoDipFit/releases. AnisoDipFit supports parallel computing and, therefore, its performance can be significantly improved when using the hardware with a large number of CPUs. For the examples given below, AnisoDipFit was run on a 64-core workstation from sys-Gen GmbH with 2.3 GHz processor frequency and 132 GB RAM.
4 Simulation of PDS Data
To get a deeper insight into how the g-anisotropy of different anisotropic spin centers influences the corresponding PDS data, the AnisoDipFit simulations were carried out. In these simulations, dipolar spectra and PDS time traces were calculated for three spin systems, namely Cu2+/organic radical, ls-Fe3+/organic radical, and hs-Fe3+/organic radical. The g-factor of the organic radical was always set to ge. The principal g-values (gxx, gyy, gzz) of the metal centers were set to their typical values, such as (2.05, 2.05, 2.25) for Cu2+, (1.56, 2.28, 2.91) for ls-Fe3+, and (6.00, 6.00, 2.00) for hs-Fe3+. For all three spin systems, the inter-spin distance distribution was assumed to have the Gaussian shape with a mean value ‹r› = 2.50 nm and a standard deviation σr = 0.10 nm. The angles ξ and φ were assumed to have a single value each. For the axial Cu2+ and hs-Fe3+ spin centers, φ was set to 0°, whereas ξ was varied in the range [0°, 90°] with a step of 15°. For the rhombic ls-Fe3+ spin center, both angles were varied in the range [0°, 90°] with a step of 30°. All simulations were done for a temperature of 10 K and a modulation depth of 0.5. The frequency axis of all simulated dipolar spectra was normalized by the average dipolar coupling constant ν0 of an isotropic spin pair with the same inter-spin distance distribution as above. After the simulations, the PDS time traces, which were calculated for different spin systems and different values of ξ and φ, were analyzed by DeerAnalysis. Although DeerAnalysis was developed exclusively for isotropic spin centers, it is still interesting to apply this program to the present case and to examine how much the DeerAnalysis-derived distance distributions deviate from the original distance distribution.
4.1 Cu2+/Organic Radical
The dipolar spectra and the PDS time traces of the spin system Cu2+/organic radical were simulated in dependence of the angle ξ and are depicted in Fig. 3a and b, respectively. Figure 3a reveals that all simulated dipolar spectra have similar shapes. Moreover, the shapes of these spectra are similar to the shape of the Pake doublet (dashed line in Fig. 3a), which is the dipolar spectrum of an isotropic spin pair. Both, the simulated spectra and the Pake doublet, show two singularities at around ± ν0 and ± 2ν0, corresponding to θ = 90° and θ = 0° in Eqs. (6) and (7), respectively. These singularities are often denoted as perpendicular (θ = 90°) and parallel (θ = 0°) components. Small differences between the simulated spectra and the Pake doublet can be described by two facts. First, the parallel component of the simulated spectra is shifted from the parallel component of the Pake doublet towards larger frequencies, especially, when ξ approaches 0°. Second, the perpendicular component of the simulated spectra is shifted from the perpendicular component of the Pake doublet towards larger frequencies, especially, when ξ approaches 90°. Both facts can be readily explained if one takes into account that the perpendicular and parallel components of the simulated spectra are scaled by the effective g-values of Cu2+ (see Eq. (6)) and that this scaling depends on the ξ angle. As an example, two particular cases, ξ = 0° and ξ = 90°, will be discussed below. In the case of ξ = 0°, \(\overrightarrow{r}\) is collinear to the gzz-axis of the Cu2+ g-tensor. Consequently, the parallel component of the spectrum is scaled by gzz, yielding a singularity at 2(gzz/ge)ν0 ≈ 2.25 ν0. Then, the other two components of the Cu2+ g-tensor give rise to two perpendicular component, which appears at (gxx/ge)ν0 = (gyy/ge)ν0 ≈ 1.02 ν0. In the case of ξ = 90°, \(\overrightarrow{r}\) is collinear to the gyy-axis of the Cu2+ g-tensor. Therefore, the parallel component is scaled by gyy and appears at 2(gyy/ge)ν0 ≈ 2.05 ν0. The perpendicular component is scaled by the Cu2+ g-values ranging from gxx to gzz. Therefore, this component spans the frequency range from (gxx/ge)ν0 ≈ 1.02 ν0 up to (gzz/ge)ν0 ≈ 1.12 ν0.

The angular dependence of the PDS data simulated for the spin system Cu2+/organic radical. a Dipolar spectra (solid lines), which were simulated for different ξ angles, are overlaid with the Pake doublet (dashed line). The magnified region around the perpendicular component is shown as an inset. b Simulated PDS time traces and c corresponding distance distributions determined by means of DeerAnalysis. The original distance distribution, which was used to simulate the data from (a) and (b), is depicted by gray shades
The similarity of the dipolar spectra, simulated for different ξ angles, resulted in the similarity of the corresponding PDS time traces (Fig. 3b). Moreover, since all simulated spectra were close in shape to the Pake doublet, the analysis of the simulated PDS time traces by DeerAnalysis yielded distance distributions, which are akin to the original distance distribution (Fig. 3c). However, few differences between the DeerAnalysis-derived distance distributions and the original distance distribution were still obtained. First, the maximum of the DeerAnalysis-derived distance distributions is shifted from the original distance (2.5 nm) towards smaller distances, especially, when ξ approaches 90°. Second, the DeerAnalysis-derived distance distributions, determined for the ξ angles below 45°, contain an additional distance peak at around 1.9 nm. The first difference stems from the fact that the perpendicular component of the simulated dipolar spectra deviates from ν0. As was shown above, this deviation depends on the angle ξ and is maximal for ξ = 90°. Note that the errors of the DeerAnalysis-derived most probable distances are small and do not exceed 0.06 nm. However, these errors can become larger if the inter-spin distance increases [70]. For the given g-tensor of Cu2+, the dependence of the DeerAnalysis distance error on the inter-spin distance is depicted in Fig. 4. As can be seen, the DeerAnalysis distance error may exceed 0.1 nm (or 1 Å) for the inter-spin distances above 4.27 nm. The addition distance peak in the DeerAnalysis-derived distance distributions is due to the fact that the parallel component of the simulated dipolar spectra is shifted from 2ν0. This shift is maximal for ξ = 0°, and DeerAnalysis interprets it as if it will be an additional perpendicular component, which corresponds to the distance of 1.9 nm.

Predicted ranges of the DeerAnalysis distance error for the spin system Cu2+/organic radical. The g-values of Cu2+ were set to gxx = gyy = 2.05 and gzz = 2.25. The upper bound of the error (solid line) is obtained when ξ = 0° and the perpendicular component of the dipolar spectrum is scaled by an average Cu2+ g-factor of (gxx + gyy)/2 = 2.05. The lower bound of the error (dashed line) is obtained when ξ = 90° and the perpendicular component of the dipolar spectrum is scaled by an average Cu2+ g-factor of (gzz + gyy)/2 = 2.15
4.2 Low-spin Fe3+/Organic Radical
The second considered spin system was the spin pair ls-Fe3+/organic radical. The dipolar spectra and the PDS time traces of this spin pair were simulated in dependence of two angles, ξ and φ (Fig. 5). As can be seen from Fig. 5a, most of the simulated dipolar spectra differ from each other, and all of them are significantly different from the Pake doublet. This differences are due to the fact that spectral components, corresponding to different θ angles, are scaled by different effective g-values of ls-Fe3+. Apparently, this g-scaling depends on the values of ξ and φ. Importantly, the effect of this g-scaling on the obtained spectral shapes is more pronounced as compared to the case of Cu2+, because all three principal g-values of ls-Fe3+ have a larger deviation from ge as compared to the principal g-values of Cu2+. The obtained differences between the simulated spectra are further inherited by the simulated PDS time traces (Fig. 5b) and the corresponding DeerAnalysis-derived distance distributions (Fig. 5c). All DeerAnalysis-derived distance distributions are significantly different from the original distance distribution and, instead of being unimodal, have three prominent components, denoted as I, II, and III in Fig. 5c. The components I and II partially overlay and correspond to the perpendicular component of the dipolar spectra, which is scaled by a number of different effective g-values of ls-Fe3+. Since most of these g-values differ from ge, the maxima of the components I and II are shifted from the original distance (2.5 nm). The component III originates from the parallel component of the simulated dipolar spectra. Since the parallel component is also scaled by an effective g-value of ls-Fe3+ and, therefore, does not necessarily appear exactly at the doubled frequency of the parallel component, DeerAnalysis erroneously interpreters it as an additional perpendicular component, corresponding to shorter distances. These results reveal that DeerAnalysis has a significant distance error when used for the spin system ls-Fe3+/organic radical. Therefore, the specialized data analyses, such as AnisoDipFit, should be used for interpreting the experimental PDS data of this spin system.

The angular dependence of the PDS data simulated for the spin system ls-Fe3+/organic radical. a Dipolar spectra (solid lines), which were simulated for different ξ and φ angles, are overlaid with the Pake doublet (dashed line). b Simulated PDS time traces and c corresponding distance distributions determined by means of DeerAnalysis. The original distance distribution, which was used to simulate the data from (a) and (b), is depicted by gray shades. Three different components of the DeerAnalysis-derived distance distributions are marked by Roman letters
4.3 High-spin Fe3+/Organic Radical
The last spin system, for which the PDS data was simulated, was the spin pair hs-Fe3+/organic radical. Due to the high axiality of the hs-Fe3+ g-tensor, the simulated dipolar spectra and PDS time traces show the strong dependence on the angle ξ (Fig. 6a, b). This dependence, together with the fact that two out of three principal g-values of hs-Fe3+ are three times larger than ge, result in the substantial difference between the shapes of the simulated spectra and the shape of Pake doublet. Consequently, the analysis of simulated PDS time traces by DeerAnalysis yields distance distributions, which is totally different from the original distance distribution (Fig. 6c). Thus, like in the case of the spin system ls-Fe3+/organic radical, only specialized data analyses, such as AnisoDipFit, can be used to analyze PDS data of the spin system ls-Fe3+/organic radical.

The angular dependence of the PDS data simulated for the spin system hs-Fe3+/organic radical. a Dipolar spectra (solid lines), which were simulated for different ξ angles, are overlaid with the Pake doublet (dashed line). b Simulated PDS time traces and c corresponding distance distributions determined by means of DeerAnalysis. The original distance distribution, which was used to simulate the data from a) and b), is depicted by gray shades
5 Fitting of Experimental PDS Data
The key function of AnisoDipFit is the data analysis, in which the distributions P(r), P(ξ), and P(φ) are extracted from the experimental PDS data. As described above, the fitting mode of AnisoDipFit is used for this purpose. Here, the fitting mode of AnisoDipFit was tested on the previously reported experimental RIDME data. This data was chosen such that the spin systems with different degree of g-anisotropy were covered. In analogy to the simulations above, the considered spin systems included Cu2+/nitroxide, ls-Fe3+/trityl, and hs-Fe3+/nitroxide. The details of the AnisoDipFit-based fitting for each of these spin systems are described below.
5.1 Cu2+/Nitroxide
Model compound 1 was used as an example of the spin system Cu2+/nitroxide (Fig. 7a). Note that 1 contains three spin centers, a Cu2+ ion and two nitroxides. Both nitroxides have the same linker to the Cu2+ ion and are located almost symmetrically with respect to that ion. Thus, the distance and the mutual orientation of both nitroxides with respect to Cu2+ can be assumed to be identical. The Q-band RIDME measurements on 1 were performed by Meyer et al. [56]. Due to the significant rigidity of 1, the RIDME time traces of 1 were orientation-selective. In order to get rid of this orientation selectivity, the RIDME time traces were acquired at several positions across the nitroxide spectrum and then averaged out. The obtained average time trace was converted into distance distribution by means of DeerAnalysis. Here, the same RIDME time trace was analyzed by means of AnisoDipFit. For this purpose, the g-values of Cu2+ were set to gxx = 2.254, gyy = 2.093, gzz = 2.042 [56], and the g-values of the nitroxides to ge. The geometric model, which was used in the fitting of the RIDME time trace, was described by the distributions P(r), P(ξ), and P(φ). All three distributions were approximated by Gaussians and the corresponding mean values and standard deviations were used as fitting parameters. During the fitting, the values of these parameters were iteratively optimized by the genetic algorithm. The total number of optimization steps was set to 500. The results of the AnisoDipFit data analysis for 1 are summarized in Fig. 7b–d. Figure 7b shows how the goodness of fit, given by χ2, changed across optimization steps. As can be seen, χ2 fell rapidly down during the first 100 optimization steps and, after this, did not change anyhow significantly during the last 400 optimization steps. This reveals that the minimum of the optimization problem was reached. Since the genetic algorithm is capable to find the global minimum even for optimization problems with several local minima [71], the obtained minimum is most likely global. This is supported by fact that a good fit to the RIDME data was indeed obtained (Fig. 7c). The optimized values of fitting parameters, which deliver this fit, are shown by white dots in Fig. 7d. In addition, Fig. 7d depicts the two-dimensional error surfaces for three pairs of fitting parameters: ‹r› and Δr, ‹ξ› and Δξ, and ‹φ› and Δφ. The uncertainty ranges of the fitting parameters are depicted as dark red regions. For the distance parameters ‹r› and Δr, the uncertainty ranges are well-defined and appear at ± 0.02 nm around the optimized values. In contrast, the uncertainty ranges of all four angular parameters span over their entire ranges, meaning that these parameters could not be determined from the present data analysis. The latter result can have several reasons. First, the g-anisotropy of Cu2+ might be still too small to provide a good resolution for ξ and φ. Second, the signal-to-noise ratio (SNR) of the RIDME time trace might be insufficient to allow the detection of small changes in the RIDME time trace caused by the g-anisotropy of Cu2+. Third, the bending motion of 1 [56] may broaden the distributions P(ξ) and P(φ).

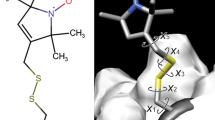
The AnisoDipFit data analysis of the Q-band RIDME time trace acquired on the model system 1. a The crystal structure of 1. The carbon atoms are shown in gray, oxygen in red, nitrogen in blue, and copper in orange. b The dependence of χ2 on the optimization step. c The RIDME time trace (black line) is overlaid with the corresponding fit (red line). d The dependence of χ2 on different pairs of fitting parameters. Dark red regions correspond to the parameters’ uncertainty intervals, which lie below the χ2 threshold. The optimized values of the fitting parameters are depicted by white dots (colour figure online)
Finally, the values of ‹r› and Δr were compared between the AnisoDipFit, DeerAnalysis, and the crystal structure of 1 (Table 1). As expected (Fig. 4), the difference between the AnisoDipFit- and DeerAnalysis-derived values of ‹r› is small, only 0.07 nm. Both values are about 0.1 nm smaller than the crystallographic value of ‹r›. The standard deviation Δr is identical for both AnisoDipFit and DeerAnalysis. Thus, the use of DeerAnalysis for the RIDME data of 1 yields quite accurate distance distribution. This can also be true for other Cu2+/nitroxide spin systems, although it is important to remember that the error of the DeerAnalysis-based data analysis can increase for larger distances (Fig. 4) and that some distance artifacts might appear in the DeerAnalysis-derived distance distributions (Fig. 3c).
5.2 Low-spin Fe3+/Trityl
To test the AnisoDipFit data analysis for the ls-Fe3+/trityl spin system, the previously reported Q-band RIDME time trace of the model compound 2 (Fig. 8a, [60]) was used. Similar to the previous example, the geometric model, which was used in the fitting of the RIDME time trace, was described by the distributions P(r), P(ξ), and P(φ). All three distributions were again approximated by Gaussians and the corresponding mean values and standard deviations were used as fitting parameters. The g-values of ls-Fe3+ were set to gxx = 1.56, gyy = 2.28, gzz = 2.91 [60], and the g-values of the trityl to ge. The results of the AnisoDipFit data analysis for 2 are summarized in Fig. 8b–d. Figure 8b reveals that the genetic algorithm has converged to the global minimum after 270 optimization steps. Figure 8c shows that the obtained global minimum corresponds to a fit, which quite exactly reproduces the shape of the RIDME time trace. Figure 8d overlays the optimized values of the fitting parameters (white dots) with their error surfaces. The dark red regions in the error surfaces depict the uncertainty ranges of the fitting parameters. In contrast to the previous example, the uncertainty ranges of all six fitting parameters, including the angular parameters, are well-defined. This result can be attributed to the fact that ls-Fe3+ has the larger g-factor anisotropy as compared to Cu2+. Based on Fig. 8d, the errors of individual fitting parameters were estimated and are listed in Table 2. It reveals the parameters of P(r) were determined with a sub-angstrom precision, whereas the parameters of P(ξ) and P(φ) with an average precision of ± 29°.

The AnisoDipFit data analysis of the Q-band RIDME time trace acquired on the model system 2. a The chemical structure of 2. b The dependence of χ2 on the optimization step. c The RIDME time trace (black line) is overlaid with the corresponding fit (red line). d The dependence of χ2 on different pairs of fitting parameters. Dark red regions correspond to the parameters’ uncertainty intervals, which lie below the χ2 threshold. The optimized values of the fitting parameters are depicted by white dots (colour figure online)
Note that the RIDME time trace of 2 was already analyzed by AnisoDipFit in the previous publication [60], but the error analysis was done differently. The main difference between the former and the present error analyses is the way of determining the uncertainty ranges of the fitting parameters. In the former error analysis, the uncertainty ranges were determined at 110% of the minimal root-mean-square deviation (RMSD) between the experimental RIDME time trace and its best fit. Recent tests have revealed that the numerical error of the calculated RMSD values (ΔRMSD ~ 3·10–4) can be as large as 10% of the minimal RMSD (RMSDmin ~ 3.0·10–3) and, therefore, the chosen threshold has to be either increased or replaced by a more adaptive threshold. The second approach was taken; the new error analysis sets the value of the threshold based on the estimated numerical error and the desired confidence level (3σ confidence level was used here). Consequently, the new error analysis yielded more accurate error estimates as compared to the former analysis.
Despite the difference in the threshold value, it is still interesting to compare the optimized fitting parameters between the present analysis and the analysis published earlier. This comparison is shown in Table 2. It reveals that the parameters of P(r) are identical and have similar error bars. In contrast, the parameters of P(ξ) and P(φ) take different values, but the corresponding error intervals overlap significantly. As can be seen, the error intervals, which were obtained for the angular parameters in the present analysis, are larger and include the significant part of the corresponding error intervals, which were determined in the previous analysis. This has two reasons. First, the new threshold yields larger uncertainty intervals than the old one, and, consequently, the corresponding error intervals are larger too. Second, there is a difference between two error analyses in the Δξ and Δφ intervals, which were used to record the error surfaces. In the previous analysis, both intervals were set to [0°, 30°]. In the present study, it was realized that these intervals are not sufficient to take into account broad distributions of ξ and φ and, therefore, both intervals were increased up to [0°, 90°]. Figure 8c reveals that the uncertainty intervals of all angular parameters indeed propagate to the region with Δξ > 30° and Δφ > 30°, which explains why the errors of the angular parameters from the previous analysis are smaller than the ones obtained here. Thus, the errors of the angular parameters were underestimated in the earlier study.
In addition, the optimized fitting parameters of 2 were compared to their MD predictions [60]. Table 2 reveals overall agreement between them. The only significant difference is obtained for the parameters of P(φ). Possible reasons of this difference were discussed in detail previously [60].
5.3 High-spin Fe3+/Nitroxide
The last test of the AnisoDipFit data analysis was done for the RIDME spectrum acquired on the nitroxide-labeled mutant of the hemeprotein met-myoglobin [42], denoted here as met-MbQ8R1 (Fig. 9a). Met-MbQ8R1 contains two electron spin centers, a nitroxide and a hs-Fe3+ ion. The hs-Fe3+ has a large axial ZFS tensor (D ~ 9.26 cm−1, E = 0.0023 cm−1 [72]), which allows considering this ion as an effective S = 1/2 center at Q-band and at temperatures below 3 K [42]. The g-factor of this center is very anisotropic and has the principal components gxx = 5.93, gyy = 5.94, and gzz = 2.00 [42]. In contrast, the g-anisotropy of the nitroxide center is small and, therefore, was neglected in the data analysis by setting all g-values to ge. Since the g-factor of the hs-Fe3+ is almost axial, the RIDME spectrum of met-MbQ8R1 does not depend on the angle φ. Therefore, this angle was excluded from the fitting and fixed at a constant value of 0°. Two other distributions, P(r) and P(ξ), were approximated by Gaussians and their mean values and standard deviations were used as fitting parameters. In addition, the temperature of the RIDME experiment was used as a fitting parameter. This was done because of two reasons. First, the RIDME experiments on met-MbQ8R1 were done at the temperature of ~ 3 K, which is low enough to have an influence of the spectral simulations in accordance to Eq. (10). Second, the error of the experimentally measured temperature was above the precision required for accurate spectral simulations. The results of the AnisoDipFit analysis for met-MbQ8R1 are given in Fig. 9b–d. Figure 9b reveals that the genetic algorithm has again converged to the global minimum. Figure 9c shows that a good fit to the RIDME spectrum has been obtained. Finally, Fig. 9d depicts the optimized values of the fitting parameters together their error surfaces. The uncertainty ranges of the fitting parameters, shown as dark red regions in Fig. 9d, were used to estimate the errors of fitting parameters (Table 3). Similar to the case of 2, the parameters of P(r) were determined with a sub-angstrom precision. The parameters of P(ξ) were determined with a precision of ± 1°, which significantly exceeds that precision of the same parameters obtained for 2. This result can be attributed to the fact that the g-anisotropy of the hs-Fe3+ is much larger than the g-anisotropy of the ls-Fe3+. Thus, as could be expected, the precision of the angular parameters strongly depends on the g-anisotropy of the anisotropic spin center. The optimized value of the temperature equals to 2.1 ± 0.2 K. This value deviates from the experimental temperature by ~ 1 K, which is in agreement with the reported experimental error of the temperature measurement [42].

The AnisoDipFit data analysis of the Q-band RIDME spectrum acquired on the model system met-MbQ8R1. a The cartoon model of met-MbQ8R1. The protein structure of met-myoglobin (PDB 1WLA) is shown as a gray cartoon model, the R1 side chain is shown as pink sticks with oxygen atoms colored red, and the Fe3+ ion is shown as a blue sphere. b The dependence of χ2 on the optimization step. c The RIDME spectrum (black line) is overlaid with the corresponding fit (red line). d The dependence of χ2 on different pairs of fitting parameters. Dark red regions correspond to the parameters’ uncertainty intervals, which lie below the χ2 threshold. The optimized values of the fitting parameters are depicted by white dots (colour figure online)
Like in the case of 2, the RIDME spectrum of met-MbQ8R1 was already analyzed by AnisoDipFit in the previous study [42], in which the error analysis was done using a fixed threshold set to 110% of the minimal RMSD. The reason for replacing this threshold by a more adaptive one was the same as in the case of 2. The numerical error of the calculated RMSD was estimated at 3.2·10–3, whereas 10% of the minimal RMSD were only 2.6·10–3. The new threshold, which is calculated based on the estimated numerical error and the desired confidence level (3σ confidence level), allowed to avoid this pitfall. Interestingly, the substitution of the threshold had only minor effect of the optimized fitting parameters and their errors (Table 3). Moreover, the optimized fitting parameters, obtained from the both AnisoDipFit analyses, show in a good agreement with the corresponding parameters predicted by MD [73] (Table 3).
6 Conclusion
The AnisoDipFit program was introduced as a tool to perform the PDS data analysis for spin systems consisting of one isotropic and one anisotropic electron spin centers. The theoretical background, the operation principles, and the different modes of AnisoDipFit were described for the first time in detail. This description, together with the prepared manual of the program, should make the use of the program more convenient.
To identify anisotropic spin centers, for which the use of this program is especially relevant, AnisoDipFit was used to simulate the PDS data of the spin systems with different anisotropic spin centers, such as Cu2+, ls-Fe3+, and hs-Fe3+. For the spin system Cu2+/organic radical, the simulated PDS data revealed only minor deviations from the PDS data corresponding to isotropic spin centers. Thus, except few outlined limitations, the program DeerAnalysis can be applied for Cu2+ as an alternative to AnisoDipFit. In contrast, the simulated PDS data of the spin systems ls-Fe3+/organic radical and hs-Fe3+/organic radical showed the fundamental difference to the PDS data of isotropic spin centers. Consequently, AnisoDipFit is currently the only available program, which allows accurate PDS data analysis for ls-Fe3+, hs-Fe3+, and other spin centers with comparable g-anisotropy.
The AnisoDipFit data analysis was successfully tested on the experimental RIDME data of the Cu2+/nitroxide and ls-Fe3+/trityl model compounds and the met-myoglobin mutant. For all three test systems, the distance distribution was determined with the sub-angstrom precision. In addition, the spatial orientation of the inter-spin vector with respect to the g-frame of the metal center was determined for two out of three test systems. The precision of this information was shown to depend on the g-anisotropy of the metal center. For the most anisotropic hs-Fe3+, the precision of the azimuthal and polar angles was ± 2°. For the less anisotropic ls-Fe3+, the precision of both angles was reduced down to ± 31°, whereas, for the least anisotropic Cu2+, both angles were undefined.
In general, AnisoDipFit extends the arsenal of available tools for PDS data analysis and should facilitate further PDS-based distance measurements on a highly relevant class of metalloproteins, including hemoproteins and many other proteins.
References
A.D. Milov, K.M. Salikhov, M.D. Shchirov, Fiz. Tverd. Tela 23, 975 (1981)
R.E. Martin, M. Pannier, F. Diederich, V. Gramlich, M. Hubrich, H.W. Spiess, Angew. Chem. Int. Ed. 37, 2833 (1998)
S. Saxena, J.H. Freed, Chem. Phys. Lett. 251, 102 (1996)
G. Jeschke, M. Pannier, A. Godt, H.W. Spiess, Chem. Phys. Lett. 331, 243 (2000)
L.V. Kulik, S.A. Dzuba, I.A. Grigoryev, Y.D. Tsvetkov, Chem. Phys. Lett. 343, 315 (2001)
S. Milikisyants, F. Scarpelli, M.G. Finiguerra, M. Ubbink, M. Huber, J. Magn. Reson. 201, 48 (2009)
P.P. Borbat, J.H. Freed, EMagRes 6, 465 (2017)
G. Jeschke, EMagRes. 5, 1459 (2016)
A.M. Bowen, E.O.D. Johnson, F. Mercuri, N.J. Hoskins, R. Qiao, J.S.O. McCullagh, J.E. Lovett, S.G. Bell, W. Zhou, C.R. Timmel, L.L. Wong, J.R. Harmer, J. Am. Chem. Soc. 140, 2114 (2018)
O. Duss, M. Yulikov, G. Jeschke, F.H.-T. Allain, Nat. Commun. 5, 3669 (2014)
M. Teucher, H. Zhang, V. Bader, K.F. Winklhofer, A.J. García-Sáez, A. Rajca, S. Bleicken, E. Bordignon, Sci. Rep. 9, 13013 (2019)
B. Joseph, A. Sikora, D.S. Cafiso, J. Am. Chem. Soc. 138, 1844 (2016)
J. Glaenzer, M.F. Peter, G.H. Thomas, G. Hagelueken, Biophys. J. 112, 109 (2017)
M.C. Puljung, H.A. DeBerg, W.N. Zagotta, S. Stoll, Proc. Natl. Acad. Sci. USA 111, 9816 (2014)
D. Abdullin, N. Florin, G. Hagelueken, O. Schiemann, Angew. Chem. Int. Ed. 54, 1827 (2015)
E.G.B. Evans, M.J. Pushie, K.A. Markham, H.-W. Lee, G.L. Millhauser, Structure 24, 1057 (2016)
A. Gamble Jarvi, T.F. Cunningham, S. Saxena, Phys. Chem. Chem. Phys. 21, 10238 (2019)
C. Altenbach, T. Marti, H.G. Khorana, W.L. Hubbell, Science 248, 1088 (1990)
L.J. Berliner, J. Grunwald, H.O. Hankovszky, K. Hideg, Anal. Biochem. 119, 450 (1982)
E.G. Bagryanskaya, O.A. Krumkacheva, M.V. Fedin, S.R.A. Marque, Methods Enzymol. 563, 365 (2015)
G.W. Reginsson, N.C. Kunjir, S.T. Sigurdsson, O. Schiemann, Chem. Eur. J. 18, 13580 (2012)
A.M. Raitsimring, C. Gunanathan, A. Potapov, I. Efremenko, J.M.L. Martin, D. Milstein, D. Goldfarb, J. Am. Chem. Soc. 129, 14138 (2007)
D. Goldfarb, Phys. Chem. Chem. Phys. 16, 9685 (2014)
A. Feintuch, G. Otting, D. Goldfarb, Methods Enzymol. 563, 415 (2015)
D. Banerjee, H. Yagi, T. Huber, G. Otting, D. Goldfarb, J. Phys. Chem. Lett. 3, 157 (2012)
A. Martorana, Y. Yan, Y. Zhao, Q.-F. Li, X.-C. Su, D. Goldfarb, Dalton Trans. 44, 20812 (2015)
T.F. Cunningham, M.R. Putterman, A. Desai, W.S. Horne, S. Saxena, Angew. Chem. Int. Ed. 54, 6330 (2015)
H. Sameach, S. Ghosh, L. Gevorkyan-Airapetov, S. Saxena, S. Ruthstein, Angew. Chem. Int. Ed. 58, 3053 (2019)
G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C.R. Timmel, D. Hilger, H. Jung, Appl. Magn. Reson. 30, 473 (2006)
C.J. López, Z. Yang, C. Altenbach, W.L. Hubbell, Proc. Natl. Acad. Sci. USA 110, E4306 (2013)
R.A. Stein, A.H. Beth, E.J. Hustedt, Methods Enzymol. 563, 531 (2015)
B. Endeward, J.A. Butterwick, R. MacKinnon, T.F. Prisner, J. Am. Chem. Soc. 131, 15246 (2009)
C. Abé, D. Klose, F. Dietrich, W.H. Ziegler, Y. Polyhach, G. Jeschke, H.-J. Steinhoff, J. Magn. Reson. 216, 53 (2012)
O. Schiemann, P. Cekan, D. Margraf, T.F. Prisner, S.T. Sigurdsson, Angew. Chem. Int. Ed. 48, 3292 (2009)
A. Marko, V. Denysenkov, D. Margraf, P. Cekan, O. Schiemann, S.T. Sigurdsson, T.F. Prisner, J. Am. Chem. Soc. 133, 13375 (2011)
A. Dalaloyan, M. Qi, S. Ruthstein, S. Vega, A. Godt, A. Feintuch, D. Goldfarb, Phys. Chem. Chem. Phys. 17, 18464 (2015)
S. Razzaghi, M. Qi, A.I. Nalepa, A. Godt, G. Jeschke, M. Yulikov, J. Phys. Chem. Lett. 5, 3970 (2014)
K. Keller, V. Mertens, M. Qi, A.I. Nalepa, A. Godt, A. Savitsky, G. Jeschke, M. Yulikov, Phys. Chem. Chem. Phys. 19, 17856 (2017)
A. Meyer, O. Schiemann, J. Phys. Chem. A 120, 3463 (2016)
D. Akhmetzyanov, H.Y.V. Ching, V. Denysenkov, P. Demay-Drouhard, H.C. Bertrand, L.C. Tabares, C. Policar, T.F. Prisner, S. Un, Phys. Chem. Chem. Phys. 18, 30857 (2016)
K. Keller, M. Zalibera, M. Qi, V. Koch, J. Wegner, H. Hintz, A. Godt, G. Jeschke, A. Savitsky, M. Yulikov, Phys. Chem. Chem. Phys. 18, 25120 (2016)
D. Abdullin, H. Matsuoka, M. Yulikov, N. Fleck, C. Klein, S. Spicher, G. Hagelueken, S. Grimme, A. Luetzen, O. Schiemann, Chem. Eur. J. 25, 8820 (2019)
B.E. Bode, J. Plackmeyer, T.F. Prisner, O. Schiemann, J. Phys. Chem. A 112, 5064 (2008)
B.E. Bode, J. Plackmeyer, M. Bolte, T.F. Prisner, O. Schiemann, J. Organomet. Chem. 694, 1172 (2009)
J.E. Lovett, A.M. Bowen, C.R. Timmel, M.W. Jones, J.R. Dilworth, D. Caprotti, S.G. Bell, L.L. Wong, J. Harmer, Phys. Chem. Chem. Phys. 11, 6840 (2009)
Z. Yang, D. Kise, S. Saxena, J. Phys. Chem. B 114, 6165 (2010)
Z. Yang, M. Ji, S. Saxena, Appl. Magn. Reson. 39, 487 (2010)
Z. Yang, M.R. Kurpiewski, M. Ji, J.E. Townsend, P. Mehta, L. Jen-Jacobson, S. Saxena, Proc. Natl. Acad. Sci. USA 109, E993 (2012)
J. Sarver, K.I. Silva, S. Saxena, Appl. Magn. Reson. 44, 583 (2013)
A.M. Bowen, M.W. Jones, J.E. Lovett, T.G. Gaule, M.J. McPherson, J.R. Dilworth, C.R. Timmel, J.R. Harmer, Phys. Chem. Chem. Phys. 18, 5981 (2016)
M.M. Roessler, M.S. King, A.J. Robinson, F.A. Armstrong, J. Harmer, J. Hirst, Proc. Natl. Acad. Sci. USA 107, 1930 (2010)
A. Doll, S. Pribitzer, R. Tschaggelar, G. Jeschke, J. Magn. Reson. 230, 27 (2013)
F.D. Breitgoff, K. Keller, M. Qi, D. Klose, M. Yulikov, A. Godt, G. Jeschke, J. Magn. Reson. 308, 106560 (2019)
A.V. Astashkin, B.O. Elmore, W. Fan, J.G. Guillemette, C. Feng, J. Am. Chem. Soc. 132, 12059 (2010)
D. Abdullin, F. Duthie, A. Meyer, E.S. Müller, G. Hagelueken, O. Schiemann, J. Phys. Chem. B 119, 13539 (2015)
A. Meyer, D. Abdullin, G. Schnakenburg, O. Schiemann, Phys. Chem. Chem. Phys. 18, 9262 (2016)
A. Giannoulis, M. Oranges, B.E. Bode, Chem. Phys. Chem. 18, 2318 (2017)
J.J. Jassoy, A. Berndhäuser, F. Duthie, S.P. Kühn, G. Hagelueken, O. Schiemann, Angew. Chem. Int. Ed. 56, 177 (2017)
I. Ritsch, H. Hintz, G. Jeschke, A. Godt, M. Yulikov, Phys. Chem. Chem. Phys. 21, 9810 (2019)
D. Abdullin, P. Brehm, N. Fleck, S. Spicher, S. Grimme, O. Schiemann, Chem. A Eur. J. 25, 14388 (2019)
A. Abragam, B. Bleaney, Electron Paramagnetic Resonance of Transition Ions (Oxford University Press, Oxford, 1970)
A.F. Bedilo, A.G. Maryasov, J. Magn. Reson. 116, 87 (1995)
A.M. Bowen, C.E. Tait, C.R. Timmel, J.R. Harmer, Struct. Bond. 152, 283 (2013)
W.H. Press, S.A. Teukolsky, W.T. Vetterling, B.P. Flannery, Numerical Recipes in C (Cambridge University Press, Cambridge, 1992)
B. Filipič, J. Štrancar, Appl. Soft Comput. 1, 83 (2001)
T. Spałek, P. Pietrzyk, Z. Sojka, J. Chem. Inf. Model. 45, 18 (2005)
S. Stoll, A. Schweiger, J. Magn. Reson. 178, 42 (2006)
D. Abdullin, G. Hagelueken, R.I. Hunter, G.M. Smith, O. Schiemann, Mol. Phys. 113, 544 (2015)
T.H. Edwards, S. Stoll, J. Magn. Reson. 270, 87 (2016)
D. Abdullin, O. Schiemann, Chem. Plus Chem. 85, 353 (2020)
B. Hartke, J. Phys. Chem. 97, 9973 (1993)
M. Fittipaldi, I. García-Rubio, F. Trandafir, I. Gromov, A. Schweiger, A. Bouwen, S. Van Doorslaer, J. Phys. Chem. B 112, 3859 (2008)
S. Spicher, S. Grimme, Angew. Chem. Int. Ed. (2020). https://doi.org/10.1002/ange.202004239
Acknowledgements
Open Access funding provided by Projekt DEAL. The author thanks Prof. Dr. Olav Schiemann for critical discussion of the program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abdullin, D. AnisoDipFit: Simulation and Fitting of Pulsed EPR Dipolar Spectroscopy Data for Anisotropic Spin Centers. Appl Magn Reson 51, 725–748 (2020). https://doi.org/10.1007/s00723-020-01214-0
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-020-01214-0