
Abstract
Background and objective
Coronavirus disease (COVID-19) is an ongoing pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Due to the incessant spread of the disease with substantial morbidity and mortality rates, there is an urgent demand for effective therapeutics and vaccines to control and diminish this pandemic. A critical step in the crosstalk between the virus and the host cell is the binding of SARS-CoV-2 spike protein to the angiotensin-converting enzyme 2 (ACE2) receptor present on the surface of the host cells. Hence, inhibition of this interaction could be a promising strategy to combat the SARS-CoV-2 infection.
Methods
Docking and Molecular Dynamics (MD) simulation studies revealed that designed peptide maintains their secondary structure and provide a highly specific and stable binding (blocking) to SARS-CoV-2.
Results
We have designed a novel peptide that could inhibit SARS-CoV-2 spike protein interaction with ACE2, thereby blocking the cellular entry of the virus.
Conclusion
Our findings suggest that computationally developed inhibitory peptide may be developed as an anti-SARS-CoV-2 agent for the treatment of SARS-CoV-2 infection. We further plan to pursue the peptide in cell-based assays and eventually for clinical trials.
Similar content being viewed by others

1. COVID-19 is an ongoing pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). |
2. SARS-CoV-2 spike protein interacts with the angiotensin-converting enzyme 2 (ACE2) receptor present on the surface of the host cells. |
3. A novel peptide has been designed to inhibit SARS-CoV-2 S-glycoprotein interaction with ACE2, thereby blocking the cellular entry of the virus. |
1 Introduction
The recent severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreak has posed a great challenge to human health. During the past 2 decades, we have encountered the outbreak of many deadly viruses, such as Ebola [1], Zika [2] and Nipah [3, 4], as well as the evolution of various strains of coronaviruses (CoV), mainly SARS-CoV [5] and MERS-CoV [6], which resulted in high morbidity and mortality. After almost 100 years of the deadly influenza virus (H1N1, or Spanish flu) pandemic, with millions of deaths worldwide (approximately 40 million) [7, 8], the recent outbreak of a novel CoV, or SARS-CoV-2, has left the entire world in helplessness and misery. The clinical spectrum of COVID-19 ranges from mild fever, cough and shortness of breath, to severe clinical conditions characterized by respiratory failure [9, 10]. Old age, together with pre-existing conditions such as lung or heart disease, diabetes, or a compromised immune system, expedite the infection time and severity [11, 12]. Multiple recent reviews [13, 14] could brief-up the statistical dynamics of COVID-19 cases worldwide.
Structurally, the CoV has the largest known RNA genome (26–32 Kb) among other known viruses, characterized by non-segmented, positive-sense, single-stranded RNA. This genome encodes four major structural proteins of the virus, including the nucleocapsid (N), envelope (E), membrane (M), and spike (S) proteins [15, 16]. The membrane and envelope proteins are associated with virus assembly, while the spike protein plays the main role in facilitating virus entry via mediating its interaction with the transmembrane surface receptor on the host cells [15, 17]. The spike protein directly interacts with the peptidase domain (PD) of the angiotensin-converting enzyme 2 (ACE2) receptor [18, 19], which technically marks the virus entry inside the cells [20]. The SARS-CoV-2 shares around 80% sequence identity with the SARS-CoV genome, suggesting similarity in their host interacting functions [21, 22]. Like SARS-CoV, the spike protein of SARS-CoV-2 contains S1 and S2 subunits, which are jointly responsible for fusion and entry of the virus [23,24,25] inside the host cells. The receptor-binding domain (RBD) in the S1 subunit initiates direct binding with the ACE2 PD, whereas the S2 subunit contains basic elements needed for the membrane fusion [19, 20, 23, 24].
ACE2, a single-pass type I transmembrane metallocarboxypeptidase enzyme is primarily involved in the maturation of peptide hormone angiotensin (Ang), which in turn regulates the vasoconstriction and blood pressure [19, 26, 27]. ACE2 is primarily expressed in alveolar epithelial type II cells and serves as a viral receptor [28, 29]. Besides alveolar epithelial type II cells, it is also expressed in several extrapulmonary tissues, including the heart, kidney and intestine [26, 30]. The full-length structure of ACE2 consists of two main domains—the N-terminal PD and the collectrin-like domain (CLD) at the C-terminal end [19, 30,31,32]. In fact, the spike glycoprotein of SARS-CoV-2 binds to the homodimer of ACE2, which facilitates virus entry into the host cells [19, 33]. In addition to this, studies have been conducted in association with ACE2 and the amino acid transporter B0AT1 (or Slc6a19), and how SARS-CoV-2 may bind to the ACE2-B0AT1 complex [19, 34]. ACE2 interaction with B0AT1 could aid in producing antivirals or a vaccine that can block CoV infection by targeting ACE2 [35,36,37,38].
With the current epidemiology of SARS-CoV-2, a vaccine might be considered a highly anticipated therapy. However, given that vaccine development and production is a highly challenging and time-consuming task, the need of the hour is to develop potent therapeutic agents that could effectively curb the infection in the early stages. Several approaches, such as decoy-soluble ACE2 proteins, antibodies from the serum of infected patients, epitope-based vaccines, repurposing of drugs, and designing blocking peptides are underway [39,40,41,42,43,44,45,46]. Peptides possess several attractive features when compared with small molecules and protein therapeutics, including high structural compatibility with target proteins, the ability to disrupt protein–protein interfaces, etc.
Computational tools ease the way to reach a therapeutic solution for COVID-19. Currently, the computational analysis of structural differences in human ACE2 impact its binding to the SARS‐CoV‐2 spike protein, which thereby lays a foundation for the design and development of ACE2-based peptide inhibitors of SARS-CoV-2 [47,48,49]. In the current study, we used computational biology tools to develop a therapeutic strategy utilizing a novel peptide by exploiting the SARS-CoV-2/ACE2 interaction.
2 Material and Methods
2.1 Three-Dimensional Structural Investigation
The protein sequence of SARS-CoV-2 was retrieved from the RCSB Protein Data Bank (PDB ID: 6M17) for peptide design and development. Initially, a stretch of 23 amino acid residues (Glu23 to Leu45) was taken from ACE PD (PDB 6M17) and saved as pdb using the ‘build structure’ module in Discovery studio visualizer (BIOVIA, DS, 2019) [50]. This sequence was further optimized for geometry using the ‘minimize’ functionality in the UCSF Chimera software [51].
2.2 Alanine Scanning and Peptide Design
We performed alanine scanning to better understand the individual roles of the residues within the 23-amino acid peptide in binding with SARS-CoV-2. Alanine substitution of each residue was performed using BIOVIA Discovery Studio 2020 software [50]. Each peptide was then modeled and energy minimized using UCSF Chimera software [51]. We also truncated five residues from the N-terminal end of the 23-amino acid (23aa) peptide to get a shorter 18-amino acids peptide (Phe28 to Leu45). This peptide was further energy minimized using the method described above.
2.3 Docking and Molecular Dynamics (MD) Simulation Studies
The crystal structure of the SARS-CoV-2 spike protein-ACE2 (PDB ID: 6M17) complex was retrieved from the Protein Data Bank (https://www.rcsb.org/) and used as the starting point for the docking studies. Molecular docking was performed using PyDock (https://life.bsc.es/pid/pydockweb) [52], HADDOCK 2.4 [53], and ZDOCK [54] servers, to validate the results for each program, which in turn should be mutually correlated. The docking results were analyzed using the Chimera [51] and DS Visualizer programs [50]. The results obtained were analyzed for binding energies and peptide conformations in the SARS-CoV-2 spike protein binding interface. Molecular dynamics (MD) simulations were performed at 50 ns for the modeled protein. The coordinates of the structure were stored and examined utilizing the analytical tool in the GROMACS 4.5.5 package [55].
2.4 Physiochemical Properties of the Peptide
The toxicity and other relevant molecular mechanistic values of the peptides, such as electrostatics, desolation, and Van der Waals (VdW) force, were predicted using the ‘ToxinPred’ server (https://crdd.osdd.net/raghava/toxinpred/) [56]. ToxinPred is an in-silico method that has been developed to predict and design toxic/non-toxic peptides. The main dataset used in this method consists of 1805 toxic peptides (≤ 35 residues). The physiochemical characteristics of the peptide sequences were determined using the ProtParam tool (https://web.expasy.org/protparam/) of the ExPASy database server [57] (Fig. 1).

ACE2 is an entry receptor for SARS-CoV-2. SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, ACE2 angiotensin-converting enzyme 2
3 Results and Discussion
3.1 The N-Terminal Region of the Angiotensin-Converting Enzyme 2 Peptidase Domain is Critical for Binding to the Severe Acute Respiratory Syndrome Coronavirus 2 Spike Protein
The crystal structure of the SARS-CoV-2 spike protein with the ACE2 PD domain (PDB ID: 6M17) was retrieved from the Protein Data Bank (https://www.rcsb.org/). The interface residues between the SARS-CoV-2 spike protein and the ACE2 PD domain were visualized and interpreted using UCSF Chimera Software [51]. After a detailed analysis of interface residues, a small stretch of the ACE2 PD N-terminal region (23-amino acids: Glu23 to Leu45) was found to be interacting majorly with the SARS-CoV-2 spike protein (Fig. 2 and Table 1).

SARS-CoV-2 interaction with the ACE2 PD domain (6M17). The projected view displays the interacting residues at the interface site. SARS-CoV-2 severe acute respiratory syndrome coronavirus 2, ACE2 angiotensin-converting enzyme 2, PD peptidase domain
Furthermore, we evaluated whether the 23-amino acid chain alone and without the remainder of the ACE2 PD domain could bind to the SARS-CoV-2 spike protein. Hence, the 23aa sequence was taken from the ACE2 PD domain and docked to the SARS-CoV-2 spike protein (6M17) using the PyDock program [52]. To perform a non-biased analysis, we performed a blind docking run whereby we did not specify the binding site during the docking simulations. The obtained results were clustered and analyzed by comparing the binding energies and docked conformations of the peptide within the SARS-CoV-2 spike protein (Table 2). Interestingly, we found that the peptide binds to the SARS-CoV-2 spike protein at exactly the same place where the original ACE2 PD domain interacts, which shows that the 23-amino acid sequence independently has the potential to inhibit the interaction of the SARS-CoV-2 spike protein and ACE2 complex. After the docking analysis, the SARS-CoV-2/ACE2 peptide interface was determined and the critical interacting amino acids were identified in the ACE2 PD domain-derived peptide involved in binding to the SARS-CoV-2 spike protein (Table 2).
3.2 Computational Alanine Scanning Analysis of the Peptide
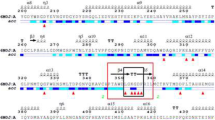
Computational alanine (A) scanning was performed to identify the critically important amino acids of the 23aa peptide inhibitor involved in binding to the SARS-CoV-2 spike protein. The approach of alanine scanning contributes to estimating the binding energy of each residue to the total binding energy of the original peptide [58]. The process involves the substitution of the amino acid with alanine and records the resulting binding energy changes by molecular docking of the substituted three-dimensional structure of the peptide. If substitution by alanine results in a significant drop in overall free binding energy, those residue(s) are considered the critically important residues for binding [59]. Hence, we performed alanine scanning of the 23aa peptide to determine the significance of each residue having the inhibitory potential of independently interacting with the ACE2 binding region of the SRAS-CoV-2 spike protein. All amino acids in the peptide were independently mutated to alanine using BIOVIA Discovery Studio 2020 software [50]. Alanine-substituted peptides were then modeled and minimized using the UCSF Chimera program [51]. Next, we performed docking studies of each mutated peptide and analyzed their binding energies and three-dimensional conformations in the ACE2 binding interface of the SARS-CoV-2 spike protein. As seen in Fig. 3, we observed that the mutated residues E23A, Q24A, A25A, K26A, T27A, L29A, D30A, K31A, N33A, E35A, A36A, E37A, D38A, L39A, Q42A, S43A, and S44A have no substantial effect on complex stability in terms of the change in total binding energy. The total binding energy of the 23aa peptide with the SARS-CoV-2 spike protein (− 55.873 kcal/mol) is comparable to the binding energy of the mutated residues (E23A, Q24A, A25A, K26A, T27A, L29A, D30A, K31A, N33A, E35A, A36A, E37A, D38A, L39A, Q42A, S43A, S44A), ranging from − 56 to − 53 kcal/mol (Fig. 3). However, the mutated residues F28A, F32A, H34A, F40A, Y41A, and L45A were found to be critically important for binding and stabilizing the peptide-SARS-CoV-2 spike protein complex (Fig. 3). We observed the change from − 55.873 kcal/mol to − 42.996 drop in the total binding score (Fig. 3). Furthermore, we deleted the non-significant (identified through alanine scanning) residues and found the deletion of any residue from F28 to L45 would make an unstable structure. Hence, we removed five residues (E23, Q24, A25, K26, T27) from the N-terminal region of the peptide and retained residues from F28 to L45, resulting in the final 18aa peptide. In addition to the molecular mechanistic values such as electrostatics and desolation, we also calculated the van der Waals (vdW) of all individual peptides (electronic supplementary Table 1). vdW is usually a weak force, a type of intermolecular force enhancing the attraction and binding of a ligand to its receptor. VdW forces are involved in the maintenance and stabilization of a drug-receptor complex.

Identification of critically important residues in the 23aa peptide by alanine scanning, and the binding energies of each peptide docked with SARS-CoV-2 spike protein predicted by pyDockWEB. The alanine scanning results provide the 18aa new peptide inhibitor with similar binding efficiency
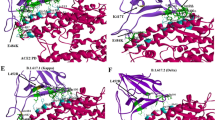
Replacing the respective amino acids (F28, F32, H34, F40, Y41, L45) with alanine significantly reduces the binding score, either by directly losing the interaction or by changing the peptide conformation, which becomes less compatible with the SARS-CoV-2 spike protein. Hence, based on the results of alanine scanning, the peptide was shortened from the extreme N-terminus due to its dispensability in SARS-CoV-2 binding. The final peptide derivative (F28, L29, D30, K31, F32, N33, H34, E35, A36, E37, D38, L39, F40, Y41, Q42, S43, S44, L45) was further docked and analyzed to validate its binding efficiency. We utilized three state-of-the-art programs (pyDock [37], ZDOCK [39] and HADDOCK 2.4 [38]) to validate our docking studies. The top poses retrieved from each software package resulted in conformations very close to the original 23-amino acid peptide, thereby validating its efficacy as a potent SARS-CoV-2 inhibitor (Fig. 4).

SARS-CoV-2 (Cyan) interaction with the 18aa-derived peptide (blue) along with the original peptide of 23-amino acid (red). Dockings were performed using different software packages: a pyDock; b ZDOCK; and c HADDOCK 2.4. SARS-CoV-2 severe acute respiratory syndrome coronavirus 2
3.3 Physiochemical Properties of the Peptide
The final 18aa peptide, with a molecular weight of 2203.39, has an amino acid composition of Ala (A) 5.6%, Asn (N) 5.6%, Asp (D) 11.1%, Gln (Q) 5.6%, Glu (E) 11.1%, His (H) 5.6%, Leu (L) 16.7%, Lys (K) 5.6%, Phe (F) 16.7%, Ser (S) 11.1% and Tyr (Y) 5.6%. The total number of negatively charged residues was four (Asp and Glu) and the total number of positively charged residues was two (Lys and His). The extinction coefficient calculated in units of M−1 cm−1, at 280 nm measured in water, was found to be 1490 ± 10. It is useful to have an estimation of this coefficient for spectrophotometrically following a protein when purifying it. The half-life is a prediction of the time it takes for half of the amount of protein in a cell to disappear after its synthesis in the cell. The presently used ExPASy tool, ProtParam, relies on the ‘N-end rule’, which relates the half-life of a protein to the identity of its N-terminal residue; the prediction is given for three model organisms (human, yeast and Escherichia coli). Considering the N-terminal of the sequence considered is F (Phe), the estimated half-life is 1.1 h (mammalian reticulocytes, in vitro). The instability index (II) is computed to be 68.65, which classifies the protein as unstable. The physical stability of peptides is influenced by both intrinsic and external factors. Prediction of peptide stability suggests precautionary steps be taken to make the therapeutically potent unstable peptides stable through certain biochemical analysis. The predicted aliphatic index of the peptide was found to be 70.56, with a grand average of hydropathicity (GRAVY) of − 0.522. ToxinPred was used to predict the toxicity of the 18aa peptide with desired toxicity by mutating the minimum number of amino acids [40]. The ‘ToxinPred’ tool allows us to submit query peptide in the FASTA format and to optimize the peptide sequence to obtain the maximum/minimum/desired toxicity based on the quantitative matrix-based position-specific scores by comparing them with the toxic/non-toxic data set. The in-silico predicted ‘ToxinPred’ toxicity results of the 18aa peptide indicate that it is non-toxic compared with the mutated peptides.
3.4 MD Simulations
The respective docked complexes of the 23aa and 18aa peptides with target protein were subjected to MD simulation studies for analysing their stability in terms of root mean square deviation (RSMD) and potential interactions at 50 ns. The stability of these two protein–peptide complexes was analyzed using the RMSD of backbone atoms. The calculated average RMSD of these complexes was in the range of 0.94–1.48 nm and 0.24–0.8 nm for the 23aa and 18aa peptide complexes, respectively (Fig. 5).

a Plots of backbone RMSD of the 23aa and 18aa peptide, shown in magenta and green, respectively. b The 23aa and 18aa peptides complexed with SARS-CoV-2. RMSD root mean square deviation, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2
By the end of the 50 ns time-scale simulation, both of the protein–peptide complexes attained their stable conformations (Fig. 5). The lower RMSD values obtained after comparing the initial and final peptide complex conformations (Fig. 5b) revealed that the docked complexes were stable and hence there was not much difference in their conformations during the course of dynamics simulation.
4 Conclusions
Several advantages of peptides, including ease of synthesis and modifications, low toxicity, and high target specificity and selectivity, led us to design a potential therapeutic candidate against COVID-19. The present in-silico study is the first step towards designing inhibitory peptides to block SARS-CoV-2 entry into the host cell. The peptide could inhibit binding of the SARS-CoV-2 spike protein with the ACE2 PD domain, which is the earliest stage of SARS-CoV-2 infection. The findings of this study suggest that a peptide of 18-amino acids could be considered as a selective therapeutic candidate for COVID-19. The study conducted by Han and Kral shows similar aspects towards designing a peptide inhibitor [49]. The identification of 15 interacting residues of an ACE2 PD with an RBD of the SARS-CoV-2 spike protein at a 3 Å region in the crystal structure (PDB 6M17) is in agreement with the findings of Han and Kral [49]. However, in comparison with our structurally stable 18aa peptide inhibitor comprising the ACE2 PD residues ranging from positions 28–45, their inhibitor 1 peptide comprising residues 21–55 is structurally unstable and non-specific; hence, they made multiple modifications to it, which thereby changes its properties as well as its similarity to our peptide. Out of a total of 15 interacting residues of ACE2 PD, 10 residues are in the stretch of Q24 to L45. Utilizing this fact in our study, each residue in the 23–45 stretch (23aa peptide) of the ACE2 PD was estimated for functional significance and structural stability based on their binding energy and conformational stability. In contrast to inhibitor 1, we obtained a stable 18aa peptide inhibitor where non-significant residues were truncated and the non-interacting residues within the 18aa peptide at the interfacial site were found important, both for binding and stability of the peptide. Hence, inhibitor 1 from Han and Kral differs in structural stability, composition and specificity due to the presence of non-significant residues leading to deformity in its helical structure. Provision of experimental investigations of the desired synthesized peptide in combination with our computational studies will be needed to confirm and complete this study. It is recommended to compare our computational results with the designed peptides in vitro and in vivo, and eventually to clinical studies.
References
Lewnard JA. Ebola virus disease: 11 323 deaths later, how far have we come? Lancet. 2018;392(10143):189–90.
Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, et al. Zika virus: history, emergence, biology, and prospects for control. Antiviral Res. 2016;130:69–80.
Kulkarni DD, Tosh C, Venkatesh G, Senthil KD. Nipah virus infection: current scenario. Indian J Virol. 2013;24(3):398–408.
Chatterjee P. Nipah virus outbreak in India. Lancet. 2018;391(10136):2200.
Anderson RM, Fraser C, Ghani AC, Donnelly CA, Riley S, Ferguson NM, et al. Epidemiology, transmission dynamics and control of SARS: the 2002–2003 epidemic. Philos Trans R Soc Lond B Biol Sci. 2004;359(1447):1091–105.
Oboho IK, Tomczyk SM, Al-Asmari AM, Banjar AA, Al-Mugti H, Aloraini MS, et al. 2014 MERS-CoV outbreak in Jeddah: a link to health care facilities. N Engl J Med. 2015;372(9):846–54.
Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 "Spanish" influenza pandemic. Bull Hist Med. 2002;76(1):105–15.
Taubenberger JK, Morens DM. 1918 Influenza: the mother of all pandemics. Emerg Infect Dis. 2006;12(1):15–22.
Wu YC, Chen CS, Chan YJ. The outbreak of COVID-19: an overview. J Chin Med Assoc. 2020;83(3):217–20.
Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Lee SS. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): diagnostic and proposed therapeutic options. Eur Rev Med Pharmacol Sci. 2020;24(7):4016–26.
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R. Features, evaluation and treatment coronavirus (COVID-19). Treasure Island: StatPearls; 2020.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395(10223):497–506.
Kissler SM, Tedijanto C, Goldstein E, Grad YH, Lipsitch M. Projecting the transmission dynamics of SARS-CoV-2 through the postpandemic period. Science. 2020;368(6493):860–8.
Uddin M, Mustafa F, Rizvi TA, Loney T, Al Suwaidi H, Al Marzouqi A, et al. SARS-CoV-2/COVID-19: Viral Genomics, Epidemiology, Vaccines, and Therapeutic Interventions. 2020
Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol Biol. 2015;1282:1–23.
Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92(4):424–32.
Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol. 2016;3(1):237–61.
Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309(5742):1864–8.
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–8.
Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–80e8.
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–3.
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–74.
Tai W, He L, Zhang X, Pu J, Voronin D, Jiang S, et al. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cellular & molecular immunology. Epub 19 Mar 2020. doi: 10.1038/s41423-020-0400-4
Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir Res. 2020;176:104742.
Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 Spike glycoprotein. Cell. 2020;181(2):281–92e6.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–90.
Donoghue M, Wakimoto H, Maguire CT, Acton S, Hales P, Stagliano N, et al. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J Mol Cell Cardiol. 2003;35(9):1043–53.
Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, thereceptor of SARS-CoV-2. bioRxiv; 2020. https://www.biorxiv.org/content/10.1101/2020.01.26.919985v2
Uhal BD, Dang M, Dang V, Llatos R, Cano E, Abdul-Hafez A, et al. Cell cycle dependence of ACE-2 explains downregulation in idiopathic pulmonary fibrosis. Eur Respir J. 2013;42(1):198–21010.
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417(6891):822–8.
Zhang H, Wada J, Hida K, Tsuchiyama Y, Hiragushi K, Shikata K, et al. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem. 2001;276(20):17132–9.
Zisman LS, Keller RS, Weaver B, Lin Q, Speth R, Bristow MR, et al. Increased angiotensin-(1–7)-forming activity in failing human heart ventricles: evidence for upregulation of the angiotensin-converting enzyme Homologue ACE2. Circulation. 2003;108(14):1707–12.
Zhou Q, Yan R, Zhang Y, Li Y, Xia L. Structure of dimeric full-length human ACE2 in complex with B0AT1. BioRxiv. 2020. https://www.biorxiv.org/content/10.1101/2020.02.17.951848v1.full.
Kowalczuk S, Broer A, Tietze N, Vanslambrouck JM, Rasko JE, Broer S. A protein complex in the brush-border membrane explains a Hartnup disorder allele. FASEB J. 2008;22(8):2880–7.
Jando J, Camargo SMR, Herzog B, Verrey F. Expression and regulation of the neutral amino acid transporter B0AT1 in rat small intestine. PLoS ONE. 2017;12(9):e0184845.
Bohmer C, Broer A, Munzinger M, Kowalczuk S, Rasko JE, Lang F, et al. Characterization of mouse amino acid transporter B0AT1 (slc6a19). Biochem J. 2005;389(Pt 3):745–51.
Hashimoto T, Perlot T, Rehman A, Trichereau J, Ishiguro H, Paolino M, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature. 2012;487(7408):477–81.
Camargo SM, Singer D, Makrides V, Huggel K, Pos KM, Wagner CA, et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology. 2009;136(3):872–82.
Procko E. The sequence of human ACE2 is suboptimal for binding the S spike protein of SARS coronavirus 2. bioRxiv. 2020. https://www.biorxiv.org/content/10.1101/2020.03.16.994236v1.
Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181(4):905–13.
Li Z, Yi Y, Luo X, Xiong N, Liu Y, Li S, et al. Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. Journal of medical virology. Epub 27 Feb 2020. 10.1002/jmv.25727.
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020;6:14.
Chen YW, Yiu CB, Wong KY. Prediction of the SARS-CoV-2 (2019-nCoV) 3C-like protease (3CL (pro)) structure: virtual screening reveals velpatasvir, ledipasvir, and other drug repurposing candidates. F1000Research. 2020;9:129.
Gurwitz D. Angiotensin receptor blockers as tentative SARS‐CoV‐2 therapeutics. Drug Dev Res. Epub 4 Mar 2020. doi: 10.1002/ddr.21656.
Robson B. Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput Biol Med. 2020;119:103670.
Bhattacharya M, Sharma AR, Patra P, Ghosh P, Sharma G, Patra BC, et al. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J Med Virol. 2020;92(6):618–31.
Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. Epub 6 Apr 2020. doi: 10.1002/jmv.25832.
Pant S, Singh M, Ravichandiran V, Murty USN, Srivastava HK. Peptide-like and small-molecule inhibitors against Covid-19. J Biomol Struct Dyn. Epub 6 May 2020. doi: 10.1080/07391102.2020.1757510.
Han Y, Kral P. Computational design of ACE2-based peptide inhibitors of SARS-CoV-2. ACS Nano. 2020;14(4):5143–7.
Systèmes DB. Discovery Studio Modeling Environment, Release 2019. Dassault Systèmes; 2019.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera: a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12.
Jimenez-Garcia B, Pons C, Fernandez-Recio J. pyDockWEB: a web server for rigid-body protein-protein docking using electrostatics and desolvation scoring. Bioinformatics. 2013;29(13):1698–9.
van Zundert GC, Bonvin AM. Modeling protein-protein complexes using the HADDOCK webserver "modeling protein complexes with HADDOCK". Methods Mol Biol. 2014;1137:163–79.
Chen R, Li L, Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52(1):80–7.
Pronk S, Pall S, Schulz R, Larsson P, Bjelkmar P, Apostolov R, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–54.
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Open Source Drug Discovery C, et al. In silico approach for predicting toxicity of peptides and proteins. PloS One. 2013;8(9):e73957.
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook. New York: Springer; 2005. p. 571–607.
Morrow JK, Zhang S. Computational prediction of protein hot spot residues. Curr Pharm Des. 2012;18(9):1255–65.
DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12(1):14–20.
Acknowledgements
The authors also gratefully acknowledge the Indian Institute of Technology Indore (IITI) for providing facilities and other support. The authors are thankful to BIOVIA-Dassault Systemes for providing SARS-CoV-2/2020 Discovery Studio Academic Research Suite License.
Author information
Authors and Affiliations
Contributions
MSB conceived, designed, and executed the research. MA, US, and SR compiled and analyzed the data. MSB has written the manuscript. US reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Funding
This study is supported by the Indian Institute of Technology Indore (IITI) India.
Conflict of Interest
MSB, MA, SR and US declare no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Baig, M.S., Alagumuthu, M., Rajpoot, S. et al. Identification of a Potential Peptide Inhibitor of SARS-CoV-2 Targeting its Entry into the Host Cells. Drugs R D 20, 161–169 (2020). https://doi.org/10.1007/s40268-020-00312-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40268-020-00312-5