
Abstract
The potential inhibitory effect of diverse triazolyl-ferrocene steroids on key enzymes of the estrogen biosynthesis was investigated. Test compounds were synthesized via copper-catalyzed cycloaddition of steroidal azides and ferrocenyl-alkynes using our efficient methodology published previously. Inhibition of human aromatase, steroid sulfatase (STS) and 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) activities was investigated with in vitro radiosubstrate incubations. Some of the test compounds were found to be potent inhibitors of the STS. A compound bearing ferrocenyl side chain on the C-2 displayed a reversible inhibition, whereas C-16 and C-17 derivatives displayed competitive irreversible binding mechanism toward the enzyme. 17α-Triazolyl-ferrocene derivatives of 17β-estradiol exerted outstanding inhibitory effect and experiments demonstrated a key role of the ferrocenyl moiety in the enhanced binding affinity. Submicromolar IC50 and Ki parameters enroll these compounds to the group of the most effective STS inhibitors published so far. STS inhibitory potential of the steroidal ferrocenes may lead to the development of novel compounds able to suppress in situ biosynthesis of 17β-estradiol in target tissues.
Similar content being viewed by others
Introduction
The ferrocene substituent features exceptional biomedical properties. Ferrocene-cinchona hybrids containing a triazole linker were shown to exert antiproliferative effect on HepG2 hepatoma and HT-29 colorectal adenocarcinoma human tumor cell lines [20]. Triazole-linked isatin-ferrocene conjugates exhibited selectivity against the hormone-dependent (MCF-7) cell line [42]. Steroid-ferrocene hybrids are believed to have a vast potential for medical application [2, 23, 27, 46]. Steroidal ferrocenes were aimed mainly to be applied for vectorization of the DNA damaging effect of the ferrocene entity to produce cytotoxic effect in cancerous cells [18]. C-2, C-16 and C-17 ferrocene derivatives of androgens as well as C-3, C-7, C-16 and C-17 ferrocene derivatives of 17β-estradiol and estrone demonstrated considerable antiproliferative effects (micromolar IC50 or GI50 values) against hormone-independent cancerous cell lines [18, 23, 49, 51, 53]. Concurrently, hormonal effects exerted on the specific sexual steroid receptors were also investigated. Low affinities to the androgen receptor were detected for the ferrocene derivatives of androgens [2, 53]. The C-7 and C-17 ferrocene derivatives of 17β-estradiol displayed estrogen receptor-binding and estrogenic effect comparable to those of 17β-estradiol [31, 52], whereas the C-3 derivative has a predicted estrogen receptor antagonistic effect [51]. Potential inhibitory effect of the ferrocene steroid compounds exerted on enzymes of the steroid metabolism has not yet been investigated, to the best of our knowledge.
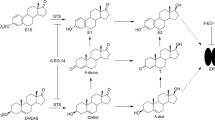
Estrogens are biosynthesized from their steroidal precursors via aromatization of their ring A (Fig. 1). The process is catalyzed by the cytochrom P450-dependent aromatase (EC 1.14.14.1). This enzyme converts mainly androst-4-ene-3,17-dione and a minor amount of testosterone into estrone and 17β-estradiol, respectively. The estrogen reservoir estrone-3-sulfate is formed from estrone, which can be then regenerated with a hydrolysis catalyzed by steroid sulfatase (STS, EC 3.1.6.2). Estrone is converted to the active hormone 17β-estradiol by the stereospecific reduction in the C-17 oxo function. This reaction is accomplished by 17β-hydroxysteroid dehydrogenases type 1 (17β-HSD1, EC 1.1.1.62), an enzyme belonging to the short-chain alcohol dehydrogenase superfamily [19]. Estrogens have a major role in the development and progression of gynecological pathologies, with hormone-dependent forms of breast cancer among them. Breast cancer tissues have been shown to express enzymes of the estrogen producing machinery in an exaggerated manner [1]. The increased expression resulted in elevated local 17β-estradiol concentration, which is believed to promote the pathophysiological cell proliferation. Estrogen ablation achieved by blocked biosynthesis can be successfully applied in the pharmacotherapy of the hormone-dependent tumors; therefore, aromatase, STS and 17β-HSD1 are interesting topics of anticancer drug development [19, 47]. The three-dimensional crystal structures of human aromatase, STS and 17β-HSD1 have been explored, and binding cavities and key amino acid residues involved in the binding have been identified [14, 15, 17]. These investigations provided valuable information on the molecular basis of substrate-binding and catalytic mechanisms; however, they proved to be less useful in the inhibitor design [19]. Majority of the inhibitors of enzymes of the estrogen biosynthesis have been developed by ligand-based approach. Experimental testing and biochemical analysis of inhibition of novel compounds are, therefore, still a feasible way of inhibitor development.

Key enzymes of the estrogen biosynthesis. Aromatase (EC 1.14.14.1). 17β-HSD1: 17β-hydroxysteroid dehydrogenases type 1 (EC 1.1.1.62). STS: steroid sulfatase (EC 3.1.6.2). 17β-HSD3: 17β-hydroxysteroid dehydrogenases type 3 (EC 1.1.1.64), not investigated. 17β-HSD5: 17β-hydroxysteroid dehydrogenases type 5 (EC 1.1.1.239), not investigated
In this study, we aimed to investigate the inhibitory effects of diverse triazolyl-ferrocene steroids exerted on the activity of key enzymes of the estrogen biosynthesis. The triazoles were obtained by copper-catalyzed azide-alkyne cycloaddition using steroidal azides and ferrocene-substituted alkynes or ferrocenyl azides and alkynyl steroids as substrates. We investigated one newly synthesized ferrocenyl steroid as well as other derivatives that were prepared previously, and the details were reported in former publications [11, 44, 45]. A related phenyl-triazolyl compound (Fehér et al. [12] and cited literature therein) and two non-ferrocenyl steroid precursors [44] were also included in the present experiments for the investigation of structure–activity aspects.
Materials and methods
Test substances
The test substances were re-synthesized according to the procedures described previously.
1—2β-(4-Ferrocenyl-1,2,3-triazol-1-yl)-3α-hydroxy-5α-androstane-17-one [11].
2—2β-(4-((E)-Ethyl 3-ferrocenylacrylate-2-methylcarbamoyl))-1,2,3-triazol-1-yl)-3α-hydroxy-5α-androstan-17-one [11].
3—6β-(4-((E)-Ethyl 3-ferrocenylacrylate-2-methylcarbamoyl)-1,2,3-triazol-1-yl)-3α,5α-dihydroxy-androstan-17-one [11].
4—16β-(4-Ferrocenyl-1,2,3-triazol-1-yl)-17α-hydroxy-5α-androstane [11].
5—16β-(4-((E)-Ethyl 3-ferrocenylacrylate-2-methylcarbamoyl)-1,2,3-triazol-1-yl)-17α-hydroxy-5α-androstane [11].
6—17-(N-(1-(1-ferrocenyl-ethyl)-1,2,3-triazol-4-yl)-methyl-carbamoyl)-4-aza-5α-androst-16-en-3-one, 1/1 mixture of two epimers [44].
7—17-(N-(prop-2-ynyl)-carbamoyl)-4-aza-5α-androst-16-en-3-one [44].
8—17-(N-(1-(1-ferrocenyl-ethyl)-1,2,3-triazol-4-yl)-methyl-carbamoyl)-3-methoxy-estra-1,3,5(10),16-tetraene, 1/1 mixture of two epimers [44].
9—17-(N-(prop-2-ynyl)-carbamoyl)-3-methoxy-estra-1,3,5(10),16-tetraene [44].
10—17α-(1-Ferrocenylmethyl-1H-1,2,3-triazol-4-yl)estradiol [12].
11—17α-(1-(1-Ferrocenyl-ethyl)-1,2,3-triazol-4-yl)-3,17β-dihydroxy-estra-1,3,5(10)-triene, 1/1 mixture of two epimers [45].
12—17α-(1-Ferrocenylmethyl-1H-1,2,3-triazol-4-yl)-17β-hydroxy-18a-homo-19-nor-androst-4-ene-3-one.
Compound 12 was synthesized via reaction of levonorgestrel and azidomethylferrocene and was purified by column chromatography. Synthesis method in detail and characterization data are described in Supplementary Material.
13—17α-(1-Benzyl-1H-1,2,3-triazol-4-yl)estradiol [12].
Enzyme experiments
Activity and inhibition of the aromatase, 17β-HSD1 and STS enzymes were assessed with radioincubation methods described in detail in our previous publications [3, 16]. These methods are based on adapted literature techniques and used human placental microsoma suspension and cytosol as source of the enzymes (references: for aromatase [48]; for 17β-HSD1 [50]; and for STS [21]. Incubation parameters were optimized in preliminary experiments. Enzyme reactions applied for the measurement of inhibitory effect satisfied conditions of the initial velocity measurements. Conversion rate in the non-inhibited control incubates was between 10 and 13%, and product formation was proportional to the enzyme concentration and the incubation time. Here, we provide short descriptions of methods applied in the enzyme experiments. (Chemicals and materials as well as the preparation of cytosol and microsomal fractions are described in Supplementary Material.)
Incubation procedures and measurement of inhibitory effect
Enzymatic incubations were carried out in 0.1 M HEPES buffer medium at a final volume of 200 µl. The substrate was added to incubate in 20 µl of 25 v/v % propylene glycol in HEPES buffer solution, whereas test compounds were applied in 10 µl of dimethyl sulfoxide solution. Incubations were performed at 37 °C and terminated by cooling and by the addition of organic solvents of the subsequent extraction procedure. The radioactivity of the isolated enzyme products was measured by means of liquid scintillation counting. Control samples with no inhibitor and blank samples were incubated simultaneously.
In incubates of the inhibition studies, substrate concentration was 1.0 µM, and test compounds were applied at 10 µM. Relative conversions compared to non-inhibited controls (100%) were determined. The assays were performed in triplicate, and the mean value and the standard deviation (SD) were calculated.
IC50 values (the inhibitor concentration that decreases the enzyme activity to 50%) were determined for the most effective test compounds and for certain reference compounds. In these cases, conversions were measured at 10–15 different concentrations in the appropriate interval between 0.001 and 50 µM. IC50 results were calculated by using unweighted iterative least squares logistic curve fitting by means of the “absolute IC50 calculation” function of the GraphPad Prism 4.0 software (GraphPad Software, Inc., San Diego, CA, USA).
Aromatase assay
In the aromatase inhibition assays, the human placental microsoma suspension (5 µl) was incubated with testosterone substrate spiked with [1,2,6,7-3H]testosterone tracer, in the presence of 0.1 mM NADPH cofactor excess. Incubation time was 40 min. The incubation mixture was extracted with 700 µl toluene, and the toluene phase was drained and washed with equal volume of HEPES buffer. Aromatase products containing phenolic hydroxy group were extracted with 1.2 M sodium hydroxide solution from the toluene extract.
17β-HSD1 assay
In the 17β-HSD1 inhibition assays, cytosol fraction of human placenta (50 µl of 25-fold diluted) was incubated using estrone substrate containing [6,7-3H]estrone tracer, in the presence of 0.1 mM NADPH cofactor excess. The incubation time was 2.5 min. The product 17β-estradiol was extracted with 100 µl ethyl acetate and isolated by a TLC method performed on silica gel plate and with dichloromethane/diisopropyl ether/ethyl acetate (75:15:10 v/v) eluent.
STS assay
STS assays were performed using human placental microsomas (10 µl of 100-fold diluted) and estrone-3-sulfate substrate with [6,7-3H]estrone-3-sulfate tracer. Incubations lasted for 20 min. Reactions were terminated by the addition of equal volume of methanol, and the product 17β-estradiol was extracted with 400 µl toluene.
Reversibility studies and kinetic analyses of STS inhibitors
In the reversibility studies, inhibitor compounds were preincubated with the human microsomal placenta fraction in a volume of 4.0 µl at 37 °C for 2.5 or 20 min. Following this procedure, samples were diluted with the incubation medium to 50-fold. Enzyme activity measurements were taken after 20 min secondary incubation time to allow dissociation. The enzymatic reactions were started by the addition of the substrate (1.0 µM final concentration of estrone-3-sulfate with tracer), and subsequent incubation procedures were then carried out as described. Conversions were compared to their respective controls, which were treated in a similar way, but without inhibitors.
Kinetic analyses were performed via measurement of the enzymatic transformation using different fixed substrate concentrations (1–5 µM) and varied inhibitor concentrations (0–0.17 µM). The KM parameter of the STS was calculated from Michaelis–Menten analysis (non-inhibited reaction velocity vs. substrate concentration) using the GraphPad Prism 4.0 software. The Dixon’s linear transformation (1/velocity vs. inhibitor concentration) was applied for evaluation, and the inhibition constant (Ki) was determined from abscissa of intersection of the lines of different substrate concentrations. Mechanism of binding was identified according to the Dixon’s graphs and the secondary plot of slopes of the Dixon’s lines vs. 1/substrate concentration.
Results
Enzyme inhibition experiments of the steroidal ferrocenes were performed by in vitro radiosubstrate incubations using human term placental microsoma suspension and cytosol as enzyme sources ([3, 16] and corresponding references therein). Aromatase inhibition was tested by the transformation of testosterone to 17β-estradiol, and STS inhibition was investigated via hydrolytic release of estrone from estrone-3-sulfate, whereas the influence on 17β-HSD1 was measured on estrone to 17β-estradiol conversion. Relative conversions compared to non-inhibited controls (100%) were measured in the presence of 10 μM concentration of the test compounds; for more efficient compounds, IC50 values were determined. Inhibitory potentials and binding affinities of the test compounds were assessed also in comparison with IC50 data of the corresponding substrate. IC50 parameters of 17β-estradiol and estrone-3-sulfamate (EMATE) as references were also determined in our STS assay. (Results are listed in Table 1.) Mechanistic and kinetic investigations were performed, and inhibitory constants (Ki) were determined for selected compounds.
Aromatase inhibition results of the test compounds
In our experiments, substances 2, 6 and 8 in the applied 10 μM test concentration suppress aromatase activity to around 80% of the relative conversions (Table 2). These results mean IC50 values exceeding 10 μM and indicate very weak inhibitory potentials. Other investigated compounds have even weaker effects, resulting in relative conversions close to 90% or higher.
17β-HSD1 inhibition results of the test compounds
Our test substances 6 and 11 inhibited 17β-HSD1 activity to relative conversions 65% and 76%, respectively (Table 2). These results show weak inhibitions even in the relatively high 10 μM test concentration. Other test compounds displayed even weaker effects, since relative conversions were not decreased below 80% in the experiments where they were applied.
STS inhibition results of the test compounds
In our experiments, the 3α-hydroxy-5α-androstane-17-one compound derivatized with triazolyl-ferrocene in position 2β (1) exerts potent inhibition, and the IC50 result 4.6 μM (Tables 2 and 3) reveals a binding affinity similar to that of the substrate estrone-3-sulfate (IC50 result 5.2 μM). The insertion of an ethylacrylate group in the side chain of the compound 2 diminishes the inhibitory effect almost completely. Another 3α-hydroxy-5α-androstane-17-one derivative bearing a similar elongated ferrocenyl side chain in position 6β and additionally a 5α-hydroxy group (3) is found to be also ineffective against STS. The 17α-hydroxy-5α-androstane compound with 16β-triazolyl-ferrocene (4) has a weak effect, but its counterpart 5 bearing the ethylacrylate elongated triazolyl-ferrocene side chain is a potent inhibitor. IC50 value of this compound (2.4 μM) indicates a twofold affinity compared to the substrate estrone-3-sulfate. Substances 6 and 8 display relative conversion results 87% and 68%, respectively. These values indicate that either isomers of the substituted 4-aza-3-oxo-androstene and the 3-methoxy-estratetraene compounds are very weak inhibitors. These triazolyl-ferrocene-derivatized substances, nevertheless, exert somewhat more potent inhibition than their carbamoyl-propene precursor compounds 7 and 9 (relative conversions are 93% and 75%, respectively).
The 17α-triazolyl-ferrocene derivative of 17β-estradiol (10) exerts highly potent inhibition. IC50 value of this compound is 0.084 μM, indicating a more than 60-fold stronger binding compared to the substrate estrone-3-sulfate (Table 2 and Fig. 2). Substance 11 (consists of the methyl-substituted isomer counterparts) displays an even more potent inhibition, and its apparent IC50 value of 0.035 μM is close to that of the reference EMATE (0.0098 μM). A related ferrocenyl compound 12 which possesses 4-ene-3-oxo structure in its ring A and an ethyl group on the C-13 has a very weak effect with a relative conversion result of 77%. The analogue compound 13 bearing phenyl group attached to the triazole ring has also potent inhibitory effect showing an IC50 result of 1.0 μM.

Concentration-dependent STS inhibition of reference EMATE and compounds 10 and 11. Inhibited relative conversion results (Rel. conv.) vs. inhibitor concentration. IC50 value, the inhibitor concentration which decreases the enzyme activity to 50%, was calculated by using unweighted iterative least squares logistic curve fitting
Potent test substances (1, 5, 10 and 11) and the reference competitive irreversible inhibitor EMATE were subjected to mechanistic and kinetic investigations. In reversibility tests performed with 1, the preincubated and diluted samples resulted in higher conversions (Fig. 3, conditions C and D), similar to the results measured for lower concentration of the test compound (Fig. 3, conditions A). This means that 1 can be released from binding by dilution and this inhibitor binds to the enzyme in a reversible manner. In case of 5, 10 and 11, results of the preincubated and diluted samples (Fig. 3, conditions C and D) were similar to that obtained for incubations with higher concentration of the inhibitor (Fig. 3, conditions B). This means that these inhibitors are not released from binding upon dilution and they are bound to the enzyme irreversibly during the preincubation period. Time course experiments reveal that the binding processes are completed within 2.5 min, in a relatively short period when compared to the time of incubation.

Investigation of reversibility of STS inhibition. Effect of the potent test substances 1, 5, 10 and 11 and the reference competitive irreversible inhibitor EMATE were assessed by dilution method. Placental microsomes were preincubated in samples of A and B in the absence of inhibitors, whereas preincubations were performed in samples of C and D with high inhibitor concentrations (2–5xIC50). Following preincubation, samples were diluted 50-fold. Samples A were then supplemented with high concentration of inhibitors (2–5xIC50), whereas 50-fold lower concentrations of the inhibitors (0.04–0.1xIC50) were added to the samples B. Inhibited activities of STS were subsequently measured by the usual radioincubation method. Samples C and D are different in time of preincubation (2.5 min and 20 min, respectively). Each column represents mean ± SD of three separate experiments. Compound 1 acts by a reversible process, as the STS activities in the samples C and D correspond to results obtained for the sample A containing low inhibitor concentration. STS activities in the samples C and D of 5, 10 and 11 and EMATE correspond to results obtained for the sample B containing high inhibitor concentration; therefore, these compounds behave as irreversible inhibitors. Experimental conditions (other details are given in Enzyme experiments part of the Materials and methods): A Preincubation in the absence of inhibitor. STS measurement with lower inhibitor conc.: EMATE 0.001 μM; 1, 5 0.2 μM; 10, 11 0.01 μM. B Preincubation in the absence of inhibitor. STS measurement with higher inhibitor conc.: EMATE 0.05 μM; 1, 5 10 μM; 10, 11 0.5 μM. C Preincubation for 2.5 min with higher inhibitor conc. EMATE 0.05 μM; 1, 5 10 μM; 10, 11 0.5 μM, STS measurement with lower inhibitor conc.: EMATE 0.001 μM; 1, 5 0.2 μM; 10, 11 0.01 μM. D Preincubation for 20 min, with higher inhibitor conc. EMATE 0.05 μM; 1, 5 10 μM; 10, 11 0.5 μM, STS measurement with lower inhibitor conc.: EMATE 0.001 μM; 1, 5 0.2 μM; 10, 11 0.01 μM
Kinetic investigations were performed with test substances 10, 11 and reference EMATE. The Dixon’s plot provided straight lines at different fixed substrate concentrations intersecting in the second quadrant characterizing the competitive inhibition mechanism (Fig. 4, part A). The replots of slopes vs. 1/substrate concentration resulted in a straight line through the origin (insets in part B of Fig. 4) which confirms the competitive binding manner [10, 40] that means that these inhibitors occupy the substrate-binding place of the enzyme. Ki parameters were found to be 0.066 μM for 10, 0.021 μM for 11 and 0.0044 μM for the reference EMATE (Fig. 4 part A). Inhibitory potentials on the basis of these Ki data were comparable to those obtained by the IC50 values. Determined Ki values are substantially lower than the KM parameter of estrone-3-sulfate (8.5 μM) that reflects the significantly higher binding affinities of these inhibitors when compared to the substrate.

Dixon’s kinetic analysis reference EMATE and compounds 10 and 11. Row A: Dixon’s plots provided straight lines at different fixed substrate concentrations. Lines intersecting in the second quadrant characterize the competitive inhibition mechanism. Row B: Secondary plot of slopes of the Dixon’s lines vs. 1/substrate concentration. Straight lines through the origin characterize the competitive binding manner (corresponding references: [10, 40])
Discussion
Inhibition of aromatase
Structural studies of aromatase have revealed that the structure of substrate-binding site provides specificity toward androgens [14]. Numerous derivatives of the preferred substrate androst-4-ene-3,17-dione have been found potent inhibitors, and the 4-hydroxy- and the 6-methylidene-1,4-diene analogues of the androst-4-ene-3,17-dione (formestane and exemestane, respectively) have been applied in the medical practice [55]. Structure–activity investigations have found that planarity of the ring A (4-ene or 1,4-diene structure), preferred anellation of the ring A and ring B junction (5α-epimers) and the presence of either apolar or polar substituents on the C-4, C-6 and C-7 carbons enhance the binding affinity of the inhibitors [7]. It has been also revealed that androst-4-ene-3,17-dione derivatives were able to bind the enzyme in the absence of 3-oxo function. Potent inhibitory effect of 16β-imidazolyl androst-4-ene-3,17-dione derivatives has been reported, too [4]. Aromatase inhibitors were also found among product-like compounds, and 2-chloro- and 6α-phenyl-estrones were the most powerful members of the series investigated [3, 28]. It is worth mentioning that conjugated aromatic systems containing triazole cycle are common structural elements of nonsteroidal aromatase inhibitors [5, 38]. Corresponding further literature references can be found in the comprehensive review articles on aromatase inhibitors published by Baston and Leroux [5], Santen et al. [38] and Yadav et al. [55].
Various structural elements of referred aromatase inhibitor examples can be recognized in our test compounds. Certain test compounds bear substrate-like 4-en-3-oxo structure or possess ring A with similar planarity to the 4-aza-3-oxo moiety, whereas others possess product-like estrane core. Some investigated compounds have side chain in position C-2, C-6 and C-16, and the ferrocenyl substituents bear a triazolyl group. Similarities to known aromatase inhibitors made our compounds reasonable targets for aromatase inhibition tests. Experiments, however, revealed that none of the test compounds exerted substantial inhibitory effect against the human aromatase activity.
Inhibition of 17β-HSD1
The substrate-binding site of 17β-HSD1 has been identified as a hydrophobic tunnel with polar residues at each end [17, 39]. Further extensive inhibitor research works demonstrated that the binding cavity is complementary to the C18 steroidal scaffold of the main natural substrate estrone. 17β-HSD1 is multispecific in terms of binding other, non-cognate steroidal substrates, e.g., C19 androstenes or androstanes, even in alternative binding orientations. Adjacent cavities and subpockets can be found around the substrate-binding site which is able to accommodate various side chains on the steroidal C-2 position or on the ring D (positions C-15, C-16 and C-17). These interactions improve binding affinity and enhance inhibitory potential considerably. Estrone and 17β-estradiol derivatives possessing aromatic substituents on C-2 or C-16 [33] and also those bearing triazolyl-aryl side chain on C-15 [24] or on a D-seco structure [43] were found to be efficient 17β-HSD1 inhibitors. Readers may find reference details of these literature data in our previous publications [3, 43] and in review articles on 17β-HSD1 inhibitors from Brožič et al. [6] and Poirier [33].
17β-HSD1 is able to bind compounds containing various steroidal cores, and suitable substituents among them aromatic side chains in position C-2, C-15, C-16 and C-17, may enhance the binding affinity of these compounds. Several structural features of our steroidal ferrocenes resemble to known 17β-HSD1 inhibitors; nevertheless, our tested compounds did not exert considerable inhibition on the 17β-HSD1 activity under our experimental conditions.
Inhibition of STS
Protein of the STS consists of a globular polar domain and a hydrophobic transmembrane stem domain, resembling to a mushroom in that overall tertiary shape of the enzyme. The substrate-binding site situated in a cavity lies at the base of the globular domain, just at the top of the stem domain [17]. STS hydrolyzes various physiological substrates to their unconjugated forms, including 3-sulfate ester of estrone, dehydroepiandrosterone, pregnenolone and cholesterol. Numerous other compounds proved to be capable of binding to the enzyme and demonstrated effective inhibition toward the sulfatase activity [22, 30, 41, 54]. A large group of these inhibitors are ester derivatives which apply the FGly75 inactivation mechanism by their acid residue. Most of these, either steroidal or nonsteroidal compounds, are sulfamates, and the first and still the best known example of these irreversible inhibitors is EMATE [36].
Various estrane-, androstane- and pregnane-based compounds exert inhibitory effect toward the STS, and their binding affinity to the enzyme can be largely modified by substituents on the steroid skeleton [22, 30, 41, 54]. Certain halogens or other small electron withdrawing groups (cyano and nitro) in position 2 and/or 4 of the steroidal ring A may enhance the inhibitory potential [3, 25, 29, 32]. Reason of the enhanced binding is not known; nevertheless, the existence of cavities able to accommodate these substituents could be postulated. Amino acids which might be involved in putative electrostatic or hydrogen-binding interactions in the proximity of position 2 (Lys134, Lys368, His136, His290 and His346) and position 4 (Lys368, Lys134 and Arg79) are also proposed [25, 32, 36].
Literature results have also revealed that appropriate side chains on the ring D might also improve binding affinity of the steroidal inhibitors [41]. 17α-Benzyl or alkyl derivatives of 17β-estradiol display enhanced inhibitor potential [13, 34]. Hydrophobic substituents on the aromatic cycle might further improve the inhibitory effect [8], and relatively rigid 17α-moieties (cyclic substituent attached with only one rotatable bond, a methylene group) were more advantageous in the binding. 16α-Benzyl- and 16β-benzyl-substituted compounds exert also augmented inhibition compared to their parent compound 17β-estradiol, although they are much less potent than their 17α counterparts. These findings indicated non-specific hydrophobic interactions of the D-ring side chains with hydrophobic residues in a long, deep and narrow pocket in the D-ring area [13]. Non-competitive or mixed type reversible inhibition of the 17α-benzyl-17β-estradiol suggested the existence of two proximate-binding sites involved in the binding of this inhibitor [8, 25]. One of them allows the steroidal part to bind, whereas another one, an allosteric-binding site, establishes non-specific hydrophobic interactions of the D-ring side chains. The latter is formed by hydrophobic residues in a long and narrow pocket located between the two hydrophobic α-helices and buried in the transmembrane domain [13, 26]. Authors Fournier and Poirier postulated that inhibitory potency might be increased by maximizing interactions with both the substrate and the allosteric-binding sites [13]. Considering enhanced inhibitor potency of further steroidal compounds bearing C-17 aryl group, a π–π-type interaction to the Phe residues present at the entrance of α-helices of the binding site have also been hypothesized [9, 41]. Large affinity difference which was observed between 17β-arylsulfonamide and 17β-arylamide derivatives of estra-1,3,5(10)-trien-3-ol indicated that other non-hydrophobic interactions of the linker which attach the aryl function might also play a key role in potent inhibition [25, 26].
STS inhibition data of the investigated steroidal ferrocene compounds reveal that the 17α-triazolyl-ferrocene derivative of 17β-estradiol (10) is a highly potent inhibitor of this enzyme. The submicromolar IC50 and Ki parameters enroll this compound to the group of the most effective steroidal STS inhibitors known to date [22, 30, 41, 54]. Inhibitory potential of this compound is similar to that of the optimized 17α-benzyl-17β-estradiol derivatives (IC50 = 0.022–0.310 μM) reported by the Poirier’s group, among which a bicyclic 4-benzyloxybenzyl derivative was the most effective inhibitor [30, 34]. Our test compound 10 also bears a bicyclic aromatic 17α substituent and both of its triazolyl and ferrocenyl moieties bear a pronounced pharmacophore character.
In order to investigate structural features contributing to the outstanding inhibitory potential of compound 10, we may compare this finding with the results of the triazolyl-benzene derivative 13 and their parent compound 17β-estradiol. Both 17α-substituted compounds have markedly enhanced effect when compared to 17β-estradiol (IC50 = 9.0 μM), and the presence of ferrocenyl moiety instead of phenyl group results in a further 12-fold increase in the inhibition. Ferrocene and phenyl ring has been used to replace each other mutually and can be regarded as bioisosteric functional groups in drug compounds. Concerning inhibition toward STS, the ferrocenyl analogue exerts a considerably higher potential, indicating a crucial and specific function of this moiety in the binding of the inhibitor to this enzyme.
Substance 11 contains methylated derivatives of 10. Although substance 11 is a mixture of epimers, it displays regular behavior in inhibition experiments (Figs. 2 and 4). Measured apparent IC50 and Ki parameters can reliably reflect the inhibitory effect, and these values indicate further enhanced potential when compared to that of the non-methylated counterpart. Structural aspects cannot be identified precisely; nevertheless, results of substance 11 emphasize that further optimization of the binding of 17α-triazolyl-ferrocene derivatives is possible even with minor modification(s) of the linker part between the triazolyl and ferrocenyl structures of the side chain.
In kinetic experiments, 10 displayed a competitive behavior. It is most likely that substrate-binding cavity is occupied by the 17β-estradiol part of the molecule. Ring A accommodated in the active center of the enzyme, and the ring D with the 17α-triazolyl-ferrocene side chain is directed toward the transmembrane domain of the STS. Literature references indicate that C-17 aryl substituents may interact with Phe and other hydrophobic amino acids of the α-helices in this domain (Fournier and Poirier 1998; [9, 41]. We may also postulate that the 17α-triazolyl-ferrocene side chain of 10 can have access to these residues, and the enhanced binding affinity of this compound can be explained by similar mechanisms, namely by π–π-type interactions toward Phe residues present at the entrance of α-helices as well as by non-specific hydrophobic interactions directed to suitable residues buried deeper in the tunnel of the α-helices. Competitive inhibition pattern also suggests that the 17α-triazolyl-ferrocene side chain is long and flexible enough to allow 17β-estradiol part to bind into the substrate-binding cavity, which means that these types of derivatives may be able to maximize interactions with both binding sites.
16β-Triazolyl-ferrocene-17α-hydroxy-5α-androstane (4) has a weak inhibitory effect, whereas 5 which bears an ethylacrylate triazolyl-ferrocene substituent on C-16 proved to be a potent inhibitor. It seems that an elongated ferrocenyl side chain is preferred for the binding of C-16-substituted derivatives. Ferrocenyl moiety attached to a longer and flexible linker may reach the binding pocket which otherwise is situated closer and positioned more appropriately for C-17 substituents [34], and this might be the reason for a better binding affinity of 5 to the STS.
In the case of the tested two 2β-substituted 3α-hydroxy-5α-androstane-17-one compounds, the shorter triazolyl-ferrocene substituent of 1 proved to be advantageous concerning the affinity to the STS. It may be postulated that this triazolyl-ferrocene side chain is capable of interacting with residues of the binding cavity supposed to exist in the proximity of the ligands’ position 2 [25, 36].
Examples of the potent test compounds (1, 5 and 10) show that various steroidal backbones may provide suitable core for ferrocenyl derivative STS inhibitors. Comparison of the highly potent 17α-triazolyl-ferrocene derivative of 17β-estradiol (10) and its 4-ene-3-oxo analogue (12), a weak inhibitor, reveals that in other cases the steroidal scaffold has a strong influence on the STS inhibitory potential.
Mechanistic experiments indicate a reversible binding mode for the C-2-substituted triazolyl-ferrocene derivative 1 and an irreversible binding mechanism for the C-16 and C-17 derivatives 5, 10 and substance 11. This binding difference approves that different interactions may be involved in the binding of the C-2 triazolyl-ferrocene derivative and the compounds substituted on their ring D. Our inhibitors do not possess arylsulfamate moiety, which is usually the active principle of the irreversible STS inhibitors; neither bear other reactive group capable of forming covalent binding to certain enzyme residues. Steroidal ferrocenes and the enzyme, therefore, are likely to establish non-covalent adducts, but the interactions can be so tight in certain cases that the inhibitors will be bound essentially irreversibly.
Biomedical relevance
In cancerous breast tissues, 17β-estradiol can be formed in situ from estrone, which is generated also locally either via the aromatase route (aromatization of adrenal androst-4-ene-3,17-dione) or the sulfatase route (desulfation of estrone-3-sulfate). It is considered that sulfatase pathway predominates, and around tenfold greater amount of estrone may be produced via the sulfatase route rather than via the aromatase pathway [19, 29]. Inhibition of STS is, therefore, a particularly attractive manner of antiestrogenic pharmacotherapy of the estrogen-dependent breast cancer [35, 37]. Numerous STS inhibitors were developed in the last decades, but none of them have been introduced into the medical practice till now [35]; Rizner [37, 47]. There still remains a considerable demand for a new class of STS inhibitor compounds. Our results on STS inhibitory potential of certain ferrocenyl steroids seem therefore to be particularly promising.
When an irreversible inhibitor is used as medicinal agent, effect can be maintained after its clearance from the system, and a de novo biosynthesis of the enzyme protein is required for the restoration of the original enzyme activity. Advantages of the resulting prolonged pharmacodynamics include lower level and frequency of dosing as well as suppressed extent of side effects. EMATE-based irreversible inhibitors, however, have the drawback that the STS inactivation process releases steroidal compounds bearing estrogenic hormonal effect [36]. Binding of steroidal ferrocenes does not transform the inhibitor molecule, and the irreversible inhibition without generating hormonally active agents makes steroidal ferrocenes superior to EMATE derivatives in a presumed pharmacological application.
Various steroidal ferrocene compounds bear cytotoxic effects which evolve hormone-independent antiproliferative effects in cancerous cells [18, 23, 49, 51, 53]. Recently tested derivatives and their analogues may also bear such a cytotoxic potential. A hypothesized synergism between the hormone-independent cytotoxicity and the suppressed estrogen-dependent cell proliferation evolved by the inhibited STS activity might be particularly advantageous in the treatment of breast cancer.
Conclusion for future biology
We investigated in vitro inhibitory effects of ten diverse steroidal ferrocene substances exerted on key enzymes of the estrogen biosynthesis. Numerous structural elements of these derivatives resemble to those of known potent aromatase and 17β-HSD1 inhibitors; nevertheless, our test compounds were found weakly effective against these enzymes. We observed a potent inhibition against STS by three triazolyl-ferrocene derivatives which display stronger affinities to the enzyme than the substrate estrone-3-sulfate itself. Compound with triazolyl-ferrocene substituent on C-2 is bound in a reversible manner, whereas the C-16 and C-17 derivatives are irreversible inhibitors. Related non-ferrocenyl compounds are found to exert lower potency, indicating that the ferrocene moiety may have a specific function in the enhanced affinity to the STS. Based on literature references, interactions of the 16β- and 17α-triazolyl-ferrocenes with Phe residues and with other hydrophobic amino acids of the transmembrane helices can be proposed. Comparison of adequate counterparts revealed a profound influence of the steroidal backbone and of the linker part in the ferrocenyl side chains on the STS inhibition. Our most potent STS inhibitor, the 17α-triazolyl-ferrocene derivative of 17β-estradiol (10) presents submicromolar IC50 and Ki parameters, and therefore, this compound belongs to the group of the most effective STS inhibitors published so far. Our results indicate that optimized combination of structural elements (steroidal core, ferrocenyl side chain, position of the substitution) may further enhance STS inhibitory potential of the steroidal ferrocenes. STS inhibitory potential of the steroidal ferrocenes may lead to the development of novel compounds able to suppress in situ 17β-estradiol production in target tissues. A presumed synergism between the hormone-independent cytotoxicity and the suppressed estrogen-dependent cell proliferation can make this type of compounds particularly promising drug candidates for the pharmacological therapy of hormone-dependent breast cancer.
Data accessibility
The data supporting Materials and methods section of this article have been uploaded as part of Supplementary Material.
References
Africander D, Storbeck KH (2018) Steroid metabolism in breast cancer: Where are we and what are we missing? Mol Cell Endocrinol 466:86–97. https://doi.org/10.1016/j.mce.2017.05.016
Astruc D (2017) Why is ferrocene so exceptional? Eur J Inorg Chem 2017:6–29. https://doi.org/10.1002/ejic.201600983
Bacsa I, Herman BE, Jójárt R, Herman KS, Wölfling J, Schneider G, Varga M, Tömböly C, Rižner T, Szécsi M, Mernyák E (2018) Synthesis and structure–activity relationships of 2- and/or 4-halogenated 13β- and 13α-estrone derivatives as enzyme inhibitors of estrogen biosynthesis. J Enzyme Inhib Med Chem 33:1271–1282
Bansal R, Guleria S, Thota S, Bodhankar SL, Patwardhan MR, Zimmer C, Hartmann RW, Harvey AL (2012) Design, synthesis and evaluation of novel 16-imidazolyl substituted steroidal derivatives possessing potent diversified pharmacological properties. Steroids 77:621–629. https://doi.org/10.1016/j.steroids.2012.02.005
Baston E, Leroux FR (2007) Inhibitors of steroidal cytochrome p450 enzymes as targets for drug development. Recent Pat Anticancer Drug Discov 2:31–58. https://doi.org/10.2174/157489207779561453
Brožič P, Rižner TL, Gobec S (2008) Inhibitors of 17β-hydroxysteroid dehydrogenase type 1. Curr Med Chem 15:137–150. https://doi.org/10.2174/092986708783330629
Cepa MM, Tavares da Silva EJ, Correia-da-Silva G, Roleira FM, Teixeira NA (2005) Structure-activity relationships of new A, D-ring modified steroids as aromatase inhibitors: design, synthesis, and biological activity evaluation. J Med Chem 48:6379–6385. https://doi.org/10.1021/jm050129p
Ciobanu LC, Boivin RP, Luu-The V, Labrie F, Poirier D (1999) Potent inhibition of steroid sulfatase activity by 3-O-sulfamate 17α-benzyl(or 4’-tert-butylbenzyl)estra-1,3,5(10)-trienes: combination of two substituents at positions C3 and C17α of estradiol. J Med Chem 42:2280–2286. https://doi.org/10.1021/jm980677l
Ciobanu LC, Boivin RP, Luu-The V, Poirier D (2001) Synthesis and steroid sulphatase inhibitory activity of C19- and C21-steroidal derivatives bearing a benzyl-inhibiting group. Eur J Med Chem 36:659–671. https://doi.org/10.1016/S0223-5234(01)01262-4
Cortés A, Cascante M, Cárdenas ML, Cornish-Bowden A (2001) Relationships between inhibition constants, inhibitor concentrations for 50% inhibition and types of inhibition: new ways of analysing data. Biochem J 357:263–268. https://doi.org/10.1042/0264-6021:3570263
Fehér K, Balogh J, Csók Z, Kégl T, Kollár L, Skoda-Földes R (2012) Synthesis of ferrocene-labeled steroids via copper-catalyzed azide-alkyne cycloaddition. Reactivity difference between 2β-, 6β- and 16β-azido-androstanes. Steroids 77:738–744
Fehér K, Nagy E, Szabó P, Juzsakova T, Srankó D, Gömöry Á, Kollár L, Skoda-Földes R (2018) Heterogeneous azide-alkyne cycloaddition in the presence of a copper catalyst supported on an ionic liquid polymer/silica hybrid material. Appl Organometal Chem 32:e4343
Fournier D, Poirier D (2011) Chemical synthesis and evaluation of 17α-alkylated derivatives of estradiol as inhibitors of steroid sulfatase. Eur J Med Chem 46:4227–4237. https://doi.org/10.1016/j.ejmech.2011.06.027
Ghosh D, Griswold J, Erman M, Pangborn W (2009) Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nature 457:219–223. https://doi.org/10.1038/nature07614
Ghosh D, Pletnev VZ, Zhu DW, Wawrzak Z, Duax WL, Pangborn W, Labri F, Lin SX (1995) Structure of human estrogenic 17β-hydroxysteroid dehydrogenase at 2.20 A resolution. Structure 3:503–513
Herman BE, Szabó J, Bacsa I, Wölfling J, Schneider G, Bálint M, Hetényi C, Mernyák E, Szécsi M (2016) Comparative investigation of the in vitro inhibitory potencies of 13-epimeric estrones and D-secoestrones towards 17β-hydroxysteroid dehydrogenase type 1. J Enzyme Inhib Med Chem 31:61–69
Hernandez-Guzman FG, Higashiyama T, Pangborn W, Osawa Y, Ghosh D (2003) Structure of human estrone sulfatase suggests functional roles of membrane association. J Biol Chem 278:22989–22997. https://doi.org/10.1074/jbc.M211497200
Hillard EA, Vessières A, Jaouen G (2010) Ferrocene functionalized endocrine modulators as anticancer agents. Top Organomet Chem 32:81–117. https://doi.org/10.1007/978-3-642-13185-1_4
Hong Y, Chen S (2011) Aromatase, estrone sulfatase, and 17β-hydroxysteroid dehydrogenase: structure-function studies and inhibitor development. Mol Cell Endocrinol 340:120–126. https://doi.org/10.1016/j.mce.2010.09.012
Kocsis L, Szabo I, Bosze S, Jernei T, Hudecz F, Csampai A (2016) Synthesis, structure and in vitro cytostatic activity of ferrocene–Cinchona hybrids. Bioorg Med Chem Lett 26:946–949
Leslie KK, Zuckerman DJ, Chruefer J, Burchell M, Smith J, Albertson BD (1994) Oestrogen modulation with parturition in the human placenta. Placenta 15:79–88
Maltais R, Poirier D (2011) Steroid sulfatase inhibitors: a review covering the promising 2000–2010 decade. Steroids 76:929–948. https://doi.org/10.1016/j.steroids.2011.03.010
Manosroi J, Rueanto K, Boonpisuttinant K, Manosroi W, Biot C, Akazawa H, Akihisa T, Issarangporn W, Manosroi A (2010) Novel ferrocenic steroidal drug derivatives and their bioactivities. J Med Chem 53:3937–3943. https://doi.org/10.1021/jm901866m
Messinger J, Schoen U, Husen B, Thole H, Koskimies P, Kallio L (2008) Therapeutically active triazoles and their use. Patent WO2008/034796
Mostafa YA, Kralt B, Rao PP, Taylor SD (2015) A-ring substituted 17β-arylsulfonamides of 17β-aminoestra-1,3,5(10)-trien-3-ol as highly potent reversible inhibitors of steroid sulfatase. Bioorg Med Chem 23:5681–5692. https://doi.org/10.1016/j.bmc.2015.07.019
Mostafa YA, Taylor SD (2012) 17β-Arylsulfonamides of 17β-aminoestra-1,3,5(10)-trien-3-ol as highly potent inhibitors of steroid sulfatase. Bioorg Med Chem 12:1535–1544. https://doi.org/10.1016/j.bmc.2011.12.036
Narvaez-Pita X, Rheingold AL, Melendez E (2017) Ferrocene-steroid conjugates: synthesis, structure and biological activity. J Organomet Chem 846:113–120. https://doi.org/10.1016/j.jorganchem.2017.06.004
Numazawa M, Ando M, Watari Y, Tominaga T, Hayata Y, Yoshimura A (2005) Structure-activity relationships of 2-, 4-, or 6-substituted estrogens as aromatase inhibitors. J Steroid Biochem Mol Biol 96:51–58. https://doi.org/10.1016/j.jsbmb.2005.01.028
Numazawa M, Tominaga T, Watari Y, Tada Y (2006) Inhibition of estrone sulfatase by aromatase inhibitor-based estrogen 3-sulfamates. Steroids 71:371–379. https://doi.org/10.1016/j.steroids.2005.12.004
Nussbaumer P, Billich A (2004) Steroid sulfatase inhibitors. Med Res Rev 24:529–576. https://doi.org/10.1002/med.20008
Osella D, Nervi C, Galeotti F, Cavigiolio G, Vessières A, Jaouen G (2001) The ferrocenyl ethynyl unit: a stable hormone tag. Helv Chim Acta 84:3289–3298. https://doi.org/10.1002/1522-2675(20011114)84:11%3c3289:AID-HLCA3289%3e3.0.CO;2-D
Phan CM, Liu Y, Kim BM, Mostafa Y, Taylor SD (2011) Inhibition of steroid sulfatase with 4-substituted estrone and estradiol derivatives. Bioorg Med Chem 19:5999–6005. https://doi.org/10.1016/j.bmc.2011.08.046
Poirier D (2011) Contribution to the development of inhibitors of 17β-hydroxysteroid dehydrogenase types 1 and 7: key tools for studying and treating estrogen-dependent diseases. J Steroid Biochem Mol Biol 125:83–94. https://doi.org/10.1016/j.jsbmb.2010.12.007
Poirier D, Boivin RP (1998) 17α-alkyl- or 17α-substituted benzyl-17β-estradiols: a new family of estrone-sulfatase inhibitors. Bioorg Med Chem Lett 8:1891–1896. https://doi.org/10.1016/S0960-894X(98)00330-8
Potter BVL (2018) SULFATION PATHWAYS: steroid sulphatase inhibition via aryl sulphamates: clinical progress, mechanism and future prospects. J Mol Endocrinol 61:T233–T252. https://doi.org/10.1530/JME-18-0045
Reed JE, Woo LW, Robinson JJ, Leblond B, Leese MP, Purohit A, Reed MJ, Potter BVL (2004) 2-Difluoromethyloestrone 3-O-sulphamate, a highly potent steroid sulphatase inhibitor. Biochem Biophys Res Commun 317:169–175. https://doi.org/10.1016/j.bbrc.2004.03.023
Rižner TL (2016) The important roles of steroid sulfatase and sulfotransferases in gynecological diseases. Front Pharmacol 7:30. https://doi.org/10.3389/fphar.2016.00030
Santen RJ, Brodie H, Simpson ER, Siiteri PK, Brodie A (2009) History of aromatase: saga of an important biological mediator and therapeutic target. Endocr Rev 30:343–375. https://doi.org/10.1210/er.2008-0016
Sawicki MW, Erman M, Puranen T, Vihko P, Ghosh D (1999) Structure of the ternary complex of human 17β-hydroxysteroid dehydrogenase type 1 with 3-hydroxyestra-1,3,5,7-tetraen-17-one (equilin) and NADP+. Proc Natl Acad Sci USA 96:840–845. https://doi.org/10.1073/pnas.96.3.840
Segel IH (1993) Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. Wiley, New York
Shah R, Singh J, Singh D, Jaggi AS, Singh N (2016) Sulfatase inhibitors for recidivist breast cancer treatment: a chemical review. Eur J Med Chem 114:170–190. https://doi.org/10.1016/j.ejmech.2016.02.054
Singh A, Saha ST, Perumal S, Kaur M, Kumar V (2018) Azide–alkyne cycloaddition en route to 1H‑1,2,3-triazole-tethered isatin–ferrocene, ferrocenylmethoxy–isatin, and isatin–ferrocenylchalcone conjugates: synthesis and antiproliferative evaluation. ACS Omega 3:1263–1268
Szabó J, Bacsa I, Wölfling J, Schneider G, Zupkó I, Varga M, Herman BE, Kalmár L, Szécsi M, Mernyák E (2016) Synthesis and in vitro pharmacological evaluation of N-[(1-benzyl-1,2,3-triazol-4-yl)methyl]-carboxamides on d-secoestrone scaffolds. J Enzyme Inhib Med Chem 31:574–579
Szánti-Pintér E, Balogh J, Csók Z, Kollár L, Gömöry A, Skoda-Földes R (2011) Synthesis of steroid-ferrocene conjugates of steroidal 17-carboxamides via a palladium-catalyzed aminocarbonylation–copper-catalyzed azide-alkyne cycloaddition reaction sequence. Steroids 76:1377–1382
Szánti-Pintér E, Csók Z, Kollár L, Vékey K, Skoda-Földes R (2012) Synthesis of ferrocene-labelled steroid derivatives via homogeneous catalytic methods. J Organomet Chem 718:105–107
Szánti-Pintér E, Wouters J, Gömöry Á, Sághy É, Szőke É, Helyes Z, Kollár L, Skoda-Földes R (2015) Synthesis of novel 13α-18-norandrostane-ferrocene conjugates via homogeneous catalytic methods and their investigation on TRPV1 receptor activation. Steroids 104:284–293
Thomas MP, Potter BVL (2015) Estrogen O-sulfamates and their analogues: clinical steroid sulfatase inhibitors with broad potential. J Steroid Biochem Mol Biol 153:160–169. https://doi.org/10.1016/j.jsbmb.2012.12.014
Thompson EA Jr, Siiteri PK (1974) Utilization of oxygen and reduced nicotinamide adenine dinucleotide phosphate by human placental microsomes during aromatization of androstenedione. J Biol Chem 249:5364–5372
Top S, Thibaudeau C, Vessières A, Brulè E, Bideau FL, Joerger JM, Plamont MA, Samreth S, Edgar A, Marrot J, Herson P, Jaouen G (2009) Synthesis and structure activity relationship of organometallic steroidal androgen derivatives. Organometallics 28:1414–1424. https://doi.org/10.1021/om800698y
Tremblay MR, Poirier D (1998) Overview of a rational approach to design type I 17β-hydroxysteroid dehydrogenase inhibitors without estrogenic activity: chemical synthesis and biological evaluation. J Steroid Biochem Mol Biol 66:179–191
Vera J, Gao LM, Santana A, Matta J, Meléndez E (2011) Vectorized ferrocenes with estrogens and vitamin D2: synthesis, cytotoxic activity and docking studies. Dalton Trans 40:9557–9565. https://doi.org/10.1039/c1dt10995b
Vessières A, Jaouen G, Gruselle M, Rossignol JL, Savignac M, Top S, Greenfield S (1998) Synthesis and receptor binding of polynuclear organometallic estradiol derivatives. J Steroid Biochem 30:301–306. https://doi.org/10.1016/0022-4731(88)90111-2
Vessières A, Spera D, Top S, Misterkiewicz B, Heldt JM, Hillard EA, Huchè M, Plamont MA, Napolitano E, Fiaschi R, Jaouen G (2006) The presence of a ferrocenyl unit on an estrogenic molecule is not always sufficient to generate in vitro cytotoxicity. Chem Med Chem 1:1275–1281. https://doi.org/10.1002/cmdc.200600176
Williams SJ (2013) Sulfatase inhibitors: a patent review. Expert Opin Ther Pat 23:79–98. https://doi.org/10.1517/13543776.2013.736965
Yadav MR, Barmade MA, Tamboli RS, Murumkar PR (2015) Developing steroidal aromatase inhibitors-an effective armament to win the battle against breast cancer. Eur J Med Chem 105:1–38. https://doi.org/10.1016/j.ejmech.2015.09.038
Acknowledgements
Open access funding provided by University of Szeged.
Funding
This work was supported by the National Research, Development and Innovation Office (OTKA 120014).
Author information
Authors and Affiliations
Contributions
BEH and MS conceptualized and designed the study, and interpreted the results; BEH, JG, CT, ES-P, KF, RS-F and MS obtained and analyzed the data; BEH, JJ, ES-P, KF; RS-F and MS drafted and revised the manuscript.
Corresponding authors
Ethics declarations
Conflict if interests
The authors declare no competing interests
Ethical statement
Human placenta specimens were applied with the permission of the Human Investigation Review Board of the University of Szeged no. 3262.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Herman, B.E., Gardi, J., Julesz, J. et al. Steroidal ferrocenes as potential enzyme inhibitors of the estrogen biosynthesis. BIOLOGIA FUTURA 71, 249–264 (2020). https://doi.org/10.1007/s42977-020-00023-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42977-020-00023-7