Antioxidant Therapies and Oxidative Stress in Friedreich’s Ataxia: The Right Path or Just a Diversion?

 , ,
, ,
Abstract
:1. Introduction
2. The Clinical Spectrum of FRDA
2.1. Neurological Features
2.2. Non-neurological Features
3. Oxidative Stress Markers in FRDA Patients and Models
4. Reducing Oxidative Stress as a Therapeutic Avenue to Stop FRDA Progression
4.1. ROS Scavengers
4.1.1. Coenzyme Q10 and Idebenone
4.1.2. A0001
4.1.3. EGb-761
4.1.4. VP-20629
4.1.5. (+)-Epicatechin
4.1.6. Thiamine
4.2. Promotion of Antioxidant Response
4.2.1. NRF2 Inducers
Omaveloxolone
Resveratrol
4.2.2. Mitochondrial Metabolism
Pioglitazone and Leriglitazone
Acetyl-l-Carnitine
4.3. Counteracting ROS Production
4.3.1. Iron Chelators
Deferiprone
4.3.2. Lipid Metabolism
EPI-743
Deuterated Fatty Acids: RT001
5. Discussion and Conclusions
6. Future Directions and Prospects
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALCAR—l-Acetyl-carnitine | ARE—Antioxidant response element |
| dPUFAs—Deuterated PUFAs | DRG—Dorsal root ganglia |
| ER - Endoplasmic Reticulum | FARS—Friedreich Ataxia Rating Scale |
| FRDA—Friedreich’s ataxia | GPX4—Glutathione peroxidase 4 |
| ICARS—International Cooperative Ataxia Rating Scale | ISC—Iron-sulphur clusters |
| KEAP1—Kelch-like ECH-associated protein 1 | KIKO—Knock-in/knock out |
| mFARS—Modified Friedreich Ataxia Rating Scale | NRF2—Nuclear factor erythroid 2-related factor 2 |
| NRFs—Nuclear respiratory factors | OXPHOS—Oxidative phosphorylation |
| PGC1-α—Peroxisome proliferator-activated receptor gamma coactivator 1-alpha | PPARγ—Peroxisome proliferator-activated receptor gamma |
| PPARs—Peroxisome proliferator-activated receptors | PUFAs—Polyunsaturated fatty acids |
| P-VEPs—Visual evoked potentials | ROS—Reactive oxygen species |
| SARA—Scale of Assessment and Rating of Ataxia | SIRT1—NAD-dependent deacetylase sirtuin-1 |
| SOD2—Superoxide dismutase 2 | SREBP1—Sterol-responsive element-binding protein 1 |
| SWJs—Square-wave jerks | TZDs—Thiazolidinediones |
References
- Vankan, P. Prevalence gradients of Friedreich’s ataxia and R1b haplotype in Europe co-localize, suggesting a common Palaeolithic origin in the Franco-Cantabrian ice age refuge. J. Neurochem. 2013, 126 (Suppl. s1), 11–20. [Google Scholar] [CrossRef] [PubMed]
- Labuda, M.; Labuda, D.; Miranda, C.; Poirier, J.; Soong, B.W.; Barucha, N.E.; Pandolfo, M. Unique origin and specific ethnic distribution of the Friedreich ataxia GAA expansion. Neurology 2000, 54, 2322–2324. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Galea, C.A.; Huq, A.; Lockhart, P.J.; Tai, G.; Corben, L.A.; Yiu, E.M.; Gurrin, L.C.; Lynch, D.R.; Gelbard, S.; Durr, A.; et al. Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 2016, 79, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is Reduced in Friedreich Ataxia Patients and is Associated with Mitochondrial Membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Cossée, M.; Schmitt, M.; Campuzano, V.; Reutenauer, L.; Moutou, C.; Mandel, J.L.; Koenig, M. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: Founder effect and premutations. Proc. Natl. Acad. Sci. USA 1997, 94, 7452–7457. [Google Scholar] [CrossRef] [Green Version]
- Dürr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C.; Mandel, J.-L.; Brice, A.; Koenig, M. Clinical and Genetic Abnormalities in Patients with Friedreich’s Ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef]
- Filla, A.; De Michele, G.; Cavalcanti, F.; Pianese, L.; Monticelli, A.; Campanella, G.; Cocozza, S. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 1996, 59, 554–560. [Google Scholar]
- Calap-Quintana, P.; Navarro, J.A.; González-Fernández, J.; Martínez-Sebastián, M.J.; Moltó, M.D.; Llorens, J.V. Drosophila melanogaster Models of Friedreich’s Ataxia. BioMed Res. Int. 2018, 2018, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Condò, I.; Ventura, N.; Malisan, F.; Rufini, A.; Tomassini, B.; Testi, R. In vivo maturation of human frataxin. Hum. Mol. Genet. 2007, 16, 1534–1540. [Google Scholar] [CrossRef] [Green Version]
- Rotig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Casis, G.; Cote, M.; Barbeau, A. Pathology of the heart in Friedreich’s ataxia: Review of the literature and report of one case. Can. J. Neurol. Sci. 1976, 3, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.-L.; Barondeau, D.P. Human Frataxin Is an Allosteric Switch That Activates the Fe−S Cluster Biosynthetic Complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donzé, M.; Reutenauer, L.; Puccio, H. Mammalian Frataxin: An Essential Function for Cellular Viability through an Interaction with a Preformed ISCU/NFS1/ISD11 Iron-Sulfur Assembly Complex. PLoS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Müller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Cortopassi, G. Frataxin knockdown causes loss of cytoplasmic iron–sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 2007, 457, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Cermeño, A.; Obis, È.; Bellí, G.; Cabiscol, E.; Ros, J.; Tamarit, J. Frataxin Depletion in Yeast Triggers Up-regulation of Iron Transport Systems before Affecting Iron-Sulfur Enzyme Activities. J. Biol. Chem. 2010, 285, 41653–41664. [Google Scholar] [CrossRef] [Green Version]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef]
- Ast, T.; Meisel, J.D.; Patra, S.; Wang, H.; Grange, R.M.H.; Kim, S.H.; Calvo, S.E.; Orefice, L.L.; Nagashima, F.; Ichinose, F.; et al. Hypoxia Rescues Frataxin Loss by Restoring Iron Sulfur Cluster Biogenesis. Cell 2019, 177, 1507–1521. [Google Scholar] [CrossRef]
- Gakh, O.; Adamec, J.; Gacy, A.M.; Twesten, R.D.; Owen, W.G.; Isaya, G. Physical evidence that yeast frataxin is an iron storage protein. Biochemistry 2002, 41, 6798–6804. [Google Scholar] [CrossRef]
- Ristow, M.; Pfister, M.F.; Yee, A.J.; Schubert, M.; Michael, L.; Zhang, C.Y.; Ueki, K.; Michael, M.D., 2nd; Lowell, B.B.; Kahn, C.R. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12239–12243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Cabo, P.; Vazquez-Manrique, R.P.; Garcia-Gimeno, M.A.; Sanz, P.; Palau, F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum. Mol. Genet. 2005, 14, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, A.-L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolinches-Amorós, A.; Mollá, B.; Pla-Martí-n, D.; Palau, F.; González-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [CrossRef] [Green Version]
- Mollá, B.; Muñoz-Lasso, D.C.; Riveiro, F.; Bolinches-Amorós, A.; Pallardó, F.V.; Fernandez-Vilata, A.; de la Iglesia-Vaya, M.; Palau, F.; Gonzalez-Cabo, P. Reversible axonal dystrophy by calcium modulation in frataxin-deficient sensory neurons of YG8R mice. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Abeti, R.; Brown, A.F.; Maiolino, M.; Patel, S.; Giunti, P. Calcium Deregulation: Novel Insights to Understand Friedreich’s Ataxia Pathophysiology. Front. Cell. Neurosci. 2018, 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mincheva-Tasheva, S.; Obis, E.; Tamarit, J.; Ros, J. Apoptotic cell death and altered calcium homeostasis caused by frataxin depletion in dorsal root ganglia neurons can be prevented by BH4 domain of Bcl-xL protein. Hum. Mol. Genet. 2014, 23, 1829–1841. [Google Scholar] [CrossRef] [Green Version]
- Friedreich, N. Ueber degenerative Atrophie der spinalen Hinterstränge. Arch. Pathol. Anat. Physiol. Klin. Med. 1863, 26, 391–419. [Google Scholar] [CrossRef]
- Schöls, L.; Amoiridis, G.; Przuntek, H.; Frank, G.; Epplen, J.T.; Epplen, C. Friedreich’s ataxia. Revision of the phenotype according to molecular genetics. Brain 1997, 120, 2131–2140. [Google Scholar] [CrossRef] [Green Version]
- Delatycki, M.B.; Paris, D.B.; Gardner, R.J.; Nicholson, G.A.; Nassif, N.; Storey, E.; MacMillan, J.C.; Collins, V.; Williamson, R.; Forrest, S.M. Clinical and genetic study of Friedreich ataxia in an Australian population. Am. J. Med. Genet. 1999, 87, 168–174. [Google Scholar] [CrossRef]
- Lecocq, C.; Charles, P.; Azulay, J.-P.; Meissner, W.; Rai, M.; N’Guyen, K.; Pereon, Y.; Fabre, N.; Robin, E.; Courtois, S.; et al. Delayed-onset Friedreich’s ataxia revisited. Mov. Disord. 2016, 31, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Tsou, A.Y.; Paulsen, E.K.; Lagedrost, S.J.; Perlman, S.L.; Mathews, K.D.; Wilmot, G.R.; Ravina, B.; Koeppen, A.H.; Lynch, D.R. Mortality in Friedreich ataxia. J. Neurol. Sci. 2011, 307, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Trouillas, P.; Takayanagi, T.; Hallett, M.; Currier, R.D.; Subramony, S.H.; Wessel, K.; Bryer, A.; Diener, H.C.; Massaquoi, S.; Gomez, C.M.; et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J. Neurol. Sci. 1997, 145, 205–211. [Google Scholar] [CrossRef]
- Cano, S.J.; Hobart, J.C.; Hart, P.E.; Korlipara, L.V.P.; Schapira, A.H.V.; Cooper, J.M. International cooperative ataxia rating scale (ICARS): Appropriate for studies of Friedreich’s ataxia? Mov. Disord. 2005, 20, 1585–1591. [Google Scholar] [CrossRef]
- Subramony, S.H.; May, W.; Lynch, D.; Gomez, C.; Fischbeck, K.; Hallett, M.; Taylor, P.; Wilson, R.; Ashizawa, T. Measuring Friedreich ataxia: Interrater reliability of a neurologic rating scale. Neurology 2005, 64, 1261–1262. [Google Scholar] [CrossRef]
- Schmitz-Hubsch, T.; du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.-S.; et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef]
- Patel, M.; Isaacs, C.J.; Seyer, L.; Brigatti, K.; Gelbard, S.; Strawser, C.; Foerster, D.; Shinnick, J.; Schadt, K.; Yiu, E.M.; et al. Progression of Friedreich ataxia: Quantitative characterization over 5 years. Ann. Clin. Transl. Neurol. 2016, 3, 684–694. [Google Scholar] [CrossRef] [Green Version]
- Rummey, C.; Corben, L.A.; Delatycki, M.B.; Subramony, S.H.; Bushara, K.; Gomez, C.M.; Hoyle, J.C.; Yoon, G.; Ravina, B.; Mathews, K.D.; et al. Psychometric properties of the Friedreich Ataxia Rating Scale. Neurol. Genet. 2019, 5, 371. [Google Scholar] [CrossRef] [Green Version]
- Delatycki, M.B.; Corben, L.A. Clinical Features of Friedreich Ataxia. J. Child Neurol. 2012, 27, 1133–1137. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich Ataxia: Neuropathology Revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Ackroyd, R.S.; Finnegan, J.A.; Green, S.H. Friedreich’s ataxia. A clinical review with neurophysiological and echocardiographic findings. Arch. Dis. Child. 1984, 59, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morral, J.A.; Davis, A.N.; Qian, J.; Gelman, B.B.; Koeppen, A.H. Pathology and pathogenesis of sensory neuropathy in Friedreich’s ataxia. Acta Neuropathol. 2010, 120, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Mazurkiewicz, J.E. Dorsal root ganglia in Friedreich ataxia: Satellite cell proliferation and inflammation. Acta Neuropathol. Commun. 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachbauer, W.; Boesch, S.; Reindl, M.; Eigentler, A.; Hufler, K.; Poewe, W.; Lo, W.; Wanschitz, J. Skeletal Muscle Involvement in Friedreich Ataxia and Potential Effects of Recombinant Human Erythropoietin Administration on Muscle Regeneration and Neovascularization. Am. Assoc. Neuropatol. 2012, 71, 708–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, A.H.; Davis, A.N.; Morral, J.A. The cerebellar component of Friedreich’s ataxia. Acta Neuropathol. 2011, 122, 323–330. [Google Scholar] [CrossRef] [Green Version]
- França, M.C., Jr.; D’Abreu, A.; Yasuda, C.L.; Cardoso Bonadia, L.; Santos da Silva, M.; Nucci, A.; Lopez-Cendes, I.; Cendes, F. A combined voxel-based morphometry and 1 H-MRS study in patients with Friedreich’s ataxia. J. Neurol. 2009, 256, 1114–1120. [Google Scholar] [CrossRef]
- Della Nave, R.; Ginestroni, A.; Giannelli, M.; Tessa, C.; Salvatore, E.; Salvi, F.; Dotti, M.T.; De Michele, G.; Piacentini, S.; Mascalchi, M. Brain structural damage in Friedreich’s ataxia. J. Neurol. Neurosurg. Psychiatry 2008, 79, 82–85. [Google Scholar] [CrossRef]
- Folker, J.; Murdoch, B.; Cahill, L.; Delatycki, M.; Corben, L.; Vogel, A. Dysarthria in Friedreich’s ataxia: A perceptual analysis. Folia Phoniatr. Logop. 2010, 62, 97–103. [Google Scholar] [CrossRef]
- Mantovan, M.C.; Martinuzzi, A.; Squarzanti, F.; Bolla, A.; Silvestri, I.; Liessi, G.; Macchi, C. Exploring mental status in Friedreich’s ataxia: A combined neuropsychological, behavioral and neuroimaging study. Eur. J. Neurol. 2006, 13, 827–835. [Google Scholar] [CrossRef]
- Rance, G.; Fava, R.; Baldock, H.; Chong, A.; Barker, E.; Corben, L.; Delatycki, M.B. Speech perception ability in individuals with Friedreich ataxia. Brain 2008, 131, 2002–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortuna, F.; Barboni, P.; Liguori, R.; Valentino, M.L.; Savini, G.; Gellera, C.; Mariotti, C.; Rizzo, G.; Tonon, C.; Manners, D.; et al. Visual system involvement in patients with Friedreich’s ataxia. Brain 2009, 132, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keage, M.J.; Delatycki, M.B.; Gupta, I.; Corben, L.A.; Vogel, A.P. Dysphagia in Friedreich Ataxia. Dysphagia 2017, 32, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Rummey, C.; Bijnens, B.; Störk, S.; Jasaityte, R.; Dhooge, J.; Baltabaeva, A.; Sutherland, G.; Schulz, J.B.; Meier, T. The heart in Friedreich ataxia: Definition of cardiomyopathy, disease severity, and correlation with neurological symptoms. Circulation 2012, 125, 1626–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Yang, X.; Feustel, P.J.; Mazurkiewicz, J.E. The Pathogenesis of Cardiomyopathy in Friedreich Ataxia. PLoS ONE 2015, e0116396. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, B.; Francis, J.M.; Cooke, F.; Korlipara, L.V.P.; Blamire, A.M.; Schapira, A.H.V.; Madan, J.; Neubauer, S.; Cooper, J.M. Analysis of the factors influencing the cardiac phenotype in Friedreich’s ataxia. Mov. Disord. 2010, 25, 846–852. [Google Scholar] [CrossRef]
- Schadt, K.A.; Friedman, L.S.; Regner, S.R.; Mark, G.E.; Lynch, D.R.; Lin, K.Y. Cross-Sectional Analysis of Electrocardiograms in a Large Heterogeneous Cohort of Friedreich Ataxia Subjects. J. Child Neurol. 2012, 27, 1187–1192. [Google Scholar] [CrossRef] [Green Version]
- Bourke, T.; Keane, D. Friedreich’s Ataxia: A review from a cardiology perspective. Ir. J. Med. Sci. 2011, 180, 799–805. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Baio, G.; Micossi, P.; Pozza, G.; Di Donato, S. Glucose metabolism alterations in Friedreich’s ataxia. Neurology 1988, 38, 1292–1296. [Google Scholar] [CrossRef]
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126, 94–102. [Google Scholar] [CrossRef]
- Coppola, G.; Marmolino, D.; Lu, D.; Wang, Q.; Cnop, M.; Rai, M.; Acquaviva, F.; Cocozza, S.; Pandolfo, M.; Geschwind, D.H. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet. 2009, 18, 2452–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, S.V.; Phatak, K.; Hoyle, J.C.; Pennell, M.L.; McCarthy, B.; Tran, T.; Prior, T.W.; Olesik, J.W.; Lutton, A.; Rankin, C.; et al. Impaired myocardial perfusion reserve and fibrosis in Friedreich ataxia: A mitochondrial cardiomyopathy with metabolic syndrome. Eur. Heart J. 2010, 32, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delatycki, M.B.; Holian, A.; Corben, L.; Rawicki, H.B.; Blackburn, C.; Hoare, B.; Toy, M.; Churchyard, A. Surgery for Equinovarus Deformity in Friedreich’s Ataxia Improves Mobility and Independence. Clin. Orthop. Relat. Res.® 2005, 430, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, J.B.; Côté, M.; Lemieux, B. The Cardiomyopathy of Friedreich’s Ataxia Morphological Observations in 3 Cases. Can. J. Neurol. Sci./J. Can. Sci. Neurol. 1980, 7, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Meneghini, R. Iron homeostasis, oxidative stress, and DNA damage. Free Radic. Biol. Med. 1997, 23, 783–792. [Google Scholar] [CrossRef]
- Lodi, R.; Cooper, J.M.; Bradley, J.L.; Manners, D.; Styles, P.; Taylor, D.J.; Schapira, A.H. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc. Natl. Acad. Sci. USA 1999, 96, 11492–11495. [Google Scholar] [CrossRef] [Green Version]
- Llorens, J.V.; Soriano, S.; Calap-Quintana, P.; Gonzalez-Cabo, P.; Moltó, M.D. The Role of Iron in Friedreich’s Ataxia: Insights From Studies in Human Tissues and Cellular and Animal Models. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef]
- Schulz, J.B.; Dehmer, T.; Schols, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Burk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Tozzi, G. Antioxidant enzymes in blood of patients with Friedreich’s ataxia. Arch. Dis. Child. 2002, 86, 376–379. [Google Scholar] [CrossRef] [Green Version]
- Jasoliya, M.J.; McMackin, M.Z.; Henderson, C.K.; Perlman, S.L.; Cortopassi, G.A. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum. Mol. Genet. 2017, 26, 2627–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchère, F.; Lönn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Lasso, D.C.; Mollá, B.; Calap-Quintana, P.; García-Giménez, J.L.; Pallardo, F.V.; Palau, F.; Gonzalez-Cabo, P. Cofilin dysregulation alters actin turnover in frataxin-deficient neurons. Sci. Rep. 2020, 10, 5207. [Google Scholar] [CrossRef] [Green Version]
- Navarro, J.A.; Ohmann, E.; Sanchez, D.; Botella, J.A.; Liebisch, G.; Molto, M.D.; Ganfornina, M.D.; Schmitz, G.; Schneuwly, S.; Moltó, M.D.; et al. Altered lipid metabolism in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2010, 19, 2828–2840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson, M.H.; Schulz, J.B.; Giunti, P. Co-enzyme Q10 and idebenone use in Friedreich’s ataxia. J. Neurochem. 2013, 126, 125–141. [Google Scholar] [CrossRef] [Green Version]
- Lynch, D.R.; Willi, S.M.; Wilson, R.B.; Cotticelli, M.G.; Brigatti, K.W.; Deutsch, E.C.; Kucheruk, O.; Shrader, W.; Rioux, P.; Miller, G.; et al. A0001 in Friedreich ataxia: Biochemical characterization and effects in a clinical trial. Mov. Disord. 2012, 27, 1026–1033. [Google Scholar] [CrossRef]
- Shire Safety and Pharmacology Study of VP 20629 in Adults with Friedreich’s Ataxia. Available online: https://www.clinicaltrials.gov/ct2/show/study/NCT01898884 (accessed on 1 April 2020).
- Efficacy of EGb761 in Patients Suffering from Friedreich Ataxia—Tabular View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT00824512 (accessed on 2 April 2020).
- Qureshi, M.Y.; Patterson, M.C.; Clark, V.; Johnson, J.N.; Moutvic, M.A.; Driscoll, S.W.; Kemppainen, J.L.; Huston, J.; Anderson, J.R.; Badley, A.D.; et al. Safety and Efficacy of (+)-Epicatechin in Subjects with Friedreich’s Ataxia: A Phase II, Open-Label, Prospective Study. J. Inherit. Metab. Dis. 2020, jimd.12285. [Google Scholar] [CrossRef]
- Costantini, A.; Laureti, T.; Pala, M.I.; Colangeli, M.; Cavalieri, S.; Pozzi, E.; Brusco, A.; Salvarani, S.; Serrati, C.; Fancellu, R. Long-term treatment with thiamine as possible medical therapy for Friedreich ataxia. J. Neurol. 2016, 263, 2170–2178. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26. [Google Scholar] [CrossRef]
- RTA 408 Capsules in Patients With Friedreich’s Ataxia—MOXIe—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02255435 (accessed on 13 July 2020).
- Reata Announces Positive Topline Results from the MOXIe Registrational Trial of Omaveloxolone in Patients with Friedreich’s Ataxia—Reata Pharmaceuticals. Available online: https://www.reatapharma.com/press-releases/reata-announces-positive-topline-results-from-the-moxie-registrational-trial-of-omaveloxolone-in-patients-with-friedreichs-ataxia/ (accessed on 14 July 2020).
- Yiu, E.M.; Tai, G.; Peverill, R.E.; Lee, K.J.; Croft, K.D.; Mori, T.A.; Scheiber-Mojdehkar, B.; Sturm, B.; Praschberger, M.; Vogel, A.P.; et al. An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J. Neurol. 2015, 262, 1344–1353. [Google Scholar] [CrossRef]
- Micronised Resveratrol as a Treatment for Friedreich Ataxia—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03933163 (accessed on 13 May 2020).
- Effect of Pioglitazone Administered to Patients With Friedreich’s Ataxia: Proof of Concept—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT00811681 (accessed on 16 April 2020).
- A Clinical Study to Evaluate the Effect of MIN-102 on the Progression of Friedreich’s Ataxia in Male and Female Patients—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03917225 (accessed on 13 July 2020).
- Schöls, L.; Zange, J.; Abele, M.; Schillings, M.; Skipka, G.; Kuntz-Hehner, S.; Van Beekvelt, M.C.P.; Colier, W.N.J.M.; Müller, K.; Klockgether, T.; et al. L-carnitine and creatine in Friedreich’s ataxia. A randomized, placebo-controlled crossover trial. J. Neural Transm. 2005, 112, 789–796. [Google Scholar] [CrossRef] [PubMed]
- An Open-label Study of the Effects of Acetyl-L-Carnitine on Cardiovascular Outcomes in Friedreich’s Ataxia—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01921868 (accessed on 3 April 2020).
- Pandolfo, M.; Arpa, J.; Delatycki, M.B.; Le Quan Sang, K.H.; Mariotti, C.; Munnich, A.; Sanz-Gallego, I.; Tai, G.; Tarnopolsky, M.A.; Taroni, F.; et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann. Neurol. 2014, 76, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Sánchez, D.; Aracil, A.; Montero, R.; Mas, A.; Jiménez, L.; O’Callaghan, M.; Tondo, M.; Capdevila, A.; Blanch, J.; Artuch, R.; et al. Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Arpa, J.; Sanz-Gallego, I.; Rodríguez-de-Rivera, F.J.; Domínguez-Melcón, F.J.; Prefasi, D.; Oliva-Navarro, J.; Moreno-Yangüela, M. Triple therapy with deferiprone, idebenone and riboflavin in Friedreich’s ataxia—Open-label trial. Acta Neurol. Scand. 2014, 129, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242. [Google Scholar] [CrossRef]
- PTC Therapeutics Provides Corporate Update and Highlights Pipeline Progress at 2020 J.P. Morgan Healthcare Conference. PTC Therapeutics, Inc. Available online: https://ir.ptcbio.com/news-releases/news-release-details/ptc-therapeutics-provides-corporate-update-and-highlights (accessed on 22 April 2020).
- Zesiewicz, T.; Heerinckx, F.; De Jager, R.; Omidvar, O.; Kilpatrick, M.; Shaw, J.; Shchepinov, M.S. Randomized, clinical trial of RT001: Early signals of efficacy in Friedreich’s ataxia. Mov. Disord. 2018, 33, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- A Study to Assess Efficacy, Long Term Safety and Tolerability of RT001 in Subjects with Friedreich’s Ataxia—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04102501 (accessed on 13 July 2020).
- Orsucci, D.; Mancuso, M.; Ienco, E.C.; LoGerfo, A.; Siciliano, G. Targeting Mitochondrial Dysfunction and Neurodegeneration by Means of Coenzyme Q10 and its Analogues. Curr. Med. Chem. 2011, 18, 4053–4064. [Google Scholar] [CrossRef]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Jauslin, M.L.; Meier, T.; Smith, R.A.J.; Murphy, P.M. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1–10. [Google Scholar] [CrossRef]
- Lodi, R.; Hart, P.E.; Rajagopalan, B.; Taylor, D.J.; Crilley, J.G.; Bradley, J.L.; Blamire, A.M.; Manners, D.; Styles, P.; Schapira, A.H.; et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann. Neurol. 2001, 49, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.E.; Lodi, R.; Rajagopalan, B.; Bradley, J.L.; Crilley, J.G.; Turner, C.; Blamire, A.M.; Manners, D.; Styles, P.; Schapira, A.H.V.; et al. Antioxidant treatment of patients with Friedreich ataxia: Four-year follow-up. Arch. Neurol. 2005, 62, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Korlipara, L.V.P.; Hart, P.E.; Bradley, J.L.; Schapira, A.H.V. Coenzyme Q10 and vitamin e deficiency in Friedreich’s ataxia: Predictor of efficacy of vitamin e and coenzyme Q10 therapy. Eur. J. Neurol. 2008, 15, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J.C.; Benfield, P.; McTavish, D. Idebenone: A Review of its Pharmacodynamic and Pharmacokinetic Properties, and Therapeutic Use in Age-Related Cognitive Disorders. Drugs Aging 1994, 5, 133–152. [Google Scholar] [CrossRef]
- Soriano, S.; Llorens, J.V.; Blanco-Sobero, L.; Gutiérrez, L.; Calap-Quintana, P.; Morales, M.P.; Moltó, M.D.; Martínez-Sebastián, M.J. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 2013, 521, 274–281. [Google Scholar] [CrossRef] [Green Version]
- Seznec, H. Idebenone delays the onset of cardiac functional alteration without correction of Fe-S enzymes deficit in a mouse model for Friedreich ataxia. Hum. Mol. Genet. 2004, 13, 1017–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustin, P.; Von Kleist-Retzow, J.C.; Chantrel-Groussard, K.; Sidi, D.; Munnich, A.; Rötig, A. Effect of idebenone on cardiomyopathy in Friedreich’s ataxia: A preliminary study. Lancet 1999, 354, 477–479. [Google Scholar] [CrossRef]
- Di Prospero, N.A.; Baker, A.; Jeffries, N.; Fischbeck, K.H. Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: A randomised, placebo-controlled trial. Lancet Neurol. 2007, 6, 878–886. [Google Scholar] [CrossRef] [Green Version]
- Lynch, D.R.; Perlman, S.L.; Meier, T. A phase 3, double-blind, placebo-controlled trial of idebenone in Friedreich Ataxia. Arch. Neurol. 2010, 67, 941–947. [Google Scholar] [CrossRef]
- Lagedrost, S.J.; Sutton, M.S.J.; Cohen, M.S.; Satou, G.M.; Kaufman, B.D.; Perlman, S.L.; Rummey, C.; Meier, T.; Lynch, D.R. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA). Am. Heart J. 2011, 161, 639–645.e1. [Google Scholar] [CrossRef]
- A Study of Efficacy, Safety and Tolerability of Idebenone in the Treatment of Friedreich’s Ataxia (FRDA) Patients—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00905268 (accessed on 31 March 2020).
- Cook, A.; Boesch, S.; Heck, S.; Brunt, E.; Klockgether, T.; Schöls, L.; Schulz, A.; Giunti, P. Patient-reported outcomes in Friedreich’s ataxia after withdrawal from idebenone. Acta Neurol. Scand. 2019, 139, 533–539. [Google Scholar] [CrossRef]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Pandolfo, M. Antioxidants and other pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2009. [Google Scholar] [CrossRef]
- Hawi, A.; Heald, S.; Sciascia, T. Use of an adaptive study design in single ascending-dose pharmacokinetics of A0001 (α-tocopherylquinone) in healthy male subjects. J. Clin. Pharmacol. 2012, 52, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Srivastav, S.; Castellani, R.J.; Plascencia-Villa, G.; Perry, G. Neuroprotective and Antioxidant Effect of Ginkgo biloba Extract Against AD and Other Neurological Disorders. Neurotherapeutics 2019, 16, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer β-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, I.K.; Yoo, K.Y.; Li, H.; Park, O.K.; Lee, C.H.; Choi, J.H.; Jeong, Y.G.; Lee, Y.L.; Kim, Y.M.; Kwon, Y.G.; et al. Indole-3-propionic acid attenuates neuronal damage and oxidative stress in the ischemic hippocampus. J. Neurosci. Res. 2009, 87, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Bendheim, P.E.; Poeggeler, B.; Neria, E.; Ziv, V.; Pappolla, M.A.; Chain, D.G. Development of indole-3-propionic acid (OXIGONTM) for Alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 213–217. [Google Scholar] [CrossRef]
- Bernatoniene, J.; Kopustinskiene, D.M. The Role of Catechins in Cellular Responses to Oxidative Stress. Molecules 2018, 23, 965. [Google Scholar] [CrossRef] [Green Version]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Luigi, M.; Salvo, D.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; et al. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5, 12583. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Ke, Z.; Luo, J. Thiamine Deficiency and Neurodegeneration: The Interplay Among Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. Mol. Neurobiol. 2017, 54, 5440–5448. [Google Scholar] [CrossRef]
- Pedraza, O.L.; Botez, M.I. Thiamine status in inherited degenerative ataxias. J. Neurol. Neurosurg. Psychiatry 1992, 55, 136–137. [Google Scholar] [CrossRef]
- Botez, M.I.; Young, S.N. Biogenic amine metabolites and thiamine in cerebrospinal fluid in heredo-degenerative ataxias. Can. J. Neurol. Sci. 2001, 28, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Jia, Z.; Zhu, H. Regulation of Nrf2 Signaling. React. Oxyg. Species (Apex, NC) 2019, 8, 312–322. [Google Scholar]
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L.H. Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3β Axis. Am. J. Pathol. 2017, 187, 2858–2875. [Google Scholar] [CrossRef] [Green Version]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [Green Version]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s Ataxia YG8R mouse model. Antioxidants Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [Green Version]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [Green Version]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Cabo, P.; Palau, F. Mitochondrial pathophysiology in Friedreich’s ataxia. J. Neurochem. 2013, 126, 53–64. [Google Scholar] [CrossRef]
- Reisman, S.A.; Gahir, S.S.; Lee, C.Y.I.; Proksch, J.W.; Sakamoto, M.; Ward, K.W. Pharmacokinetics and pharmacodynamics of the novel NrF2 activator omaveloxolone in primates. Drug Des. Devel. Ther. 2019, 13, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Robledinos-Antón, N.; Fernández-Ginés, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-inducer prevents mitochondrial defects and oxidative stress in friedreich’s ataxia models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef] [PubMed]
- Pannu, N.; Bhatnagar, A. Resveratrol: From enhanced biosynthesis and bioavailability to multitargeting chronic diseases. Biomed. Pharmacother. 2019, 109, 2237–2251. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Voullaire, L.; Sandi, C.; Pook, M.A.; Ioannou, P.A. Pharmacological Screening Using an FXN-EGFP Cellular Genomic Reporter Assay for the Therapy of Friedreich Ataxia. PLoS ONE 2013, 8, e0055940. [Google Scholar] [CrossRef] [Green Version]
- Georges, P.; Boza-Moran, M.G.; Gide, J.; Pêche, G.A.; Forêt, B.; Bayot, A.; Rustin, P.; Peschanski, M.; Martinat, C.; Aubry, L. Induced pluripotent stem cells-derived neurons from patients with Friedreich ataxia exhibit differential sensitivity to resveratrol and nicotinamide. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Eder, K.; Kish, S.J.; Kirchgessner, M.; Ross, B.M. Brain phospholipids and fatty acids in Friedreich’s ataxia and spinocerebellar atrophy type-1. Mov. Disord. 1998, 13, 813–819. [Google Scholar] [CrossRef]
- Worth, A.J.; Basu, S.S.; Deutsch, E.C.; Hwang, W.-T.; Snyder, N.W.; Lynch, D.R.; Blair, I.A. Stable isotopes and LC-MS for monitoring metabolic disturbances in Friedreich’s ataxia platelets. Bioanalysis 2015, 7, 1843–1855. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.J.; Bouchard, R.; Gauthier, G.L.; Bouchard, J.P.; Barbeau, A. Quantitative metabolic profiling of alpha-keto acids in Friedreich’s ataxia. Can. J. Neurol. Sci. 1982, 9, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Davignon, J.; Huang, Y.S.; Wolf, J.P.; Barbeau, A. Fatty acid profile of major lipid classes in plasma lipoproteins of patients with Friedreich’s ataxia-demonstration of a low linoleic acid content most evident in the cholesterol-ester fraction. Can. J. Neurol. Sci. 1979, 6, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Obis, È.; Irazusta, V.; Sanchís, D.; Ros, J.; Tamarit, J. Frataxin deficiency in neonatal rat ventricular myocytes targets mitochondria and lipid metabolism. Free Radic. Biol. Med. 2014, 73, 21–33. [Google Scholar] [CrossRef]
- Coppola, G.; Burnett, R.; Perlman, S.; Versano, R.; Gao, F.; Plasterer, H.; Rai, M.; Saccá, F.; Filla, A.; Lynch, D.R.; et al. A gene expression phenotype in lymphocytes from friedreich ataxia patients. Ann. Neurol. 2011, 70, 790–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, G.R.; Pride, P.M.; Babbey, C.M.; Payne, R.M. Friedreich’s ataxia reveals a mechanism for coordinate regulation of oxidative metabolism via feedback inhibition of the SIRT3 deacetylase. Hum. Mol. Genet. 2012, 21, 2688–2697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Yang, T.; Liu, H.; Han, L.; Zhang, K.; Hu, X.; Zhang, X.; Yin, K.J.; Gao, Y.; Bennett, M.V.L.; et al. Peroxisome proliferator-activated receptor γ (PPARγ): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163–164, 27–58. [Google Scholar] [CrossRef]
- Handschin, C.; Spiegelman, B.M. Peroxisome Proliferator-Activated Receptor γ Coactivator 1 Coactivators, Energy Homeostasis, and Metabolism. Endocr. Rev. 2006, 27, 728–735. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of Reactive Oxygen Species and Neurodegeneration by the PGC-1 Transcriptional Coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Marmolino, D.; Manto, M.; Acquaviva, F.; Vergara, P.; Ravella, A.; Monticelli, A.; Pandolfo, M. PGC-1alpha down-regulation affects the antioxidant response in Friedreich’s ataxia. PLoS ONE 2010, 5, e10025. [Google Scholar] [CrossRef]
- Corona, J.C.; Duchen, M.R. PPARγ as a therapeutic target to rescue mitochondrial function in neurological disease. Free Radic. Biol. Med. 2016, 100, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Soccio, R.E.; Chen, E.R.; Lazar, M.A. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014, 20, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Minoryx Leriglitazone. Available online: https://www.minoryx.com/leriglitazone/ (accessed on 16 April 2020).
- Poli, S.; Rodríguez-Pascau, L.; Britti, E.; Ros, J.; González-Cabo, P.; Lynch, D.; Martinell, M.; Pizcueta, P. MIN102 (Leriglitazone), a Brain Penetrant PPAR Gamma Agonist for the Treatment of Friedreich’s Ataxia. In Proceedings of the American Academy of Neurology 2020 Annual Meeting, Toronto, ON, Canada, 25 April–1 May 2020; p. 4147. [Google Scholar]
- Garcia-Gimenez, J.L.; Sanchis-Gomar, F.; Pallardo, F.V. Could thiazolidinediones increase the risk of heart failure in Friedreich’s ataxia patients? Mov. Disord. 2011, 26, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, G.C.; Mckenna, M.C. L-Carnitine and acetyl-L-carnitine roles and neuroprotection in developing brain. Neurochem. Res. 2017, 42, 1661–1675. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.L.-H.; Sivagurunathan, S.; Ting, S.; Jansson, P.J.; Austin, C.J.D.; Kelly, M.; Semsarian, C.; Zhang, D.; Richardson, D.R. Molecular and functional alterations in a mouse cardiac model of Friedreich ataxia: Activation of the integrated stress response, eIF2α phosphorylation, and the induction of downstream targets. Am. J. Pathol. 2013, 183, 745–757. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The dentate nucleus in Friedreich’s ataxia: The role of iron-responsive proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.; Puccio, H. Dysregulation of cellular iron metabolism in Friedreich ataxia: From primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front. Pharmacol. 2014, 5, 130. [Google Scholar] [CrossRef] [Green Version]
- Whitnall, M.; Suryo Rahmanto, Y.; Sutak, R.; Xu, X.; Becker, E.M.; Mikhael, M.R.; Ponka, P.; Richardson, D.R. The MCK mouse heart model of Friedreich’s ataxia: Alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc. Natl. Acad. Sci. USA 2008, 105, 9757–9762. [Google Scholar] [CrossRef] [Green Version]
- Bonilha da Silva, C.; Bergo, F.P.G.; D’Abreu, A.; Cendes, F.; Lopes-Cendes, I.; França, M.C. Dentate nuclei T2 relaxometry is a reliable neuroimaging marker in Friedreich’s ataxia. Eur. J. Neurol. 2014, 21, 1131–1136. [Google Scholar] [CrossRef]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.L.-H.; Becker, E.M.; Whitnall, M.; Suryo Rahmanto, Y.; Ponka, P.; Richardson, D.R. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef] [Green Version]
- Whitnall, M.; Suryo Rahmanto, Y.; Huang, M.L.-H.; Saletta, F.; Lok, H.C.; Gutiérrez, L.; Lázaro, F.J.; Fleming, A.J.; St Pierre, T.G.; Mikhael, M.R.; et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. USA 2012, 109, 20590–20595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Reichmann, H. Role of iron in neurodegenerative diseases. J. Neural Transm. 2016, 123, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef]
- Botzenhardt, S.; Li, N.; Chan, E.W.; Sing, C.W.; Wong, I.C.K.; Neubert, A. Safety profiles of iron chelators in young patients with haemoglobinopathies. Eur. J. Haematol. 2017, 98, 198–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.-S.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008, 111, 1690–1699. [Google Scholar] [CrossRef]
- Pandolfo, M.; Hausmann, L. Deferiprone for the treatment of Friedreich’s ataxia. J. Neurochem. 2013, 126 (Suppl), 142–146. [Google Scholar] [CrossRef]
- Kakhlon, O.; Manning, H.; Breuer, W.; Melamed-Book, N.; Lu, C.; Cortopassi, G.; Munnich, A.; Cabantchik, Z.I. Cell functions impaired by frataxin deficiency are restored by drug-mediated iron relocation. Blood 2008, 112, 5219–5227. [Google Scholar] [CrossRef]
- Goncalves, S.; Paupe, V.; Dassa, E.P.; Rustin, P. Deferiprone targets aconitase: Implication for Friedreich’s ataxia treatment. BMC Neurol. 2008, 8, 20. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavi, A.; Torgovnick, A.; Kell, A.; Megalou, E.; Castelein, N.; Guccini, I.; Marzocchella, L.; Gelino, S.; Hansen, M.; Malisan, F.; et al. Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp. Gerontol. 2013, 48, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.S.; Nestruck, A.C.; Barbeau, A.; Bouchard, J.P.; Davignon, J. Plasma lipids and lipoproteins in Friedreich’s ataxia and familial spastic ataxia—Evidence for an abnormal composition of high density lipoproteins. Can. J. Neurol. Sci. 1978, 5, 149–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.L.; Chamberlain, S.; Robinson, N. Lipids and lipoproteins in Friedreich’s ataxia. J. Neurol. Neurosurg. Psychiatry 1980, 43, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolson, G.L. Metabolic syndrome and mitochondrial function: Molecular replacement and antioxidant supplements to prevent membrane peroxidation and restore mitochondrial function. J. Cell. Biochem. 2007, 100, 1352–1369. [Google Scholar] [CrossRef]
- Wu, J.-R.; Tuo, Q.-Z.; Lei, P. Ferroptosis, a Recent Defined Form of Critical Cell Death in Neurological Disorders. J. Mol. Neurosci. 2018, 66, 197–206. [Google Scholar] [CrossRef]
- Abeti, R.; Uzun, E.; Renganathan, I.; Honda, T.; Pook, M.A.; Giunti, P.; Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; et al. Targeting lipid peroxidation and mitochondrial imbalance in Friedreich’s ataxia. Pharmacol. Res. 2015, 99, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Auchere, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.-M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet. 2008, 17, 2790–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulteau, A.L.; Planamente, S.; Jornea, L.; Dur, A.; Lesuisse, E.; Camadro, J.M.; Auchère, F. Changes in mitochondrial glutathione levels and protein thiol oxidation in ∆yfh1 yeast cells and the lymphoblasts of patients with Friedreich’s ataxia. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Tan, G. Decreased expression of genes involved in sulfur amino acid metabolism in frataxin-deficient cells. Hum. Mol. Genet. 2003, 12, 1699–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calap-Quintana, P.; Soriano, S.; Llorens, J.V.; Al-Ramahi, I.; Botas, J.; Moltó, M.D.; Martínez-Sebastián, M.J. TORC1 Inhibition by Rapamycin Promotes Antioxidant Defences in a Drosophila Model of Friedreich’s Ataxia. PLoS ONE 2015, 10, e0132376. [Google Scholar] [CrossRef] [PubMed]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Investig. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Hider, R.C.; Kong, X.L. Glutathione: A key component of the cytoplasmic labile iron pool. BioMetals 2011, 24, 1179–1187. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Grazia Cotticelli, M.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a novel therapeutic target for Friedreich’s ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Li, Y.; Xia, J.; Chen, Y.; Chen, S.; Wang, X.; Sun, W.; Wang, T.; Ren, X.; et al. Identification of Frataxin as a regulator of ferroptosis. Redox Biol. 2020, 32. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Banthiya, S.; van Leyen, K. Mammalian lipoxygenases and their biological relevance. Biochim. Biophys. Acta 2015, 1851, 308–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Kahn-Kirby, A.H.; Amagata, A.; Maeder, C.I.; Mei, J.J.; Sideris, S.; Kosaka, Y.; Hinman, A.; Malone, S.A.; Bruegger, J.J.; Wang, L.; et al. Targeting ferroptosis: A novel therapeutic strategy for the treatment of mitochondrial disease-related epilepsy. PLoS ONE 2019, 14, e0214250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrader, W.D.; Amagata, A.; Barnes, A.; Enns, G.M.; Hinman, A.; Jankowski, O.; Kheifets, V.; Komatsuzaki, R.; Lee, E.; Mollard, P.; et al. α-Tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg. Med. Chem. Lett. 2011, 21, 3693–3698. [Google Scholar] [CrossRef]
- Enns, G.M.; Kinsman, S.L.; Perlman, S.L.; Spicer, K.M.; Abdenur, J.E.; Cohen, B.H.; Amagata, A.; Barnes, A.; Kheifets, V.; Shrader, W.D.; et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012, 105, 91–102. [Google Scholar] [CrossRef]
- Lepretti, M.; Martucciello, S.; Alberto Burgos Aceves, M.; Putti, R.; Lionetti, L. Omega-3 Fatty Acids and Insulin Resistance: Focus on the Regulation of Mitochondria and Endoplasmic Reticulum Stress. Nutrients 2018, 10, 350. [Google Scholar] [CrossRef] [Green Version]
- Cotticelli, M.G.; Crabbe, A.M.; Wilson, R.B.; Shchepinov, M.S. Insights into the role of oxidative stress in the pathology of friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013, 1, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.; Siah, K.W.; Lo, A.W. Estimation of clinical trial success rates and related parameters. Biostatistics 2019, 20, 273–286. [Google Scholar] [CrossRef]
- Pandolfo, M. Neurologic outcomes in Friedreich ataxia. Neurol. Genet. 2020, 6, e415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, G.; Jasoliya, M.; Sahdeo, S.; Saccà, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; et al. Dimethyl Fumarate Mediates Nrf2-Dependent Mitochondrial Biogenesis in Mice and Humans. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6251607/ (accessed on 23 March 2020).
- Seco-Cervera, M.; González-Rodríguez, D.; Ibáñez-Cabellos, J.S.; Peiró-Chova, L.; González-Cabo, P.; García-López, E.; Vílchez, J.J.; Sanz-Gallego, I.; Pallardó, F.V.; García-Giménez, J.L. Circulating miR-323-3p is a biomarker for cardiomyopathy and an indicator of phenotypic variability in Friedreich’s ataxia patients. Sci. Rep. 2017, 7, 5237. [Google Scholar] [CrossRef] [PubMed]
- Seco-Cervera, M.; González-Rodríguez, D.; Ibáñez-Cabellos, J.S.; Peiró-Chova, L.; Pallardó, F.V.; García-Giménez, J.L. Small RNA-seq analysis of circulating miRNAs to identify phenotypic variability in Friedreich’s ataxia patients. Sci. Data 2018, 5, 180021. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Wang, Q.; Weng, L.; Hauser, L.A.; Strawser, C.J.; Rocha, A.G.; Dancis, A.; Mesaros, C.; Lynch, D.R.; Blair, I.A. Liquid Chromatography-High Resolution Mass Spectrometry Analysis of Platelet Frataxin as a Protein Biomarker for the Rare Disease Friedreich’s Ataxia. Anal. Chem. 2018, 90, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- McMackin, M.Z.; Durbin-Johnson, B.; Napierala, M.; Napierala, J.S.; Ruiz, L.; Napoli, E.; Perlman, S.; Giulivi, C.; Cortopassi, G.A. Potential biomarker identification for Friedreich’s ataxia using overlapping gene expression patterns in patient cells and mouse dorsal root ganglion. PLoS ONE 2019, 14, e0223209. [Google Scholar] [CrossRef] [PubMed]
- Zeitlberger, A.M.; Thomas-Black, G.; Garcia-Moreno, H.; Foiani, M.; Heslegrave, A.J.; Zetterberg, H.; Giunti, P. Plasma Markers of Neurodegeneration Are Raised in Friedreich’s Ataxia. Front. Cell. Neurosci. 2018, 12, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, I.A.; Farmer, J.; Hersch, S.; Larkindale, J.; Lynch, D.R.; Napierala, J.; Napierala, M.; Payne, R.M.; Subramony, S.H. The current state of biomarker research for Friedreich’s ataxia: A report from the 2018 FARA biomarker meeting. Futur. Sci. OA 2019, 5, FSO398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef]
- Lefevre, S.; Sliwa, D.; Rustin, P.; Camadro, J.-M.; Santos, R. Oxidative stress induces mitochondrial fragmentation in frataxin-deficient cells. Biochem. Biophys. Res. Commun. 2012, 418, 336–341. [Google Scholar] [CrossRef]
- Llorens, J.V.; Navarro, J.A.; Martínez-Sebastián, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Moltó, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007, 21, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Shoichet, S.A. Frataxin promotes antioxidant defense in a thiol-dependent manner resulting in diminished malignant transformation in vitro. Hum. Mol. Genet. 2002, 11, 815–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases frataxin expression: A potential therapy for Friedreich’s Ataxia. PLoS ONE 2019, 14, e0217776. [Google Scholar] [CrossRef]
- Cortopassi, G.; Danielson, S.; Alemi, M.; Zhan, S.S.; Tong, W.; Carelli, V.; Martinuzzi, A.; Marzuki, S.; Majamaa, K.; Wong, A. Mitochondrial disease activates transcripts of the unfolded protein response and cell cycle and inhibits vesicular secretion and oligodendrocyte-specific transcripts. Mitochondrion 2006, 6, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Edenharter, O.; Schneuwly, S.; Navarro, J.A. Mitofusin-Dependent ER Stress Triggers Glial Dysfunction and Nervous System Degeneration in a Drosophila Model of Friedreich’s Ataxia. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J. Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal. 2018, 2, NS20180061. [Google Scholar] [CrossRef] [Green Version]
- Purroy, R.; Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J. Mitochondrial pore opening and loss of Ca2+ exchanger NCLX levels occur after frataxin depletion. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 618–631. [Google Scholar] [CrossRef]
- Crombie, D.E.; Curl, C.L.; Raaijmakers, A.J.; Sivakumaran, P.; Kulkarni, T.; Wong, R.C.; Minami, I.; Evans-Galea, M.V.; Lim, S.Y.; Delbridge, L.; et al. Friedreich’s ataxia induced pluripotent stem cell-derived cardiomyocytes display electrophysiological abnormalities and calcium handling deficiency. Aging (Albany NY) 2017, 9, 1440–1452. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [Green Version]
- Monnier, V.; Llorens, J.; Navarro, J. Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia. Int. J. Mol. Sci. 2018, 19, 1989. [Google Scholar] [CrossRef] [Green Version]
- Soriano, S.; Calap-Quintana, P.; Llorens, J.V.; Al-Ramahi, I.; Gutiérrez, L.; Martínez-Sebastián, M.J.; Botas, J.; Moltó, M.D. Metal Homeostasis Regulators Suppress FRDA Phenotypes in a Drosophila Model of the Disease. PLoS ONE 2016, 11, e0159209. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Yu, D.; Collins, S.E.; Qian, J.; Parsons, P.J.; Yang, K.X.; Chen, Z.; Mazurkiewicz, J.E.; Feustel, P.J. Friedreich’s ataxia causes redistribution of iron, copper, and zinc in the dentate nucleus. Cerebellum 2012, 11, 845–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdomini, M.; Belbellaa, B.; Monassier, L.; Reutenauer, L.; Messaddeq, N.; Cartier, N.; Crystal, R.G.; Aubourg, P.; Puccio, H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 2014, 20, 542–547. [Google Scholar] [CrossRef]
- Piguet, F.; de Montigny, C.; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. 2018, 26, 1940–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans-Galea, M.V.; Pébay, A.; Dottori, M.; Corben, L.A.; Ong, S.H.; Lockhart, P.J.; Delatycki, M.B. Cell and Gene Therapy for Friedreich Ataxia: Progress to Date. Hum. Gene Ther. 2014, 25, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Shroff, G. A novel approach of human embryonic stem cells therapy in treatment of Friedrich’s Ataxia. Int. J. Case Rep. Images 2015, 6, 261. [Google Scholar] [CrossRef] [Green Version]


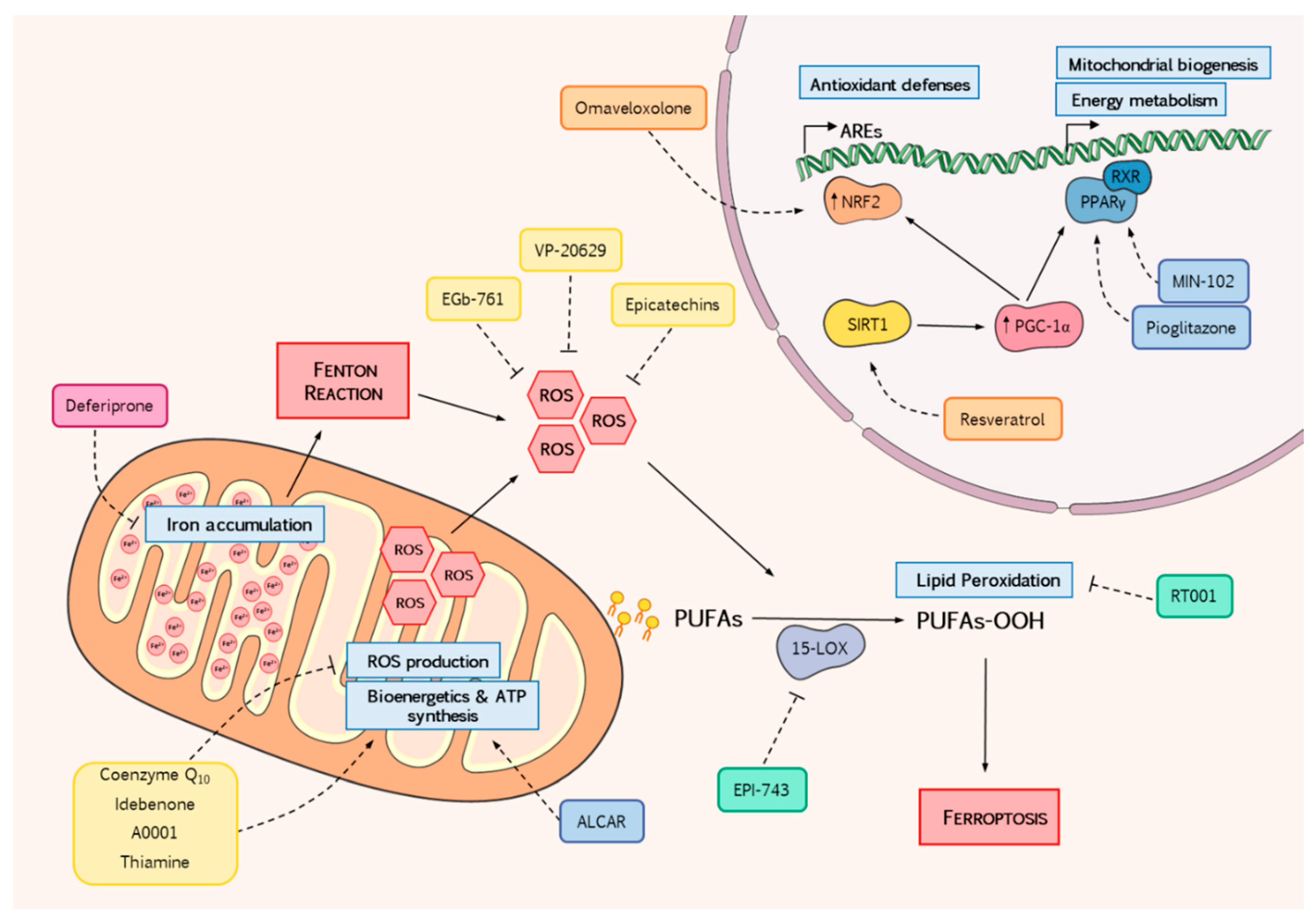{kind=link}
| Compound | Mechanisms of Action | Doses | Clinical Trials | Study Outcomes | Current State |
|---|---|---|---|---|---|
| Direct Reactive Oxygen Species (ROS) Scavengers | |||||
| CoQ10 | Mitochondrial cofactor Radical scavenger Maintains antioxidant molecules | 30–600 mg/day | Several clinical trials have been conducted. Reviewed by Parkinson et al. [75] | Combined therapy of CoQ10-vitamin E improved neurological function and bioenergetics in patients with deficiency in CoQ10 or vitamin E | No further investigation |
| Idebenone | Structural analogue of CoQ10 with enhanced bioavailability | 2.5–75 mg/kg/day 360–2250 mg/day | Variable results in neurological and cardiac function. Inconclusive benefits | No further investigation | |
| A0001 | Enhanced version of CoQ10 High antioxidant potential Alleviates mitochondrial ROS Protects from lipid peroxidation Electron donor to the respiratory complex | 1–1.5 g/day | Phase II, double-blind, placebo-controlled 4-week study with two doses [76] | No differences in primary outcomes (glucose-handling) but significant improvements in Friedreich Ataxia Rating Scale (FARS) score | No further investigation |
| VP-20629 | ROS scavenger Alleviates lipid peroxidation Neuroprotective | 150–1200 mg/day | Phase I, randomized, double-blind, placebo controlled with single or multiple dose for 7 days [77] | Safe and well tolerated. No major benefits reported | No further investigation |
| EGb-761 | Neuroprotective Antioxidant | 240 mg/day | Phase II, randomized, placebo-controlled, 3-months, double blind study [78] | No significant differences were found. Insufficient number of individuals | No further investigation |
| Epicatechins | ROS scavenger Metal ion chelator Promotes antioxidant activity Inhibits Por-oxidant activity | 75–150 mg/day | Phase II, open-label, prospective, 24-weeks, single center study [79] | Improvement in cardiac structure and function. No significant changes in neurological outcomes | No further investigation |
| Thiamine | Specific cofactor in energetic metabolism ROS Scavenger | 200 mg/week | Open-label trial for 2 years [80] | Improvements in Scale for the Assessment and Rating of Ataxia (SARA) score, cardiological outcomes and recovery of motor skills | No further investigation |
| Nrf2 Inducers | |||||
| Omaveloxolone (RTA-408) | Increases antioxidant defenses Reduces Lipid peroxidation and mitochondrial ROS production Ameliorates mitochondrial energy imbalance Prevents from oxidative stress | 2.5–300 mg/day | Phase II/III, multicenter, randomized, placebo-controlled, double-blind, 12-weeks, dose-escalation trial (Part 1 of MOXIe) [81] | No differences in primary outcome (peak work in exercise) but significant improvements in modified version of FARS (mFARS) were reported | NCT02255435 Phase II/III, randomized, placebo-controlled, double-blind, 48 weeks, parallel-group study is still ongoing (Part 2 of MOXIe). Announced results suggest improvement in mFARS [82,83] |
| Resveratrol | Anti-inflammatory Anti-apoptotic Neuroprotective Activates antioxidant defenses | 1–5 g/day | Phase I/II Open-label clinical 12-week pilot study with two doses [84] | Frataxin levels remained unchanged. Lipid peroxidation markers decreased. FARS and ICARS scores improved. Gastrointestinal side effects reported | NCT03933163 Phase II, double-blind, placebo controlled 2-period crossover trial is currently ongoing with an enhanced formulation of resveratrol [85] |
| Mitochondrial Metabolism | |||||
| Pioglitazone | Increases PGC-1α Activates antioxidant defenses Reduces insulin resistance Improves energetic metabolism | 15–45 mg/day | Phase III, prospective, randomized, double-blind, 2-years trial [86] | Pending publication | No further investigation |
| Leriglitazone (MIN-102) | Increases PGC-1α Restores mitochondrial function and bioenergetics Increases DRG neuron survival Improves motor function | Not specified | No previous clinical trials | NCT03917225 Phase II, randomized, double-blind, placebo-controlled study is currently ongoing (FRAMES) [87] | |
| ALCAR | Mitochondrial cofactor Improves bioenergetic dysfunction Neuroprotective Anti-inflammatory Antioxidant | 2–3 g/day | Placebo-controlled, 4-months, triple-phase crossover, creatine-ALCAR combined therapy [88] | No differences in Phosphocreatine, ICARS score or echocardiographic data compared to placebo | NCT01921868 Open-label trial of ALCAR with cardiovascular outcomes for 24 months. Results are pending publication [89] |
| Iron Chelators | |||||
| Deferiprone | Removes iron excess Enhances bioenergetic impairment Decreases ISCs damage | 5–60 mg/kg/day | Several clinical trials have been conducted either with unique or combined therapy [90,91,92] | Improvements in cardiac outcomes, but no neurological effects were reported. Mild adverse effects such as neutropenia were observed | No further investigation |
| Lipid Metabolism | |||||
| EPI-743 (PTC-743) | 15-lipoxigenase inhibitor CoQ10 improved antioxidant potential Anti-inflammatory Modulates energy metabolism | 600 and 1200 mg/day | Phase II, Double-blind, with two phases and two doses. Placebo-controlled trial for 6 months and open-label for 18 months [93] | Safe and well tolerated. First phase: no differences in primary end point (visual acuity) but significant improvements in FARS score with high dose. Second phase: deceleration in severity of the disease, enhancing neurological function | Phase III, registrational trial is planned for 2020 [94] |
| RT001 (Retrotope) | Anti-inflammatory Reduces mitochondrial dysfunction, ROS production and ER stress | 1.8–8.64 g/day | Phase I/II, double-blind placebo-controlled trial with two doses for 28 days [95] | Safe and well tolerated. Both doses improved cardiopulmonary and neurological tests. | NCT04102501 Phase III, randomized, double-blind, placebo-controlled trial is ongoing [96] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, L.R.; Lapeña, T.; Calap-Quintana, P.; Moltó, M.D.; Gonzalez-Cabo, P.; Navarro Langa, J.A. Antioxidant Therapies and Oxidative Stress in Friedreich’s Ataxia: The Right Path or Just a Diversion? Antioxidants 2020, 9, 664. https://doi.org/10.3390/antiox9080664
Rodríguez LR, Lapeña T, Calap-Quintana P, Moltó MD, Gonzalez-Cabo P, Navarro Langa JA. Antioxidant Therapies and Oxidative Stress in Friedreich’s Ataxia: The Right Path or Just a Diversion? Antioxidants. 2020; 9(8):664. https://doi.org/10.3390/antiox9080664
Chicago/Turabian StyleRodríguez, Laura R., Tamara Lapeña, Pablo Calap-Quintana, María Dolores Moltó, Pilar Gonzalez-Cabo, and Juan Antonio Navarro Langa. 2020. "Antioxidant Therapies and Oxidative Stress in Friedreich’s Ataxia: The Right Path or Just a Diversion?" Antioxidants 9, no. 8: 664. https://doi.org/10.3390/antiox9080664