
Abstract
DNase I hypersensitive sites (DHSs) are highly sensitive active chromatin regions to DNase I enzymes, which provide the basis for the study of gene transcriptional regulation mechanism and play an important role in the analysis of gene expression regulatory elements. The identification of DHSs has contributed to biomedical research and genome analysis. There are already southern blotting technology and high-throughput sequencing technology to identify DHSs, but these experimental methods are often time-consuming and expensive, thus, novel and powerful computational methods are needed to predict DHSs. It is understood that researchers in related fields have proposed many feasible methods for the identification of DNase I hypersensitive sites. However, the accuracy of these methods is not satisfactory, so it is necessary to use more effective methods to predict DHSs. Therefore, on the basis of previous studies, we design a novel predictor called iDHS-DXG. First of all, we choose three sequence-derived feature representation methods to extract features, including kmer, mismatch and the dinucleotide property matrix based on Moran coefficient. Truncated singular value decomposition is selected for reducing the dimensionality of the benchmark dataset, and the optimal dimension is obtained through the test. Then, synthetic minority over-sampling technique is utilized to balance the positive and negative samples. After that, we introduce extreme gradient boosting ensemble classifier to predict DHSs. Compared with the previous research results, the main performance evaluation metrics of our method have been improved after five-fold cross-validation test. DHSs were identified on two human genome datasets with an accuracy of 90.84% and 91.27% respectively. This result shows that our method is a feasible, effective and competitive tool for the analysis of gene regulatory elements. Our research is helpful for biologists and geneticists to study genome analysis and gene regulation mechanism. Meanwhile, it is also of great significance to the development of human disease and drug design. Furthermore, the datasets and codes of iDHS-DXG can be obtained from the website: http://github.com/Xtian-696/iDHS-DXG/.








Similar content being viewed by others
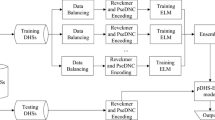

References
Basith S, Manavalan B, Shin TH, Lee G (2018) iGHBP: computational identification of growth hormone binding proteins from sequences using extremely randomised tree. Comput Struct Biotechnol 16:412–420
Cai YD, Feng KY, Lu WC, Chou KC (2006) Using LogitBoost classifier to predict protein structural classes. J Theor Biol 238:172–176
Cao DS, Xu QS, Liang YZ (2013) propy: a tool to generate various modes of Chou’s PseAAC. Bioinformatics 29:960–962
Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP (2002) SMOTE: synthetic minority over-sampling technique. J Artif Intell Res 16:321–357
Chen TQ, Guestrin C (2016) XGBoost: a scalable tree boosting system. In: ACM SIGKDD international conference on knowledge discovery & data mining, pp 785–794
Chen W, Lei TY, Jin DC, Lin H, Chou KC (2014) PseKNC: a flexible web server for generating pseudo K-tuple nucleotide composition. Anal Biochem 456:53–60
Chen W, Lin H, Chou KC (2015) Pseudo nucleotide composition or PseKNC: an effective formulation for analyzing genomic sequences. Mol BioSyst 11:2620–2634
Cheng X, Zhao SG, Lin WZ, Xiao X, Chou KC (2017) pLoc-mAnimal: predict subcellular localization of animal proteins with both single and multiple sites. Bioinformatics 33:3524–3531
Chou KC (1988) Review: low-frequency collective motion in biomacromolecules and its biological functions. Biophys Chem 30:3–48
Chou KC (2001) Prediction of protein cellular attributes using pseudo amino acid composition. PROTEINS: structure. Funct Genet 43:246–255
Chou KC (2005) Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 21:10–19
Chou KC (2009) Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr Proteomics 6:262–274
Chou KC (2011) Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 273:236–247
Chou KC (2015) Impacts of bioinformatics to medicinal chemistry. Med Chem 11:218–234
Chou KC (2017) An unprecedented revolution in medicinal chemistry driven by the progress of biological science. Curr Top Med Chem 17:2337–2358
Chou KC (2019a) Advance in predicting subcellular localization of multi-label proteins and its implication for developing multi-target drugs. Curr Med Chem 26:4918–4943
Chou KC (2019b) Impacts of pseudo amino acid components and 5-steps rule to proteomics and proteome analysis. Curr Top Med Chem 19:2283–2300
Chou KC (2019c) Progresses in predicting post-translational modification (2019). Int J Pept Res Ther. https://doi.org/10.1007/s10989-019-09893-5
Chou KC (2020a) The development of gordon life science institute: its driving force and accomplishments. Nat Sci 12:202–217
Chou KC (2020b) Other mountain stones can attack jade: the 5-steps rule. Nat Sci 12:59–64
Chou KC (2020c) Proposing 5-steps rule is a notable milestone for studying molecular biology. Nat Sci 12:74–79
Chou KC (2020d) Using similarity software to evaluate scientific paper quality is a big mistake. Nat Sci 12:42–58
Chou KC, Cai YD (2003) Prediction and classification of protein subcellular location: sequence-order effect and pseudo amino acid composition. J Cell Biochem 90:1250–1260
Chou KC, Elrod DW (2002) Bioinformatical analysis of G-protein-coupled receptors. J Proteome Res 1:429–433
Chou KC, Forsen S (1980) Diffusion-controlled effects in reversible enzymatic fast reaction system: critical spherical shell and proximity rate constants. Biophys Chem 12:255–263
Chou KC, Shen HB (2007) Recent progress in protein subcellular location prediction. Anal Biochem 370:1–16
Chou KC, Zhang CT (1995) Review: prediction of protein structural classes. Crit Rev Biochem Mol 30:275–349
Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA, Ginsburg D, Zhou DS, Luo S, Vasicek TJ et al (2006) Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16:123–131
Dehzangi A, Heffernan R, Sharma A, Lyons J, Paliwal K, Sattar A (2015) Gram-positive and Gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into Chou’s general PseAAC. J Theor Biol 364:284–294
Ding SY, Zhang SL (2016) A Gram-negative bacterial secreted protein types prediction method based on PSI-BLAST profile. Biomed Res Int 3206741:1–5
Du PF, Wang X, Xu C, Gao Y (2012) PseAAC-Builder: a cross-platform stand-alone program for generating various special Chou’s pseudo amino acid compositions. Anal Biochem 425:117–119
Du PF, Gu SW, Jiao Y (2014) PseAAC-General: fast building various modes of general form of Chou’s pseudo amino acid composition for large-scale protein datasets. Int J Mol Sci 15:3495–3506
Fan GL, Li QZ (2012) Predict mycobacterial proteins subcellular locations by incorporating pseudo-average chemical shift into the general form of Chou’s pseudo amino acid composition. J Theor Biol 304:88–95
Felsenfeld G (1992) Chromatin as an essential part of the transcriptional mechanism. Nature 355:219–224
Felsenfeld G, Groudine M (2003) Controlling the double helix. Nature 421:448–453
Feng P, Jiang N, Liu N (2014) Prediction of DNase I hypersensitive sites by using pseudo nucleotide compositions. Sci World J 2014:740506
Gross DS, Garrard WT (1988) Nuclease hypersensitive sites in chromatin. Annu Rev Biochem 57:159–197
Hu L, Huang T, Shi X, Lu WC, Cai YD, Chou KC (2011) Predicting functions of proteins in mouse based on weighted protein-protein interaction network and protein hybrid properties. PLoS ONE 6:e14556
Jia JH, Liu Z, Xiao X, Liu BX, Chou KC (2015) iPPI-Esml: an ensemble classifier for identifying the interactions of proteins by incorporating their physicochemical properties and wavelet transforms into PseAAC. J Theor Biol 377:47–56
Jia JH, Liu Z, Xiao X, Liu BX, Chou KC (2016a) iPPBS-Opt: a sequence-based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules 21:95
Jia JH, Liu Z, Xiao X, Liu BX, Chou KC (2016b) iSuc-PseOpt: identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal Biochem 497:48–56
Kabir M, Yu DJ (2017) Predicting DNase I hypersensitive sites via un-biased pseudo trinucleotide composition. Chemometr Intell Lab 167:78–84
Kabir M, Ahmad S, Iqbal M, Hayat M (2020) iNR-2L: a two-level sequence-based predictor developed via Chou’s 5-steps rule and general PseAAC for identifying nuclear receptors and their families. Genomics 112:276–285
Koohy H, Down TA, Hubbard TJ (2013) Chromatin accessibility data sets show bias due to sequence specificity of the DNase I enzyme. PLoS ONE 8:e69853
Leslie CS, Eskin E, Cohen A, Weston J, Noble WS (2004) Mismatch string kernels for discriminative protein classification. Bioinformatics 20:467–476
Lever J, Gakkhar S, Gottlieb M, Rashnavadi T, Lin S, Siu C, Jones SJM (2017) A collaborative filtering-based approach to biomedical knowledge discovery. Bioinformatics 34:652–659
Li TT, Chou KC, Forsen S (1980) The flow of substrate molecules in fast enzyme catalyzed reaction systems. Chem Scr 16:192–196
Li DF, Luo LQ, Zhang W, Liu F, Luo F (2016) A genetic algorithm-based weighted ensemble method for predicting transposon-derived piRNAs. BMC Bioinformat 17:329
Liang YY, Zhang SL (2019) iDHS-DMCAC: identifying DNase I hypersensitive sites with balanced dinucleotide-based detrending moving-average cross-correlation coefficient. SAR QSAR Environ Res 30:429–445
Liu B, Liu F, Wang X, Chen J, Fang L, Chou KC (2015a) Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res 43:W65–W71
Liu GQ, Xing YQ, Cai L (2015b) Using weighted features to predict recombination hotspots in Saccharomyces cerevisiae. J Theor Biol 382:15–22
Liu BQ, Liu YM, Jin XP, Wang XL, Liu B (2016a) iRSpot-DACC: a computational predictor for recombination hot/cold spots identification based on dinucleotide-based auto-cross covariance. Sci Rep-uk 6:33483
Liu B, Long R, Chou K-C (2016b) iDHS-EL: identifying DNase I hypersensitive sites by fusing three different modes of pseudo nucleotide composition into an ensemble learning framework. Bioinformatics 32:2411–2418
Liu B, Wu H, Chou KC (2017) Pse-in-One 2.0: an improved package of web servers for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nat Sci 9:67–91
Liu K, Chen W, Lin H (2019) XG-PseU: an eXtreme gradient boosting based method for identifying pseudouridine sites. Mol Genet Genomics 295:13–21
Madrigal P, Krajewski P (2012) Current bioinformatic approaches to identify DNase I hypersensitive sites and genomic footprints from DNase-seq data. Front Genet. https://doi.org/10.3389/fgene.2012.00230
Manavalan B, Basith S, Shin TH, Wei L, Lee G (2018a) mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 35:2757–2765
Manavalan B, Shin TH, Lee G (2018b) DHSpred: support-vector-machine- based human DNase I hypersensitive sites prediction using the optimal features selected by random forest. Oncotarget 9:1944–1956
Manavalan B, Govindaraj RG, Shin TH, Kim MO, Lee G (2018c) iBCE-EL: a new ensemble learning framework for improved linear B-cell epitope prediction. Front Immunol 9:1695
Meher PK, Sahu TK, Saini V, Rao AR (2017) Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci Rep 7:42362
Moran PA (1950) Notes on continuous stochastic phenomena. Biometrika 37:17–23
Noble WS, Kuehn S, Thurman R, Yu M, Stamatoyannopoulos J (2005) Predicting the in vivo signature of human gene regulatory sequences. Bioinformatics 21:i338–i343
Rahman MM, Davis DN (2013) Addressing the class imbalance problem in medical datasets. Int J Mach Learn Comput 3:224–228
Shen HB, Chou KC (2008) PseAAC: a flexible web-server for generating various kinds of protein pseudo amino acid composition. Anal Biochem 373:386–388
Silvério-Machado R, Couto BRGM, dos Santos MA (2014) Retrieval of Enterobacteriaceae drug targets using singular value decomposition. Bioinformatics 31:1267–1273
Song L, Crawford GE (2010) DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb Protoc 2010:pdb.prot5384
Su R, Hu J, Zou Q, Manavalan B, Wei L (2019) Empirical comparison and analysis of web-based cell-penetrating peptide prediction tools. Brief Bioinform 21:408–420
Tahir M, Tayara H, Chong KT (2019) iRNA-PseKNC(2methyl): identify RNA 2’-O-methylation sites by convolution neural network and Chou’s pseudo components. J Theor Biol 465:1–6
Wu C, Bingham PM, Livak KJ, Holmgren R, Elgin SCR (1979) The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence. Cell 16:797–806
Xiao X, Cheng X, Chen GQ, Mao Q, Chou KC (2019) pLoc_bal-mGpos: predict subcellular localization of Gram-positive bacterial proteins by quasi-balancing training dataset and PseAAC. Genomics 111:886–892
Xu ZC, Jiang SY, Qiu WR, Liu YC, Xiao X (2017) iDHSs-PseTNC: identifying DNase I hypersensitive sites with pseuo trinucleotide component by deep sparse auto-encoder. Lett Org Chem 14:655–664
Yang JY, Chen X (2011) Improving taxonomy-based protein fold recognition by using global and local features. Proteins Struct Funct Bioinformat 79:2053–2064
Zhang CT, Chou KC (1992) An optimization approach to predicting protein structural class from amino acid composition. Protein Sci 1:401–408
Zhang SL, Liang YY (2018) Predicting apoptosis protein subcellular localization by integrating auto-cross correlation and PSSM into Chou’s PseAAC. J Theor Biol 457:163–169
Zhang SX, Zhou ZP, Chen XM, Hu Y, Yang LD (2017) pDHS-SVM: a prediction method for plant DNase I hypersensitive sites based on support vector machine. J Theor Biol 426:126–133
Zhang SX, Chang MJ, Zhou ZP, Dai XF, Xu ZH (2018a) pDHS-ELM: computational predictor for plant DNase I hypersensitive sites based on extreme learning machines. Mol Genet Genomics 293:1035–1049
Zhang SX, Li JH, Su L, Zhou ZP (2018b) pDHS-DSET: prediction of DNase I hypersensitive sites in plant genome using DS evidence theory. Anal Biochem 564:54–63
Zhang SX, Zhuang WC, Xu ZH (2018c) Prediction of DNase I hypersensitive sites in plant genome using multiple modes of pseudo components. Anal Biochem 549:149–156
Zhang SL, Yu QH, He HR, Zhu F, Wu PJ, Gu LZ, Jiang SJ (2020) iDHS-DSAMS: identifying DNase I hypersensitive sites based on the dinucleotide property matrix and ensemble bagged tree. Genomics 112:1282–1289
Zhou GP, Deng MH (1984) An extension of Chou’s graphic rules for deriving enzyme kinetic equations to systems involving parallel reaction pathways. Biochem J 222:169–176
Funding
This study was funded by the National Natural Science Foundation of China (No. 11601407), the Natural Science Basic Research Plan in Shaanxi Province of China (Nos. 2018JM1037, 2019JQ-279).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no conflict of interest
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
There was no human participant and consent was not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, S., Xue, T. Use Chou’s 5-steps rule to identify DNase I hypersensitive sites via dinucleotide property matrix and extreme gradient boosting. Mol Genet Genomics 295, 1431–1442 (2020). https://doi.org/10.1007/s00438-020-01711-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00438-020-01711-8