
Abstract
Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) is a key mediator of cell death and inflammation. The unique hydrophobic pocket in the allosteric regulatory domain of RIPK1 has enabled the development of highly selective small-molecule inhibitors of its kinase activity, which have demonstrated safety in preclinical models and clinical trials. Potential applications of these RIPK1 inhibitors for the treatment of monogenic and polygenic autoimmune, inflammatory, neurodegenerative, ischaemic and acute conditions, such as sepsis, are emerging. This article reviews RIPK1 biology and disease-associated mutations in RIPK1 signalling pathways, highlighting clinical trials of RIPK1 inhibitors and potential strategies to mitigate development challenges.
Similar content being viewed by others

Introduction
Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) is a master regulator of the cellular decision between pro-survival NF-κB signalling and death in response to a broad set of inflammatory and pro-death stimuli in human diseases1,2. RIPK1 kinase activation has been demonstrated in post-mortem human pathological samples of autoimmune and neurodegenerative conditions3,4,5,6, and inhibition of RIPK1 kinase activity has shown efficacy in a wide range of animal models of human diseases. As TNFR1-mediated RIPK1 activation is the most comprehensively characterized paradigm, RIPK1 inhibitors were originally considered to primarily offer a small-molecule alternative to anti-TNF antibody therapies for TNF-driven autoimmune conditions. However, as researchers continued to delve into the mechanisms governed by RIPK1, it has become apparent that RIPK1 inhibitors may offer key therapeutic options that anti-TNF therapies do not: first, RIPK1 inhibitors are safe in the central nervous system (CNS) as RIPK1 kinase does not signal through TNFR2 which has a protective role in the CNS7; second, RIPK1 participates in a broader set of pro-inflammatory activities than those restricted to TNF8; third, RIPK1 is regulated by a distinct set of signalling molecules that are genetically implicated in human autoimmune and autoinflammatory diseases, as discussed below, and thus patient stratification may be important in conducting clinical trials of RIPK1 inhibitors.
Necrostatin-1s (Nec-1s) was the first small-molecule inhibitor of RIPK1 kinase to be developed and has been widely used to investigate the role of RIPK1 in mechanistic studies and animal models of human diseases1,8,9,10,11. Broad therapeutic applications of RIPK1 inhibitors for the treatment of a wide range of human diseases are being investigated in clinical trials. The peripherally restricted GSK′772 is being developed for peripheral autoimmune diseases, including psoriasis, rheumatoid arthritis (RA) and ulcerative colitis12,13,14. The brain-penetrant DNL747 is in human clinical trial phase Ib/IIa for amyotrophic lateral sclerosis (ALS)15,16. These trials have laid the groundwork for advancing clinical applications of RIPK1 inhibitors.
The important role of RIPK1 in driving cell death and inflammation, the established safety of inhibiting RIPK1 kinase activity in humans and the ability to develop selective small-molecule kinase inhibitors of RIPK1 due to the presence of a unique kinase-regulating allosteric pocket are the major factors that have contributed to RIPK1’s prominence as a therapeutic target. In this Review, we outline the current understanding of RIPK1 biology in activating cell death and NF-κB signalling, systematically review monogenic and polygenic variants of known RIPK1 regulators and discuss how these mutations may contribute to disease pathology. The involvement of RIPK1 in sepsis and acute ischaemic conditions is also discussed. We postulate that the improved understanding of genetic and mechanistic data may be beneficial in segmenting patients in clinical trials, particularly for neurodegenerative and inflammatory diseases. Finally, we summarize the current state of RIPK1 inhibitors in the clinic, including disease indications, small-molecule chemotypes, as well as RIPK1 target engagement and pharmacodynamic biomarkers.
Distinct kinase and scaffold functions of RIPK1
RIPK1 is a 76-kDa protein with an amino-terminal (N-terminal) kinase domain, a carboxy-terminal (C-terminal) death domain and an intermediate domain with a RHIM (receptor-interacting protein homotypic interacting motif) that can bind to other RHIM-containing proteins2,17. Whereas the C-terminal death domain mediates homodimerization as well as heterodimerization with other death domain-containing proteins, such as FADD, TNFR1 and Fas, the N-terminal kinase domain mediates autophosphorylation in trans to promote its own activation18,19. The scaffold function of RIPK1 is essential for mediating pro-survival NF-κB signalling and mouse perinatal survival: Ripk1–/– mice are born normally but die at postnatal day 1–3 (ref.20). RIPK1 deficiency reduces NF-κB-mediated transcription of pro-survival proteins, such as cellular FLICE-like inhibitory protein (cFLIP), cIAP1 and A20 (ref.21). The perinatal lethality of Ripk1–/– mice can be rescued by inhibition of both apoptotic and regulated necrotic cell death (necroptosis) in Ripk1–/–Fadd–/–Ripk3–/–, Ripk1–/–Casp8–/–Ripk3–/– or Ripk1–/–Fadd–/–Mlkl–/– mice22,23,24,25,26.
In contrast to the phenotype of Ripk1–/– mice, mice harbouring RIPK1 knock-in kinase dead mutations, including D138N and K45A, or a K584R death domain mutation that blocks the dimerization-mediated activation of RIPK1 are normal19,27,28. In fact, these mutant mice are resistant to various inflammatory and degenerative conditions in mouse models of disease. Interestingly, both genetic and pharmacological inhibition of RIPK1 kinase activity, such as treatment with Nec-1s or GNE684 (refs10,29), offer complete resistance against the TNF-induced animal model of septic shock30,31,32. Thus, the kinase activity of RIPK1 promotes cell death and inflammation whereas the scaffold function of RIPK1 supports postnatal survival.
RIPK1 signalling pathway mediated by TNFR1
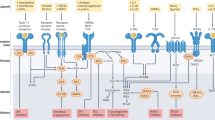
Activation of RIPK1 kinase mediated by TNFR1 signalling promotes most of the deleterious effects activated by TNF in human disease2. In TNF-stimulated cells, the activation of RIPK1 is regulated in a transient multimeric complex associated with the intracellular domain of TNFR1, known as complex I (Fig. 1). The recruitment and activity of these proteins determines the balance between inhibited RIPK1 kinase activity, where NF-κB pro-survival signalling is activated, and activated RIPK1 kinase activity, which leads to inflammation and cell death. Mutations that impact the recruitment to complex I or activity of these proteins leads to aberrant RIPK1 kinase activity in disease, most commonly autoimmunity or inflammation (Box 1).

Complex I: stimulation of TNFR1 by TNF promotes the formation of an intracellular signalling complex associated with the death domain of trimerized TNFR1 that recruits two death domain-containing proteins: adaptor protein TRADD and receptor-interacting serine/threonine-protein kinase 1 (RIPK1). TRADD recruits E3 ubiquitin ligases cIAP1/2 and XIAP to perform K63 ubiquitylation of complex I, including RIPK1 K377, which in turn recruits the LUBAC complex, comprised of HOIP, HOIL1 and SHARPIN. LUBAC mediates linear (M1) ubiquitylation of RIPK1. Deubiquitinase CYLD and its adaptor protein SPATA2 modulate M1/K63 ubiquitylation of RIPK1. M1 deubiquitinase OTULIN activates LUBAC. K63 ubiquitin chains on RIPK1 recruit TAB2/3 and TAK1. M1 ubiquitin chains on RIPK1 recruit the NEMO–IκB kinase (IKK) complex, TBK1, A20, ABIN1 (A20 binding inhibitor of NF-κB-1) and OPTN. A20 in complex I suppresses the activation of RIPK1 kinase. NF-κB activation: activation of TAK1 and the IKKs promote NF-κB pathway activation to mediate transcription of both pro-inflammatory and pro-survival genes, including A20, which modulates the ubiquitylation of RIPK1 to control its activation, and cellular FLICE-like inhibitory protein (cFLIP), which modulates activation of caspase 8 (Casp8). RIPK1-dependent apoptosis (RDA): activation of TNFR1 under A20, ABIN1, cIAP1/2, NEMO, TBK1, IKK or TAK1-deficient conditions leads to RIPK1 kinase activation. Activated RIPK1 binds to FADD and Casp8 to form complex IIa and promote activation of caspases and apoptosis. Increased levels of A20 promote the activation of RIPK1 in complex IIa. Necroptosis: inhibition of Casp8-mediated cleavage of RIPK1 promotes RIPK1 dimerization via the C-terminal death domain, which leads to its activation and the subsequent formation of the necrosome (complex IIb) comprised of RIPK1, FADD, Casp8, RIPK3 and mixed-lineage kinase domain-like pseudokinase (MLKL), which in turn executes necroptosis. RIPK1-independent apoptosis: when the NF-κB pathway is inhibited, TNF stimulation can promote the formation of a cytosolic complex with FADD and Casp8 to mediate apoptosis independent of RIPK1. P, phosphate; RIPK1i, RIPK1 inhibitor; Ub, ubiquitin. Adapted from ref.1, Springer Nature Limited.
RIPK1 and TRADD, another death domain-containing adaptor protein, are rapidly recruited into complex I by binding to the death domain of TNFR1. Activation of RIPK1 in complex I, as measured by the well-established phosphorylation of S166, can enact either apoptotic or necroptotic forms of cell death3,9. In complex I, the activation of RIPK1 is determined by a code including complex ubiquitylation, phosphorylation and other modification events on RIPK1, which include those directly organized by TRADD and those mediated by proteins transcribed and translated downstream of the NF-κB pathway that are also recruited into the complex (for example, A20)8. This post-translational RIPK1 code, which may be both cell type-specific and stimulus-specific, modulates the extent of RIPK1 kinase activation, which in turn determines the mode of cell death. There is an extensive body of literature identifying cell type-specific roles for RIPK1 activity using conditional mouse models that we and others have summarized previously, and so do not cover in detail in this article1,2. For example, in certain cell types, such as oligodendrocytes, TNF stimulation alone may promote the activation of RIPK1 and cell death3,4; in other cell types, such as fibroblasts, sustained activation of RIPK1 kinase can only be achieved when TNF stimulation is combined with chemical inactivators of RIPK1 repressors, such as cIAP1/2 (inhibited by SM-164), TAK1, TBK1 or IKKs, that are both mediators of the NF-κB pathway and inhibitors of RIPK1 kinase activation18,33,34,35.
Ubiquitylation of RIPK1 in complex I, which is organized by TRADD, is essential for suppressing the aberrant activation of RIPK1 kinase. TRADD recruits TRAF2 and the E3 ubiquitin ligases cIAP1 and cIAP2 into complex I to mediate RIPK1 K63 ubiquitylation. K63 ubiquitylation of RIPK1, in turn, mediates the recruitment and activation of TAK1 kinase through the polyubiquitin binding adaptors TAB2 and TAB3 (ref.36). K63 ubiquitylation of complex I facilitates the recruitment of the LUBAC complex containing HOIP, HOIL1 and SHARPIN, which in turn performs M1 ubiquitylation of RIPK1 and TNFR1. M1 ubiquitylation of complex I is important for the recruitment of the trimeric IKK complex through the polyubiquitin-binding adaptor subunit IKKγ/NEMO2,37, as well as other ubiquitin binding proteins such as ABIN1 (A20 binding and inhibitor of NF-κB-1) and the kinase TBK1 (refs18,38). The activation of RIPK1 is suppressed by direct inhibitory phosphorylation mediated by TAK1, IKKα/β and ΤΒΚ1 (ref.1). cIAP1 may also mediate K48 ubiquitylation of RIPK1 to promote its degradation by the proteasome39. In addition, TNFAIP3, which is transcriptionally induced by the NF-κB pathway, encodes the ubiquitin editing enzyme A20, which controls the activation of RIPK1 by modulating its ubiquitylation pattern40.
TNF stimulation of fibroblasts derived from either wild-type mice or healthy patients does not lead to cell death owing to the suppression of RIPK1 activation in complex I by inhibitory ubiquitylation and phosphorylation. Stimulation with TNF in combination with factors that reduce the inhibition of RIPK1 leads to necroptosis, RIPK1-dependent apoptosis (RDA) and inflammation (Box 1). In experimental paradigms, RIPK1 kinase activity and necroptosis can be activated by treatment with a combination of TNF, cycloheximide (CHX), which blocks the activation of NF-κB-mediated transcription and translation, and zVAD.fmk, which blocks the activation of caspases10. Inhibition of caspases strongly sensitizes to RIPK1 activation, as the cleavage of RIPK1 by Casp8 provides an important inhibitory mechanism to block the overactivation of RIPK1 (refs41,42,43). RDA can be activated with a combination of TNF and the TAK1 inhibitor (5Z)-7-oxozeanol (known as 5Z7), or TNF and SM-164, which blocks inhibitory phosphorylation and ubiquitylation of RIPK1, respectively, and blocks activation of pro-survival NF-κB signalling33. As detailed in this Review, these experimental manipulations mimic some aspects of human genetic deficiencies in various regulators of NF-κB activation that lead to aberrant RIPK1 kinase activation and provide guidance for possible indications where RIPK1 inhibitors may be efficacious (Table 1).
RIPK1 in immune and autoinflammatory diseases
Mutations in the genes that encode RIPK1 and multiple proteins that regulate RIPK1 signalling can lead to immune and autoinflammatory diseases. These clinically identified mutations highlight the important role of RIPK1 in regulating the innate immune response and provide mechanistic insights into the functional role of RIPK1 in disease. The spectrum of immune and autoinflammatory diseases presents as a continuum between autoimmune disorders involving primarily the adaptive immune system and autoinflammatory conditions involving primarily the innate immune system, all of which can be found in diseases involving RIPK1.
RIPK1 mutations in humans
Rare mutations leading to loss of function (LoF) and gain of function (GoF) of RIPK1 have been identified in individuals with immunodeficiencies and autoinflammatory diseases (Table 2).
Biallelic loss-of-function RIPK1 mutations
Rare biallelic LoF mutations in RIPK1, including missense, nonsense and frameshift mutations, have been identified in patients with combined immunodeficiency and inflammatory bowel disease (IBD)44,45. Whereas Ripk1–/– mice die perinatally, patients with LoF mutations in RIPK1 exhibit paediatric onset of primary immunodeficiency characterized by an increased susceptibility to infections and early-onset IBD20,44,45. At the cellular level, the loss of RIPK1 in human skin fibroblasts impairs activation of the NF-κB pathway, MAPKs and Jun in response to TNF or poly(I:C) and increases the activation of necroptosis mediated by RIPK3 and mixed-lineage kinase domain-like pseudokinase (MLKL), but not apoptosis44,45. The production of pro-inflammatory cytokines, including TNF, IL-6 and IL-10, in response to LPS stimulation is severely impaired in peripheral blood mononuclear cells (PBMCs) from patients with RIPK1 LoF compared with control PBMCs44. Dysregulation of cytokine production and the host defence response towards the gut microbiome likely play a key role in promoting early-onset IBD and progressive polyarthritis in these patients.
Heterozygous non-cleavable RIPK1 mutations
Casp8 inactivates RIPK1 by cleaving human and mouse RIPK1 after residues D324 and D325, respectively, which separates the RIPK1 kinase domain from the intermediate and death domains46. Mice with a D325A knock-in mutation that prevents cleavage by Casp8 die embryonically, and can be rescued by cis-inactivation of RIPK1 by D138N mutation, the loss of TNFR1, or inactivating both necroptosis and apoptosis by double knockout of Ripk3 and Fadd, or Mlkl and FADD31,47,48. Rare variants of RIPK1, such as D324V and D324H, that block cleavage by Casp8 have been identified in individuals with an autosomal dominant autoinflammatory disease41,42. In contrast to the phenotype observed in patients with RIPK1 LoF mutations, patients with heterozygous non-cleavable RIPK1 mutations develop autoinflammatory disease characterized by recurrent fevers and lymphadenopathy. Marked increases in pro-inflammatory cytokines and chemokines, such as IL-6, TNF and interferon-γ (IFNγ), were found in sera from patients. Impaired cleavage of RIPK1 D324 variants by Casp8 hypersensitized patient PBMCs to RIPK1 activation, including both apoptosis and necroptosis induced by TNF, which can be blocked by the RIPK1 inhibitor Nec-1s (ref.42).
Although PBMCs from patients with non-cleavable RIPK1 mutations are more susceptible to inflammatory stimulation, fibroblasts from one such patient showed resistance to necroptosis and ferroptosis and reduced expression of pro-inflammatory cytokines in response to stimuli42. Considerable changes in the gene expression patterns were found in these fibroblasts, including downregulated expression of RIPK1, RIPK3 and TNFR1, as well as elevated levels of the anti-oxidative glutathione and genes that offer resistance to ferroptosis. Such compensatory gene expression might be necessary to overcome the deleterious effects of non-cleavable RIPK1 and allow for patient survival, although these findings need to be expanded to additional patients.
Together, these data suggest that autoinflammatory disease caused by non-cleavable RIPK1 variants may represent a canonical human RIPK1 hyperactivating disease that can respond to RIPK1 inhibitor treatment.
Disease-associated mutations in regulators of RIPK1
Dysregulation of RIPK1 signalling is involved in a heterogeneous group of monogenic immune and autoinflammatory diseases that can present with either episodic or chronic symptoms (Table 2). As many of these disease-associated genes are also involved in regulating NF-κB signalling49, some amount of disease pathology can be attributed to altered NF-κB signalling. Interestingly, these genes, including TNFAIP3 (encoding A20), TNIP1 (encoding ABIN1), IKBKG (encoding NEMO), OTULIN and members of the LUBAC complex, are also direct regulators of RIPK1 activation (Table 1). Thus, exogenous triggers that lead to transient inflammation in healthy subjects may promote sustained inflammation and cell death involving different tissues and organs in individuals with aberrant RIPK1 regulation. Traditional treatment for autoimmune diseases has focused on managing immune hyperactivity by dampening non-specific inflammatory responses and immune cell proliferation. However, this approach renders patients vulnerable to opportunistic infections that can be life-threatening. Understanding the inflammatory mechanisms regulated by RIPK1 may help to develop therapies that can specifically target the disease pathology in these rare diseases. Furthermore, understanding the contribution of RIPK1 in these rare diseases may also help to elucidate roles for RIPK1 in autoimmune and inflammatory diseases that are not genetically linked to RIPK1.
A20 deficiency
A20, encoded by the TNFAIP3 gene, is an inducible ubiquitin-editing enzyme that restricts both Toll-like receptor (TLR) and TNF-induced inflammatory responses by regulating the ubiquitylation of key signalling proteins, including RIPK1, TRAF6 and NEMO40. Mouse models with cell lineage-specific A20 deficiency phenocopy different human inflammatory diseases, suggesting an important role for A20 in restricting RIPK1 activation in multiple tissues (Table 3). A20 contains an N-terminal ovarian tumour (OTU) domain that can deubiquitylate K63/K48-linked polyubiquitin chains from its substrates, and C-terminal zinc finger domains that can bind and modulate linear (M1) ubiquitin chains. Multiple heterozygous LoF mutations in the TNFAIP3 gene, including early nonsense and frameshift mutations, have been identified in a paediatric systemic inflammatory disease or systemic blood vessel inflammation similar to Behçet’s disease50,51. These patients showed increased levels of pro-inflammatory cytokines, such as TNF, IL-1β and IL-6, and demonstrated clinical improvement after treatment with anti-TNF or anti-IL-1β therapy.
In addition, low-penetrance coding and non-coding variants in TNFAIP3 have been suggested to underlie multiple autoimmune diseases, including Crohn’s disease, psoriasis, RA, type 1 diabetes mellitus and susceptibility to allergy and asthma52,53,54,55. Reduced A20 expression may even contribute to atopic eczema (atopic dermatitis)56, one of the most common inflammatory skin disorders, affecting up to 7% of adults and 25% of children globally. These studies suggest that dysregulated A20 may be a common underlying factor in the pathogenesis of inflammatory diseases.
NEMO deficiency syndrome
NEMO, the scaffolding subunit of the IKK holocomplex comprising IKKα and IKKβ, is critical for regulating activation of the NF-κB pathway during canonical inflammatory responses57. NEMO contains an N-terminal dimerization domain involved in binding with IKKα and IKKβ, and two distinct ubiquitin binding domains, including a UBAN domain that preferentially binds to M1 over K63 ubiquitin chains58. NEMO deficiency syndrome is a complex disease caused by LoF or hypomorphic mutations in the X-linked IKBKG gene, which encodes NEMO, and includes clinical definitions of incontinentia pigmenti and anhidrotic ectodermal dysplasia with immunodeficiency. A case study demonstrated that anti-TNF therapy (infliximab) successfully treated inflammatory colitis in an infant with NEMO deficiency59.
Nemo–/– mice and epidermis-specific knockout of NEMO (NemoEKO) reproduced incontinentia pigmenti phenotypes, including embryonic lethality in males and inflammatory skin lesions in heterozygous females60,61. The skin inflammation of NemoEKO mice can be delayed by crossing them with Tnfr1–/–62, suggesting that RIPK1 kinase may be involved in mediating inflammatory response in incontinentia pigmenti.
Mutant mice with specific loss of NEMO in intestinal epithelial cells (IECs) (NemoIEC-KO mice) show apoptosis of Paneth cells and colonocytes, and microbiota-driven chronic colitis63. The colitis of NemoIEC-KO mice was blocked by genetic or pharmacological inhibition of RIPK1 kinase activity, suggesting that the pathology in this mutant line may be driven more by RIPK1-dependent cell death and inflammatory mechanisms than failure to activate NF-κB30,63.
Taken together, these studies suggest that RIPK1 kinase activity may play an important role in colitis and excessive inflammation in NEMO syndrome. Thus, RIPK1 kinase inhibitors may be beneficial for the treatment of colitis and other inflammatory issues in patients deficient in NEMO. However, as patients with NEMO deficiency syndrome have an inability to mount a successful immune response and are hypersensitive to infection, reminiscent of patients deficient in RIPK1, the inability to properly activate the NF-κB pathway may also contribute to the symptoms of NEMO deficiency syndrome.
OTULIN deficiency
OTULIN is the only known deubiquitylating enzyme that specifically removes M1 ubiquitin chains64. Patients with biallelic hypomorphic mutations in OTULIN develop a severe form of autoinflammatory disease, known as OTULIPENIA or OTULIN-related autoinflammatory syndrome (ORAS)65,66. In OTULIN-deficient cells, levels of M1 ubiquitylation on multiple target proteins, such as NEMO, TNFR1 and RIPK1, are generally increased. As activity of the LUBAC activity complex is negatively regulated by M1 ubiquitylation, OTULIN-deficient cells have reduced LUBAC activity67. Cells derived from patients with OTULIN deficiencies have a strong inflammatory signature including overproduction of pro-inflammatory cytokines, such as IL-1β, IL-6, IL-18 and IFNγ, in response to LPS, TNF or IL-1β. These OTULIN-deficient cells are also sensitized to TNF-induced cell death68. Consistent with the important role of TNF in this disease, these patients responded well to treatment with anti-TNF antibodies (infliximab)66.
Otulin–/– mice and OtulinC129A catalytically inactive knock-in mice die during embryonic development65,67. Embryonic lethality of the OtulinC129A mice can be delayed by Ripk1D138N/D138N or Tnfr1–/–, whereas combined loss of Ripk3 and Casp8 prolongs survival to the perinatal stage, suggesting that necroptosis is activated when OTULIN is not functional.
LUBAC deficiency
The LUBAC complex is composed of HOIP, HOIL1 and SHARPIN. Patients identified with LoF or hypomorphic mutations in HOIP or HOIL1 develop immunodeficiencies and autoinflammatory disease characterized by frequent viral and bacterial infections and multi-organ autoinflammation69,70,71. These hypomorphic mutations in HOIP and biallelic LoF mutations in HOIL1 lead to a reduction in levels of the LUBAC complex driven by a loss of stability. LUBAC-deficient cells show reduced levels of IKK phosphorylation and compromised NF-κB activation. LUBAC is critical for suppressing RIPK1 activation, and, as a consequence, enhanced RIPK1 activity may be responsible for autoinflammatory symptoms in patients harbouring LUBAC mutations72.
Mice deficient for the gene encoding the LUBAC regulatory subunit SHARPIN, also known as chronic proliferative dermatitis mice (cpdm) (Sharpincpdm/cpdm mice), suffer from severe multi-organ inflammation, particularly in the skin with similarity to atopic dermatitis and psoriasis in humans28,73. Interestingly, the skin inflammation of cpdm mice can be blocked by reducing or knocking out TNF74, which points to a key role for this signalling node in the cpdm phenotype. Inhibition of RIPK1 by GNE684 or RIPK1-D138N protects against dermatitis in cpdm mice28,30,75.
Polygenic diseases associated with RIPK1 regulators
Polygenic human diseases, including several autoimmune and inflammatory diseases, are associated with mutations or risk variants in multiple genes, which can include regulators of RIPK1. When developing RIPK1 kinase inhibitors for the treatment of these diseases, patient stratification may be important to identify those with pathogenic activation of RIPK1.
Inflammatory bowel disease
Crohn’s disease and ulcerative colitis are the two primary forms of IBD that are both characterized by intestinal inflammation and epithelial cell loss. Genome-wide association studies (GWAS) of Crohn’s disease and ulcerative colitis have identified more than 200 loci that are above the thresholds of significance76. However, non-genetic factors likely play a major role in conferring susceptibility as the concordance rate in monozygotic twins is only 16% in ulcerative colitis and 30–35% in Crohn’s disease77. TNFAIP3, the gene encoding A20, is a recognized risk factor for both Crohn’s disease and ulcerative colitis. Both increased and decreased expression of A20 has been associated with IBD, suggesting the importance of balancing ubiquitin editing activity in complex I. Whereas mice with enterocyte-specific A20 deletion are hypersensitive to experimental colitis and TNF-induced epithelial apoptosis78, excessive A20 can dimerize, which sensitizes IECs to undergo RIPK1-dependent apoptosis when stimulated by TNF79 (Table 3). IECs with elevated A20 in the mucosa of patients with IBD are associated with Casp3 activation. Inhibition of RIPK1 has been shown to ameliorate pathology in both A20-deficient and elevated conditions79,80. As specific single-nucleotide polymorphisms (SNPs) in TNFAIP3 have been found to be correlated with primary therapeutic response to anti-TNF (infliximab) therapy81, changes in intestinal A20 expression may be used as a biomarker for RIPK1-dependent pathology to stratify patients with IBD for treatment with RIPK1 inhibitors.
GWAS have also identified ATG16L1 and other autophagy-related genes as risk factors for IBD82. ATG16L1 is a component in the macromolecular complex that lipidates LC3/ATG8 to promote the formation of the canonical double-membrane autophagosome. ATG16L1T300A, a risk allele associated with IBD susceptibility, introduces a caspase cleavage site that destabilizes the ATG16L1 protein product and reduces autophagy in the presence of TNF83,84. ATG16L1-deficient intestinal organoids show increased sensitivity to TNF-induced necroptosis, which can be effectively inhibited by Nec-1s (ref.85). Both ATG16L1 IEC conditional knockout mice and A20 IEC conditional knockout mice show increased severity of hypothermia induced by TNF, which can be blocked by the RIPK1 inhibitor GNE684 (ref.30).
Psoriasis
GWAS identified more than 60 disease susceptibility regions, including the TNFAIP3, TNIP1 and RELA loci in psoriasis86. Reduced levels of TNFAIP3 mRNA and A20 protein have been found in the skin of patients with psoriasis, which can be directly correlated to disease severity87. Furthermore, specific SNPs in TNFAIP3 have also been found to predict the response to anti-TNF therapies88, suggesting the predictive value of TNFAIP3 in identifying patients likely to respond to a RIPK1 inhibitor treatment for psoriasis.
TNIP1, encoding the ABIN1 protein, is one of the highest scoring non-MHC genes associated with various autoimmune and autoinflammatory diseases, including psoriasis, psoriatic arthritis, systemic sclerosis and systemic lupus erythematosus (SLE) across multiple GWAS54. ABIN1, a ubiquitin binding protein with a UBAN domain that binds preferentially to linear ubiquitin chains, interacts with A20 to negatively regulate the activation of MAPK and NF-κB-mediated gene transcription downstream of TNFR1 and TLRs89,90,91. ABIN1, which is recruited into the TNFR1 signalling complex by binding linear ubiquitin chains, in turn promotes the recruitment of A20 (ref.38). ABIN1-deficient cells and A20-deficient cells are both sensitized to RIPK1 activation and necroptosis. Tnip1–/– mice die embryonically owing to liver necrosis that can be rescued by Ripk3–/– or Ripk1D138N/D138N. TNIP1 heterozygosity sensitizes cells to a hyperactive antiviral response mediated by an NF-κB-dependent and RIPK1-independent mechanism that promotes the expression of pattern recognition receptors, such as TLR3, RIG-I and MDA5 (ref.92). Inhibition of RIPK1 blocks the increased production of pro-inflammatory cytokines in Tnip1+/– mice in response to viral stimuli. Thus, ABIN1 regulates the innate immune response through the NF-κB pathway, utilizing both RIPK1-dependent and independent signalling pathways. Inhibition of RIPK1 may reduce the exaggerated inflammatory response in patients with TNIP1 mutations without affecting their host defence responses.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a common inflammatory autoimmune disease characterized by chronic synovial inflammation, joint destruction and systemic complications. The HLA-DRB1 alleles are the primary genetic risk factor associated with RA. In addition, GWAS have identified ~100 non-HLA loci, including TNF, associated with RA. GWAS data also suggest the involvement of multiple immune cell types, including T cells, B cells, natural killer cells, dendritic cells and monocytes, as well as the pro-inflammatory cytokines produced by these cells, in mediating RA pathogenesis93. Interestingly, SNPs and reduced levels of TNFAIP3 have been associated with RA94. Inhibition of RIPK1 by GNE684 improved pathology in a rodent model of collagen-induced arthritis30.
Multiple sclerosis
Multiple sclerosis (MS) is an autoimmune disease of the CNS resulting in neurodegeneration that involves the loss of myelinating oligodendrocytes that normally insulate the axons of neurons. TNF is strongly linked with the aetiology of MS95,96. Oligodendrocytes can undergo necroptosis in the presence of TNF stimulation alone3. In lesions of brain samples from patients with MS or mouse models of MS, activation of RIPK1, RIPK3 and MLKL and formation of the necrosome were observed3. cFLIP is transcribed in response to NF-κB activation and can suppress the formation of apoptotic signalling complexes97. In the lesions from patients with MS, levels of cFLIP were found to be decreased, which may be the result of chronic neuroinflammation. Decreased cFLIP and reduced levels of activated Casp8 in patients with MS, and thus decreased Casp8 inactivating cleavage of RIPK1, likely explain the increased RIPK1 kinase activity and necroptosis seen in MS. Genetic (Ripk1D138N/D138N) and pharmacological (Nec-1s) inhibition of RIPK1 kinase ameliorated disease pathology, improved animal behaviour, attenuated the production of pro-inflammatory cytokines and decreased recruitment of immune cells in two animal models of MS3. Another inhibitor of RIPK1 kinase developed by Takeda also showed efficacy in an animal model of MS98.
The contribution of peripheral immune cells and microglia to MS has been highlighted by a recent genetic association study using 47,429 MS cases and 68,374 controls99. This study identified 551 putative MS susceptibility genes that are highly expressed in cell types involved in adaptive immunity and innate immunity, including TNFAIP3, which encodes A20. As RIPK1 is highly expressed in macrophages and microglia in experimental autoimmune encephalomyelitis lesions, inhibition of RIPK1 may directly reduce inflammatory responses as RIPK1 activity is involved in mediating the inflammatory responses in these cells100,101. Thus, the beneficial effects of inhibiting RIPK1 may include modulation of both inflammation and cell death in susceptible oligodendrocytes, microglia and neurons. This raises the intriguing possibility that RIPK1 may act in both a cell-autonomous and a non-cell autonomous manner in MS. As the existing approved MS drugs all target adaptive immunity and do not directly address neuroinflammation that drives progression in MS, RIPK1 inhibition may represent a novel therapeutic approach that targets multiple aspects of disease pathology.
Neutrophil and complement-mediated autoimmune disorders
Systemic lupus erythematosus (SLE) is another heterogeneous autoimmune and inflammatory disorder characterized by excessive immune and inflammatory responses. SLE is a chronic condition that affects multiple organs and tissues. Multiple independent SNPs in the TNIP1 and TNFAIP3 genes are found to be associated with SLE102,103,104.
Assembly and activation of the necroptotic machinery, including RIPK1, has been implicated in NETosis, a programmed suicide of neutrophils in tissues involved in mounting normal immune responses against pathogens105,106,107. NETosis results in the formation of neutrophil extracellular traps (NETs); this leads to the release of autoantigens from neutrophils and contributes to autoimmune pathology108. In several diseases in which NETs drive toxicity, inhibiting RIPK1 and thereby blocking other components of the necroptosis pathway has shown efficacy in disease models, including autoimmune vasculitis, venous thrombosis and SLE108,109,110. More broadly, toxicity induced by neutrophil extravasation and NET formation has been implicated in widespread diseases, ranging from autoimmune conditions to neurodegenerative diseases where an impaired blood–brain barrier results in aberrant NET formation in the brain parenchyma111,112,113.
The canonical and alternative complement pathways are well-characterized and essential contributors to the pathogenesis of diseases such as SLE. The activation of complement in disease results in complement-dependent cytotoxicity. Complement can activate RIPK1–RIPK3–MLKL-dependent necroptosis as a contributor of complement-dependent cytotoxicity, which can be attenuated by RIPK1 inhibitors114. Furthermore, in some instances of autoimmunity where complement activation drives disease, the complement pathways can be activated by neutrophil NETosis, which is also regulated by RIPK1 activation109. Taken together, these findings suggest that RIPK1 may be involved in the pathogenesis of neutrophil and complement-mediated autoimmune conditions.
RIPK1 in chronic neurodegenerative diseases
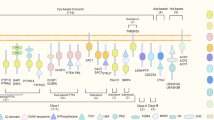
Given that RIPK1 activation and necroptosis have now been genetically and mechanistically linked with human neurodegenerative diseases, there has been a keen interest in developing CNS-penetrant RIPK1 inhibitors to alter disease progression for diseases such as ALS, frontotemporal dementia (FTD), AD and other ageing-related neurodegenerative diseases4,6,18 (Fig. 2). Beyond these indications where several independent groups and clinical data have confirmed a role for pathogenic RIPK1 kinase activation, limited data suggest that RIPK1 activation may also play a role in the pathology of Huntington disease and Duchenne muscular dystrophy; however, the mechanisms underpinning this have not yet been defined115,116.

Activation of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) in neurodegenerative diseases may promote the death of neurons and oligodendrocytes cell-autonomously, and inflammation in microglia and astrocytes that acts non-cell-autonomously to promote neurodegeneration. Neurons and oligodendrocytes: amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD)-associated loss-of-function (LoF) mutations in TBK1 and OPTN and ageing-related reduction in TAK1 can promote RIPK1-dependent apoptosis and necroptosis, which can be inhibited by RIPK1 inhibitors. Parkinson disease (PD)-associated mutations in OPA1 and DRP1, ALS-associated mutations in SOD1 and the chemical stressor MPTP lead to mitochondrial dysfunction and the production of reactive oxygen species (ROS). Cytosolic ROS can modulate cysteine residues on RIPK1, which promotes activation. Microglia and astrocytes: activation of RIPK1 leads to the production of pro-inflammatory cytokines in microglia. Activation of RIPK1 in Alzheimer disease (AD) leads to increased production of CST7 and CH25H, both of which are associated with the disease-associated microglia phenotype. PD/lysosomal storage disorder (LSD)-associated genes, such as GBA and NPC1, also result in lysosomal dysfunction, which can promote RIPK1 activation, likely by promoting the accumulation of RIPK1. ALS-associated mutations in SOD1 are also known to promote mitochondrial dysfunction and ROS production, which may promote glial activation of RIPK1 in disease. MLKL, mixed-lineage kinase domain-like pseudokinase; P, phosphate; RDA, RIPK1-dependent apoptosis; RIPK1i, RIPK1 inhibitor.
ALS and FTD
ALS is a progressive neurodegenerative disease that affects motor neurons in the brain and the spinal cord. FTD is a group of related neurodegenerative diseases that lead to the progressive degeneration of neurons in the temporal and frontal lobes of the brain, with consequences on social, emotional and language abilities. LoF mutations in two genes, OPTN and TBK1, lead to familial ALS/FTD117,118. Loss of OPTN, a monogenic cause of ALS, resulted in activation of RIPK1 and necroptosis in the spinal cords of Optn–/– mice, which was rescued by genetic and pharmacological inactivation of RIPK1 kinase4. OPTN–/– cells and the spinal cords in OPTN–/– mice show increased levels of total RIPK1 protein, due to decreased proteasomal degradation of RIPK1 protein. Furthermore, gain-of-function mutations have been found in p62 (encoded by SQSTM1), another causal ALS factor that associates with OPTN; p62 mutants have been found to interact with RIPK1 to shift the balance between necroptotic and apoptotic death119,120.
TBK1 haploinsufficiency is a monogenic cause of ALS/FTD118,121. Mice lacking both copies of Tbk1 show profound RIPK1 activation, resulting in embryonic lethality that is rescued by genetic inactivation of RIPK1 (ref.18). Furthermore, TBK1 is responsible for directly inhibiting RIPK1 by phosphorylation at T189; thus, loss of TBK1 activity increases susceptibility to RIPK1 activation. Age-dependent reduction of TAK1 expression in human brains was shown to cooperate with heterozygous loss of TBK1 to promote late-onset ALS/FTD-like pathology mediated by decreased RIPK1 inhibition18,33.
The activation of RIPK1, RIPK3 and MLKL has been found in post-mortem spinal cord samples from patients with ALS4. An ex vivo human co-culture system of astrocytes and motor neurons from patients harbouring a SOD1 familial ALS mutation showed necroptotic death of motor neurons, which could be rescued with Nec-1 (ref.122). In addition, a Sod1G93A mouse model of ALS treated orally with Nec-1s showed improvements in survival, motor activity and axonal pathology. Taken together, these observations suggest a potential approach to genetic stratification of patients in a disease where ~90% of patients exhibit idiopathic disease without known genetic identifiers.
Alzheimer disease and ageing
AD is a form of progressive dementia that leads to the irreversible loss of neurons involved in cognitive and executive functions. The neuropathology of AD includes the presence of amyloid plaques and neurofibrillary tangles. Chronically sustained neuroinflammation plays an important role in mediating neural dysfunction and neurodegeneration in AD. Similar to what is observed in the insoluble fractions of post-mortem human CNS samples from patients with MS and ALS3,4, increased activation of RIPK1, RIPK3 and MLKL was also found in the insoluble fractions of sporadic AD post-mortem brains5,6. A recent study reported the detection of all three activated components of the necrosome machinery, pRIPK1, pRIPK3 and pMLKL, in granulovacuolar degeneration lesions in degenerating neurons in AD and preclinical stages of AD pathology123. Interestingly, the presence of activated necrosome components correlated with neuronal loss in AD-affected brain regions, such as the hippocampal CA1 region and the frontal cortex layer III, and Tau pathology, but not Aβ pathology.
RIPK1 may play an important role in driving neuroinflammation in AD6. The activation of RIPK1 was shown to mediate the transcriptional induction of Cst7, which encodes an endosomal/lysosomal cathepsin inhibitor named cystatin F, in microglia of mouse models of AD. Cst7 is a biomarker for disease-associated microglia present in spatial proximity to Aβ plaques found in both post-mortem human AD brain samples and in an AD mouse model124. Inhibition of RIPK1 suppresses the expression of a set of genes, such as CH25H, which are altered in microglia in two animal models of AD, in the Sod1G93A model of ALS and in ageing microglia, which enhance the ability of microglia to uptake and degrade Aβ6. CH25H is one of the cholesterol metabolism-related genes associated with AD125. Treatment with Nec-1s has been shown to be efficacious in reducing neuroinflammation and cognitive deficits in the APP/PS1 Aβ-driven mouse model of AD6. These results suggest that RIPK1 may control a neuroinflammatory pathway involved in multiple neurodegenerative diseases and in ageing.
Similar to the loss of oligodendrocytes seen in MS, RIPK1 and necroptosis may also drive white matter loss, which has been found to be among the earliest brain pathological changes in AD, preceding the formation of neural fibrillary tangles and amyloid plaques126. In addition, age-related changes in brain white matter — which include the reduction of myelinated fibres and myelin sheath degeneration co-localized with increased ubiquitin deposits — coupled with the presence of neuroinflammation may mediate the age-related cognitive decline seen in elderly individuals without AD127. The neuroinflammation and oligodendrocyte degeneration present in normal ageing human brains suggests the possibility of modulating RIPK1 kinase to support cognitive function in ageing. In addition, ageing is associated with mitochondrial dysfunction and the subsequent production of reactive oxygen species, the latter of which has shown responsiveness to RIPK1 inhibition in animal models128,129. Although this area requires significantly more exploration, one elucidated mechanism by which ageing sensitizes to RIPK1 activation is through the reduction of TAK1 expression in the CNS resulting from cerebral hypoperfusion in ageing human brains18,130. The loss of TAK1-mediated inhibitory phosphorylation of RIPK1 at S321 may provide a biomarker for RIPK1 activation in ageing brains.
A non-synonymous variant in SHARPIN has been identified as a risk variant for late-onset AD131. As SHARPIN-deficient cpdm mice develop TNF-dependent severe dermatitis and multi-organ inflammation that can be blocked upon inhibition of RIPK1 kinase28,30,75, the reduction of SHARPIN activity might contribute to the risk of developing late-onset AD by reducing M1 ubiquitylation of RIPK1 to promote its activation.
Parkinson disease and lysosomal storage disorders
LRRK2, the most common monogenic cause of Parkinson disease (PD), which is also upregulated in sporadic disease, has been identified as a sensitizer to RIPK1-dependent apoptosis132,133. Additionally, in vitro and in vivo models of these diseases suggest that RIPK1 can be activated by mitochondrial damage or lysosomal dysfunction. Induced pluripotent stem cell-derived dopaminergic neurons from patients harbouring OPA1 mutations that sensitize to mitochondrial fragmentation undergo necroptosis, which can be inhibited by RIPK1 inhibitors. Treatment with Nec-1 can also rescue in vivo loss of dopaminergic neurons in mice treated with MPTP, a rodent model of PD134. As mitochondrial dysfunction is known to produce high levels of reactive oxygen species, it is possible that increased reactive oxygen species are directly responsible for RIPK1 activation in PD135.
Consistent with RIPK1 activation downstream of lysosomal inhibition seen in AD models, lysosomal dysfunction caused by Niemann–Pick disease, type C1 and Gaucher’s disease also results in RIPK1-dependent activation of necroptosis136,137.
RIPK1 in sepsis and acute ischaemic insults
Sepsis
Sepsis is a life-threatening condition caused by immune hyperreactivity to viral, bacterial and fungal infections138. Sepsis is characterized by dysregulated production of pro-inflammatory cytokines (known as a cytokine storm), lymphopenia, coagulopathy, increased vascular permeability and eventual organ failure and death. Both genetic inhibition and pharmacological inhibition of RIPK1 in animal models have been shown to block TNF-induced sepsis27,29. Kinase inhibition of RIPK1 leads to robust and highly reproducible protection against sepsis, including attenuation of hypothermia and complete rescue of lethality. These observations suggest that the kinase activity of RIPK1 is critical in propagating immune hyperreactivity following infection, which may happen in septic conditions associated with severe pathogen infection. The efficacy of RIPK1 inhibitors in sepsis is mechanistically attributable to the reduction of pro-inflammatory cytokines and circulating damage-associated molecular patterns, rescue of increased intestinal or vascular permeability and activation of the clotting cascade in the vascular endothelium compartment27,29,139. Interestingly, the uncontrolled cytokine storm, which occurs in both patients and animal models, is thought to be driven by the interplay of inflammatory signalling and inflammatory cell death138. This deleterious cycle could be attenuated through RIPK1 inhibition, not only through a blockade of inflammatory response but also by inhibiting necrotic cell death; the RIPK1 inhibitor Nec-1 has been shown to protect against lung injury in a neonatal model of sepsis induced by bacteria-driven caecal slurry140.
Patients with severe COVID-19 caused by the novel SARS-CoV-2 infection exhibit multiple hallmarks of sepsis with increased plasma pro-inflammatory cytokines, including IL-6, IFNγ, CCL2, IL-10, G-CSF and TNF, coagulation dysfunction and lymphopenia, which has been suggested to be a predictor for the severity of COVID-19 (refs141,142). Activation of RIPK1 has been shown to promote the production of pro-inflammatory cytokines, including IL-6 and TNF, as seen in patients with non-cleavable RIPK1 mutations42. RIPK1 has also been shown to promote the production of IL-6, TNF, IL-8 and GM-CSF in mice in response to LPS or poly(I:C) injection, which mimic bacterial or viral infections, respectively92,143. In addition, RIPK1-mediated necroptosis of HIV-infected CD4+ T cells has been implicated in the depletion of T cell populations in patients144, suggesting that, more broadly, lymphopenia seen in viral infections such as HIV and SARS-CoV-2 might be attributable to the activation of RIPK1. Activation of necroptosis in severe sepsis and septic shock was also supported by the elevated levels of RIPK3 in the plasma of these patients145.
Taken together, these data suggest that an RIPK1 inhibitor may present a novel therapeutic option to reduce the aberrant hyperinflammatory response and sepsis in the context of both viral and bacterial infections.
Cerebral acute or ischaemic insults
Nec-1 was shown to be protective in the middle cerebral artery occlusion mouse model of stroke10, which was the first example of the role of necroptosis in animal models of human diseases. Subsequently, multiple in vivo studies confirmed that RIPK1 kinase inhibition shows efficacy in middle cerebral artery occlusion models146,147,148,149,150, acute brain trauma151 and ischaemia–reperfusion injuries in the brain, retina, heart, kidneys and liver149,152,153,154,155,156.
Widespread RIPK1 activation has been seen in ischaemia–reperfusion models of stroke, with rapid activation of necroptosis in neurons and endothelial cells, which may transition to apoptosis driven by the reduced TAK1 levels as the result of cerebral hypoperfusion130. Although there are compelling in vivo data that RIPK1 inhibition is effective at rescuing pathological and behavioural attributes following acute cerebral insults, a RIPK1 inhibitor has yet to be advanced into clinical trials for these conditions, which is largely due to broad challenges seen in conducting clinical trials for acute cerebral insults157. However, since a RIPK1 inhibitor may reduce cell death under both ischaemic and haemorrhagic stroke conditions, the ability to administer a RIPK1 inhibitor to stroke patients without differential diagnosis in an ambulance is an advantage that should be considered to reduce neuronal loss and preserve vascular function in the limited time window.
Other acute or ischaemic insults
Involvement of RIPK1 has also been suggested in mediating acute kidney injuries induced by cisplatin, gadolinium contrast, sepsis or ischaemia–reperfusion156,158,159,160,161,162 and hepatitis induced by ethanol, LPS, acetaminophen or concanavalin A163,164,165,166. Interestingly, a polymorphism in the TNF promoter at position 238 (known as the TNFA allele) leading to increased levels of TNF is associated with insulin resistance and is prevalent at higher rates in patients with nonalcoholic fatty liver disease167. Similarly, patients with nonalcoholic steatohepatitis and mice fed high-fat diets also showed increased levels of TNF, RIPK3 and MLKL168. Finally, ischaemia–reperfusion injuries are commonly seen following organ transplant. In mouse models of lung, kidney and heart transplant, inhibition of RIPK1 or necroptosis in the transplanted tissue improved allograft survival169,170,171.
Clinical development of RIPK1 inhibitors
RIPK1 contains a unique hydrophobic pocket that allosterically regulates the activation of its kinase activity9,172 (Fig. 3). All RIPK1 inhibitors described to date bind to this pocket. A phenotypic screen for small molecules that can block necroptosis led to the identification of the first specific inhibitors of RIPK1, 5-(1H-indol-3-ylmethyl)-2-thiohydantoins and 5-(1H-indol-3-ylmethyl)hydantoins known as Nec-1s9,10 (Table 4). A subsequent family of benzoxazepinone RIPK1 inhibitors was published by GlaxoSmithKline (GSK), which included GSK′481 and the clinical candidate GSK′772 (refs173,174), which was followed by the identification of a dihydropyrazole chemotype of RIPK1 inhibitors including GSK′963 and GSK′547 (refs175,176). Published and patented compounds generated by Takeda, Genentech and Rigel are all from the same benzoxazepinone family as the GSK′481 series177,178,179.

Receptor-interacting serine/threonine-protein kinase 1 (RIPK1) contains a unique hydrophobic pocket located between the N terminus and C terminus of the kinase domain, which allosterically regulates kinase activation. All RIPK1 inhibitors discovered to date, such as necrostatin-1s (Nec-1s) shown here, bind to this pocket and stabilize RIPK1 in an inactive conformation (PDB: 4ITH). This pocket is created owing to the outward movement of the αC-helix, resulting in the loss of an ionic pair between catalytic Lys45 and Glu63 of the αC-helix. The other side of the pocket is formed by the DLG motif in the inactive DLG-in conformation (catalytic Asp146 facing away from the active centre) and the activation segment, which immediately follows the DLG motif. Ser161 residue in the activation segment forms a critical hydrogen bond with the indole of Nec-1s. GSK′772 and other benzoxazepinones also extend into the ATP binding pocket, which may contribute to the increased affinity. Adapted with permission from ref.172, Elsevier.
Clinical trials
A timeline of clinical trials investigating RIPK1 inhibitors is summarized in Fig. 4. GSK′772 was the first RIPK1 kinase inhibitor to enter phase Ia clinical trials, and has subsequently progressed through phase Ia, Ib and II clinical trials173,174,180. This peripherally restricted compound is currently being tested for peripheral autoimmune diseases. Following completion of the initial phase II clinical trials with GSK′772 in ulcerative colitis, RA and psoriasis, phase Ib trials in psoriasis were reinitiated at a much higher dose (960 mg versus 60 mg daily)181. GSK′772 has continued to show an excellent safety profile in a phase IIa clinical trial in mild to moderate plaque psoriasis, but additional data may be needed to demonstrate efficacy in this indication182.

Clinical trials of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) inhibitors by GlaxoSmithKline (GSK), Denali Therapeutics, in partnership with Sanofi, and Rigel Pharmaceuticals12,13,14,15,16,183,186,188,190,230,231,232. AD, Alzheimer disease; ALS, amyotrophic lateral sclerosis; PDAC, pancreatic ductal adenocarcinoma; RA, rheumatoid arthritis; UC, ulcerative colitis.
DNL104 (Denali Therapeutics) was the first brain-penetrant RIPK1 inhibitor advanced into a phase Ia clinical trial; although this programme was later discontinued owing to limited post-dosing liver toxicity deemed unrelated to RIPK1 inhibition, this study demonstrated the safety of inhibiting RIPK1 in the CNS183. Denali Therapeutics subsequently initiated a phase Ia clinical trial for DNL747, another brain-penetrant RIPK1 inhibitor, followed by phase Ib/IIa trials of this compound in ALS and AD184. As of June 2020, phase Ib/IIa clinical trials for DNL747 are ongoing for ALS; however, Denali announced that DNL788, a brain-penetrant back-up compound, will be entering phase Ia trials in early 2021 (ref.185). Due to the progressive, chronic nature of AD and that >95% RIPK1 kinase inhibition may be required for this disease, DNL747 was deemed to have potential dose-limiting toxicities that were not seen preclinically with DNL788, although no toxicity has been seen in the clinic to date. Sanofi has partnered with Denali on RIPK1 inhibitors and completed a phase Ia clinical trial of the peripherally restricted DNL758 for peripheral autoimmune indications, including MS184,186,187. Rigel has also initiated a phase Ia clinical trial targeting autoimmune diseases with R552, a benzoxazepinone presumed to be peripherally restricted based on the properties of this chemical family188.
Given that RIPK1 kinase was suggested to promote macrophage-mediated adaptive immune tolerance in pancreatic cancer176 — a concept that was disputed later by another study30 — a combination trial of GSK′095, a clinical dihydropyrazole RIPK1 inhibitor candidate, and a checkpoint inhibitor was initiated for pancreatic ductal adenocarcinoma189. However, in late 2019 it was decided not to develop this asset further. To date, this remains the only RIPK1 combination therapy that has been advanced through investigational new drug (IND)-enabling studies into the clinic. Other combination therapies with RIPK1 inhibitors could be conceptualized based on our understanding of RIPK1 biology, on a disease-specific basis.
In summary, as of June 2020, RIPK1 inhibitors have entered human clinical trials for six indications: ALS and AD in neurodegeneration; psoriasis, ulcerative colitis and RA in autoimmune disease; and pancreatic ductal adenocarcinoma in oncology12,13,14,15,16,190.
Clinical biomarkers of RIPK1 activation
Phosphorylation of S166 RIPK1 has been established as a biomarker of RIPK1 target engagement3,9. In phase Ia clinical trials of DNL104, ex vivo assay-based phosphorylation of S166 RIPK1 in human PBMCs was used to assess the inhibition of RIPK1 kinase183, suggesting clinical feasibility to determine the activation of RIPK1 in blood samples for target engagement studies. In the phase Ia study of GSK′772, target engagement was also measured in PBMCs ex vivo using a novel immunoassay based on a conformational change that occurred following inhibitor binding to RIPK1 (ref.180).
Production of several pro-inflammatory cytokines and chemokines regulated by RIPK1 kinase activity can also be used as biomarkers of RIPK1 activation191. Of these rapidly inducible, secreted factors, GSK selected the chemokines macrophage inflammatory protein 1α (MIP1α; also known as CCL3) and MIP1β (also known as CCL4) as pharmacodynamic biomarkers downstream of RIPK1 activation180. GSK′772 dose-dependently reduced MIP-1α and MIP1β production, which corresponded to their target engagement data. Denali presented data indicating that IL-1β, IL-6 and MCP1 (also known as CCL2) were also RIPK1 responsive, demonstrating that DNL747 dose-dependently reduced IL-1β in primary human cells192.
Taken together, these studies demonstrate that RIPK1 preclinical biology has provided clinically validated biomarkers of RIPK1 activation in blood samples, which may also provide value as inclusion criteria to identify patients who will most benefit from RIPK1 inhibitor therapy.
Outlook
LoF or GoF mutations in the gene encoding RIPK1 itself, as well as in those encoding multiple regulators of the NF-κB pathway and RIPK1 activation, such as TAK1, NEMO, A20, ABIN1, OTULIN and the LUBAC complex, have been identified in human inflammatory and ageing-related diseases. In addition to these monogenic diseases, dysregulation of RIPK1, such as that which occurs on down-regulation or up-regulation of A20, may also be involved in mediating polygenic diseases involving RIPK1. The activation of RIPK1-mediated neuroinflammation may provide a common basis for the role of RIPK1 in human neurodegenerative diseases. Although a specific set of genetic associations between RIPK1 activation and neurodegenerative diseases has been discovered, the presence of activated RIPK1, RIPK3 and MLKL in post-mortem human pathological samples seen in MS, ALS and AD demonstrates the direct relevance of this pathway even in sporadic cases. Notably, the role of A20 and cFLIP, which are transcriptional targets of NF-κB, in suppressing the activation of RIPK1 suggests that defects in activation of the NF-κB pathway, in genetic and sporadic settings, may promote the activation of RIPK1 from dysregulated A20-mediated RIPK1 ubiquitylation modification as well as inactivation of Casp8 due to dysregulated cFLIP expression.
Phosphorylation of RIPK1 S166, as well as downstream events (such as pRIPK3 and pMLKL) and the increased production of specific pro-inflammatory cytokines (such as CCL2, MIP1α/CCL3, MIP1β/CCL4, IL-6 and TNF), can be used as biomarkers of RIPK1 activation. However, a brain-penetrant positron-emission tomography imaging probe for RIPK1 kinase activity would be particularly helpful in the development of RIPK1 inhibitors for the treatment of neurodegenerative diseases. As pathways downstream of RIPK1, including cell death, pro-inflammatory cytokines and NF-κB activation, can be modulated directly by disease pathogenesis, it is important to include biomarkers that specifically measure RIPK1 activation when considering a clinical study. It is also important to remember the cell type specificity of RIPK1 activation and the genetic context of the diseases, as sustained activation of RIPK1 in specific cell types, such as fibroblasts, can only be achieved in certain settings.
Clinical trials of RIPK1 inhibitors in patients are still in their early days, and, to date, limited chemotypes of RIPK1 inhibitors with appropriate in vivo properties have been developed. Development of selective, potent and safe small-molecule inhibitors of RIPK1, biomarkers to reliably measure the clinical efficacy of RIPK1 kinase inhibition and patient stratification remain the key challenges facing future clinical development.
References
Yuan, J., Amin, P. & Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2019). This review summarizes what is currently known about the role of RIPK1 in neurological diseases.
Ofengeim, D. & Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14, 727–736 (2013).
Ofengeim, D. et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 10, 1836–1849 (2015). This paper provides the first evidence for the role of RIPK1 in a chronic autoimmune disease with neurodegeneration — MS.
Ito, Y. et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353, 603–608 (2016). This paper provides the first genetic connection of RIPK1 kinase and necroptosis with ALS.
Caccamo, A. et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 20, 1236–1246 (2017).
Ofengeim, D. et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 114, E8788–E8797 (2017).
Dong, Y. et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc. Natl Acad. Sci. USA 113, 12304–12309 (2016).
Degterev, A., Ofengeim, D. & Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl Acad. Sci. USA 116, 9714–9722 (2019).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008). This paper identifies RIPK1 kinase as the target of multiple necrostatins, including Nec-1.
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005). This paper isolates multiple necrostatins, including Nec-1, and uses necrostatins to define necroptosis as a regulated necrotic cell death mechanism that broke the traditional dogma that necrosis was only passive cell death.
Degterev, A., Maki, J. L. & Yuan, J. Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase. Cell Death Differ. 20, 366 (2013).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT02776033 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT02858492 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT02903966 (2016).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03757351 (2018).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03757325 (2018).
Stanger, B. Z., Leder, P., Lee, T. H., Kim, E. & Seed, B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell 81, 513–523 (1995). This paper discovers RIPK1 as a death-domain containing serine/threonine kinase that can bind to Fas.
Xu, D. et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19 (2018). This paper demonstrates that the decline of TAK1 levels in human ageing brains sensitized the activation of RIPK1 to promote neuroinflammation and degeneration and the onset of ALS/FTD in individuals heterozygous for TBK1.
Meng, H. et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc. Natl Acad. Sci. USA 115, E2001–E2009 (2018).
Kelliher, M. A. et al. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303 (1998). This paper shows that the scaffold function of RIPK1 is involved in mediating NF-κB activation.
Shan, B., Pan, H., Najafov, A. & Yuan, J. Necroptosis in development and diseases. Genes Dev. 32, 327–340 (2018).
Dillon, C. P. et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202 (2014).
Kaiser, W. J. et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl Acad. Sci. USA 111, 7753–7758 (2014).
Rickard, J. A. et al. RIPK1 regulates RIPK3–MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 (2014).
Alvarez-Diaz, S. et al. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity 45, 513–526 (2016).
Dannappel, M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014).
Polykratis, A. et al. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 193, 1539–1543 (2014).
Berger, S. B. et al. Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 192, 5476–5480 (2014).
Patel, S. et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 27, 161–175 (2020). This paper demonstrates RIPK1 activation in human RA and psoriasis samples by pS166 immunochemistry, efficacy of a RIPK1 inhibitor in animal disease models of IBD, RA and skin inflammation, and effect of RIPK1 inhibitor for pancreatic cancer metastases.
Duprez, L. et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35, 908–918 (2011).
Polykratis, A. et al. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J. Immunol. 193, 1539–1543 (2014).
Wei, R. et al. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev. 31, 1162–1176 (2017).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Lafont, E. et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018).
Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 10, 1729 (2019).
Kanayama, A. et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol. Cell 15, 535–548 (2004).
Peltzer, N., Darding, M. & Walczak, H. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol. 26, 445–461 (2016).
Dziedzic, S. A. et al. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat. Cell Biol. 20, 58–68 (2018).
Annibaldi, A. et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol. Cell 69, 566–580.e5 (2018).
Wertz, I. & Dixit, V. A20—a bipartite ubiquitin editing enzyme with immunoregulatory potential. Adv. Exp. Med. Biol. 809, 1–12 (2014).
Lalaoui, N. et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577, 103–108 (2020).
Tao, P. et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 577, 109–114 (2020). Together with Lalaoui et al. (2020) (ref. 41), this paper reports the discovery of a dominant autoinflammatory human disease caused by non-cleavable RIPK1.
Newton, K. et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 575, 679–682 (2019).
Cuchet-Lourenco, D. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018).
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 116, 970–975 (2019). Together with Cuchet-Lourenco et al. (2018) (ref. 44), this paper reports that biallelic loss-of-function mutations in RIPK1 lead to primary immunodeficiency and intestinal inflammation.
Lin, Y., Devin, A., Rodriguez, Y. & Liu, Z. G. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes. Dev. 13, 2514–2526 (1999).
Newton, K. et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574, 428–731 (2019).
Zhang, X., Dowling, J. P. & Zhang, J. RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis. 10, 245 (2019).
Aksentijevich, I. & Zhou, Q. NF-κB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front. Immunol. 8, 399 (2017).
Zhou, Q. et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 48, 67–73 (2016). This paper identifies LoF in A20 as a cause of autoinflammatory disease.
Rajamaki, K. et al. Haploinsufficiency of A20 impairs protein–protein interactome and leads into caspase-8-dependent enhancement of NLRP3 inflammasome activation. RMD Open 4, e000740 (2018).
Fung, E. Y. et al. Analysis of 17 autoimmune disease-associated variants in type 1 diabetes identifies 6q23/TNFAIP3 as a susceptibility locus. Genes Immun. 10, 188–191 (2009).
Musone, S. L. et al. Sequencing of TNFAIP3 and association of variants with multiple autoimmune diseases. Genes Immun. 12, 176–182 (2011).
Nair, R. P. et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat. Genet. 41, 199–204 (2009). This paper identifies NF-κB associated genes as risk factors for psoriasis.
Schuijs, M. J. et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 349, 1106–1110 (2015).
Devos, M. et al. Keratinocyte expression of A20/TNFAIP3 controls skin inflammation associated with atopic dermatitis and psoriasis. J. Invest. Dermatol. 139, 135–145 (2019).
Hacker, H. & Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. STKE 2006, re13 (2006).
Rahighi, S. et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 136, 1098–1109 (2009).
Artac, H. et al. Infliximab therapy for inflammatory colitis in an infant with NEMO deficiency. Immunol. Res. 67, 450–453 (2019).
Schmidt-Supprian, M. et al. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol. Cell 5, 981–992 (2000).
Makris, C. et al. Female mice heterozygous for IKKγ/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell 5, 969–979 (2000).
Nenci, A. et al. Skin lesion development in a mouse model of incontinentia pigmenti is triggered by NEMO deficiency in epidermal keratinocytes and requires TNF signaling. Hum. Mol. Genet. 15, 531–542 (2006).
Vlantis, K. et al. NEMO prevents RIP kinase 1-mediated epithelial cell death and chronic intestinal inflammation by NF-κ-dependent and -independent functions. Immunity 44, 553–567 (2016).
Keusekotten, K. et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 153, 1312–1326 (2013).
Damgaard, R. B. et al. The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 166, 1215–1230.e20 (2016).
Zhou, Q. et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl Acad. Sci. USA 113, 10127–10132 (2016). This paper identifies OTULIN mutations as a cause of early-onset autoinflammatory disease.
Heger, K. et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559, 120–124 (2018).
Damgaard, R. B. et al. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 11, e9324 (2019).
Boisson, B. et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 212, 939–951 (2015). This paper identifies mutations in the LUBAC complex as a cause of autoinflammation and immunodeficiency.
Oda, H. et al. Second case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front. Immunol. 10, 479 (2019).
Boisson, B. et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 13, 1178–1186 (2012).
Peltzer, N. & Walczak, H. Cell death and inflammation — a vital but dangerous liaison. Trends Immunol. 40, 387–402 (2019).
HogenEsch, H. et al. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol. 143, 972–982 (1993).
Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
Webster, J. D. et al. RIP1 kinase activity is critical for skin inflammation but not for viral propagation. J. Leukoc. Biol. 107, 941–952 (2020).
Jostins, L. et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Spehlmann, M. E. et al. Epidemiology of inflammatory bowel disease in a German twin cohort: results of a nationwide study. Inflamm. Bowel Dis. 14, 968–976 (2008).
Vereecke, L. et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J. Exp. Med. 207, 1513–1523 (2010).
Garcia-Carbonell, R. et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc. Natl Acad. Sci. USA 115, E9192–E9200 (2018).
Polykratis, A. et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 21, 731–742 (2019).
Vereecke, L. et al. A20 controls intestinal homeostasis through cell-specific activities. Nat. Commun. 5, 5103 (2014).
Stappenbeck, T. S. et al. Crohn disease: a current perspective on genetics, autophagy and immunity. Autophagy 7, 355–374 (2011).
Lassen, K. G. et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl Acad. Sci. USA 111, 7741–7746 (2014).
Murthy, A. et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506, 456–462 (2014).
Matsuzawa-Ishimoto, Y. et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 214, 3687–3705 (2017).
Tsoi, L. C. et al. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nat. Commun. 8, 15382 (2017).
Sahlol, N. Y., Mostafa, M. S., Madkour, L. A. E. & Salama, D. M. Low TNFAIP3 expression in psoriatic skin promotes disease susceptibility and severity. PLoS ONE 14, e0217352 (2019).
Tejasvi, T. et al. TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J. Invest. Dermatol. 132, 593–600 (2012).
Oshima, S. et al. ABIN-1 is a ubiquitin sensor that restricts cell death and sustains embryonic development. Nature 457, 906–909 (2009).
Wertz, I. E. et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430, 694–699 (2004).
Herhaus, L. et al. Molecular recognition of M1-linked ubiquitin chains by native and phosphorylated UBAN domains. J. Mol. Biol. 431, 3146–3156 (2019).
Su, Z. et al. ABIN-1 heterozygosity sensitizes to innate immune response in both RIPK1-dependent and RIPK1-independent manner. Cell Death Differ. 26, 1077–1088 (2019).
Li, P. et al. Integrative analysis of genome-wide association study and expression quantitative trait loci datasets identified various immune cell-related pathways for rheumatoid arthritis. Ann. Hum. Genet. 84, 72–79 (2020). This review summarizes risk variants for RA associated with immune signalling pathways.
Elsby, L. M. et al. Functional evaluation of TNFAIP3 (A20) in rheumatoid arthritis. Clin. Exp. Rheumatol. 28, 708–714 (2010).
Pegoretti, V., Baron, W., Laman, J. D. & Eisel, U. L. M. Selective modulation of TNF–TNFRs signaling: insights for multiple sclerosis treatment. Front. Immunol. 9, 925 (2018).
Gregory, A. P. et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 488, 508–511 (2012). Together with Tejasvi et al. (2012) (ref. 88), this paper provides the first insight that genetic variants in the TNFR1/RIPK1 signalling pathway modulate response to anti-TNF therapy in autoimmune disease.
Silke, J. & Strasser, A. The FLIP side of life. Sci. Signal. 6, pe2 (2013).
Yoshikawa, M. et al. Discovery of 7-oxo-2,4,5,7-tetrahydro-6H-pyrazolo[3,4-c]pyridine derivatives as potent, orally available, and brain-penetrating receptor interacting protein 1 (RIP1) kinase inhibitors: analysis of structure–kinetic relationships. J. Med. Chem. 61, 2384–2409 (2018).
International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188 (2019).
Kim, S. J. & Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 4, e716 (2013).
Zhu, K. et al. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 9, 500 (2018).
Gateva, V. et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 41, 1228–1233 (2009).
Kawasaki, A. et al. Association of TNFAIP3 interacting protein 1, TNIP1 with systemic lupus erythematosus in a Japanese population: a case–control association study. Arthritis Res. Ther. 12, R174 (2010).
Adrianto, I. et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat. Genet. 43, 253–258 (2011).
Naccache, P. H. & Fernandes, M. J. Challenges in the characterization of neutrophil extracellular traps: the truth is in the details. Eur. J. Immunol. 46, 52–55 (2016).
D’Cruz, A. A. et al. The pseudokinase MLKL activates PAD4-dependent NET formation in necroptotic neutrophils. Sci. Signal. 11, eaao1716 (2018).
Wang, X., Yousefi, S. & Simon, H. U. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 9, 111 (2018).
Guo, R. et al. A role for receptor-interacting protein kinase-1 in neutrophil extracellular trap formation in patients with systemic lupus erythematosus: a preliminary study. Cell Physiol. Biochem. 45, 2317–2328 (2018).
Schreiber, A. et al. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl Acad. Sci. USA 114, E9618–E9625 (2017).
Nakazawa, D. et al. Activated platelets induce MLKL-driven neutrophil necroptosis and release of neutrophil extracellular traps in venous thrombosis. Cell Death Discov. 4, 6 (2018).
Lee, K. H. et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun. Rev. 16, 1160–1173 (2017).
Zenaro, E. et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886 (2015).
Bolton, S. J., Anthony, D. C. & Perry, V. H. Loss of the tight junction proteins occludin and zonula occludens-1 from cerebral vascular endothelium during neutrophil-induced blood-brain barrier breakdown in vivo. Neuroscience 86, 1245–1257 (1998).
Lusthaus, M., Mazkereth, N., Donin, N. & Fishelson, Z. Receptor-interacting protein kinases 1 and 3, and mixed lineage kinase domain-like protein are activated by sublytic complement and participate in complement-dependent cytotoxicity. Front. Immunol. 9, 306 (2018).
Zhu, S., Zhang, Y., Bai, G. & Li, H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis. 2, e115 (2011).
Morgan, J. E. et al. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 9, 3655 (2018).
Maruyama, H. et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226 (2010).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636 (2015). Together with Maruyama et al. (2010) (ref. 117), this paper demonstrates that mutations in TBK1 and OPTN are causes of ALS/FTD.
Fecto, F. et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68, 1440–1446 (2011).
Goodall, M. L. et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev. Cell 37, 337–349 (2016).
Cirulli, E. T. et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441 (2015).
Re, D. B. et al. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron 81, 1001–1008 (2014).
Koper, M. J. et al. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 139, 463–484 (2019).
Keren-Shaul, H. et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017).
Papassotiropoulos, A. et al. Cholesterol 25-hydroxylase on chromosome 10q is a susceptibility gene for sporadic Alzheimer’s disease. Neurodegener Dis. 2, 233–241 (2005).
Butt, A. M., De La Rocha, I. C. & Rivera, A. Oligodendroglial cells in Alzheimer’s disease. Adv. Exp. Med. Biol. 1175, 325–333 (2019).
Hinman, J. D. & Abraham, C. R. What’s behind the decline? The role of white matter in brain aging. Neurochem. Res. 32, 2023–2031 (2007).
Duan, S. et al. Inhibiting RIPK1 limits neuroinflammation and alleviates postoperative cognitive impairments in d-galactose-induced aged mice. Front. Behav. Neurosci. 12, 138 (2018).
Qing, W. et al. Inhibiting RIP1 Improves chronic stress-induced cognitive impairments in d-galactose-induced aging mice. Front. Behav. Neurosci. 12, 234 (2018).
Naito, M. G. et al. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl Acad. Sci. USA 117, 4959–4970 (2020).
Asanomi, Y. et al. A rare functional variant of SHARPIN attenuates the inflammatory response and associates with increased risk of late-onset Alzheimer’s disease. Mol. Med. 25, 20 (2019).
Di Maio, R. et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 10, eaar5429 (2018).
Amin, P. et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc. Natl Acad. Sci. USA 115, E5944–E5953 (2018).
Iannielli, A. et al. Pharmacological inhibition of necroptosis protects from dopaminergic neuronal cell death in Parkinson’s disease models. Cell Rep. 22, 2066–2079 (2018).
Zhang, Y. et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 8, 14329 (2017).
Cougnoux, A. et al. Necroptosis in Niemann–Pick disease, type C1: a potential therapeutic target. Cell Death Dis. 7, e2147 (2016).
Vitner, E. B. et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat. Med. 20, 204–208 (2014).
Nedeva, C., Menassa, J. & Puthalakath, H. Sepsis: inflammation is a necessary evil. Front. Cell Dev. Biol. 7, 108 (2019).
Zelic, M. et al. RIP kinase 1-dependent endothelial necroptosis underlies systemic inflammatory response syndrome. J. Clin. Invest. 128, 2064–2075 (2018).
Bolognese, A. C. et al. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery 164, 110–116 (2018).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Tan, L. et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal. Transduct. Target. Ther. 5, 33 (2020).
Najjar, M. et al. RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by Toll-like receptor 4. Immunity 45, 46–59 (2016).
Pan, T. et al. Necroptosis takes place in human immunodeficiency virus type-1 (HIV-1)-infected CD4+ T lymphocytes. PLoS ONE 9, e93944 (2014).
Wang, B. et al. Necroptosis regulated proteins expression is an early prognostic biomarker in patient with sepsis: a prospective observational study. Oncotarget 8, 84066–84073 (2017).
Xu, X. et al. Synergistic protective effects of humanin and necrostatin-1 on hypoxia and ischemia/reperfusion injury. Brain Res. 1355, 189–194 (2010).
Deng, X. X., Li, S. S. & Sun, F. Y. Necrostatin-1 prevents necroptosis in brains after ischemic stroke via inhibition of RIPK1-mediated RIPK3/MLKL signaling. Aging Dis. 10, 807–817 (2019).
Su, X. et al. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem. Res. 40, 643–650 (2015).
Chen, Y. et al. Necrostatin-1 improves long-term functional recovery through protecting oligodendrocyte precursor cells after transient focal cerebral ischemia in mice. Neuroscience 371, 229–241 (2018).
Yang, C. et al. Inhibition of necroptosis rescues SAH-induced synaptic impairments in hippocampus via CREB–BDNF pathway. Front. Neurosci. 12, 990 (2018).
You, Z. et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 28, 1564–1573 (2008).
Hong, J. M., Kim, S. J. & Lee, S. M. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol. Appl. Pharmacol. 308, 1–10 (2016).
Zhe-Wei, S., Li-Sha, G. & Yue-Chun, L. The role of necroptosis in cardiovascular disease. Front. Pharmacol. 9, 721 (2018).
Rosenbaum, D. M. et al. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal ischemia–reperfusion injury model. J. Neurosci. Res. 88, 1569–1576 (2010).
von Massenhausen, A. et al. Phenytoin inhibits necroptosis. Cell Death Dis. 9, 359 (2018).
Newton, K. et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576 (2016).
Furlan, A. J. Challenges in acute ischemic stroke clinical trials. Curr. Cardiol. Rep. 14, 761–766 (2012).
Linkermann, A., De Zen, F., Weinberg, J., Kunzendorf, U. & Krautwald, S. Programmed necrosis in acute kidney injury. Nephrol. Dial. Transpl. 27, 3412–3419 (2012).
Linkermann, A. et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 81, 751–761 (2012).
Linkermann, A. et al. The RIP1-kinase inhibitor necrostatin-1 prevents osmotic nephrosis and contrast-induced AKI in mice. J. Am. Soc. Nephrol. 24, 1545–1557 (2013).
Muller, T. et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol. Life Sci. 74, 3631–3645 (2017).
Dong, W. et al. Necrostatin-1 attenuates sepsis-associated acute kidney injury by promoting autophagosome elimination in renal tubular epithelial cells. Mol. Med. Rep. 17, 3194–3199 (2018).
Roychowdhury, S., McMullen, M. R., Pisano, S. G., Liu, X. & Nagy, L. E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 57, 1773–1783 (2013).
Kim, S. J. & Lee, S. M. Necrostatin-1 protects against d-galactosamine and lipopolysaccharide-induced hepatic injury by preventing TLR4 and RAGE signaling. Inflammation 40, 1912–1923 (2017).
Takemoto, K. et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 4, 777–787 (2014).
Filliol, A. et al. RIPK1 protects from TNFα-mediated liver damage during hepatitis. Cell Death Dis. 7, e2462 (2016).
Valenti, L. et al. Tumor necrosis factor α promoter polymorphisms and insulin resistance in nonalcoholic fatty liver disease. Gastroenterology 122, 274–280 (2002).
Afonso, M. B. et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 129, 721–739 (2015).
Kanou, T. et al. Inhibition of regulated necrosis attenuates receptor-interacting protein kinase 1-mediated ischemia–reperfusion injury after lung transplantation. J. Heart Lung Transpl. 37, 1261–1270 (2018).
Lau, A. et al. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transpl. 13, 2805–2818 (2013).
Pavlosky, A. et al. RIPK3-mediated necroptosis regulates cardiac allograft rejection. Am. J. Transpl. 14, 1778–1790 (2014).
Xie, T. et al. Structural basis of RIP1 inhibition by necrostatins. Structure 21, 493–499 (2013).
Harris, P. A. et al. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J. Med. Chem. 60, 1247–1261 (2017).
Harris, P. A. et al. DNA-encoded library screening identifies benzo[B][1,4]oxazepin-4-ones as highly potent and monoselective receptor interacting protein 1 kinase inhibitors. J. Med. Chem. 59, 2163–2178 (2016). This paper is the first publication of a benzoxazepinone RIPK1 inhibitor, which subsequently has been explored by multiple groups.
Berger, S. B. et al. Characterization of GSK′963: a structurally distinct, potent and selective inhibitor of RIP1 kinase. Cell Death Discov. 1, 15009 (2015).
Wang, W. et al. RIP1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 34, 757–774.e7 (2018).
Yoshikawa, M. et al. Discovery of 7-oxo-2,4,5,7-tetrahydro-6H-pyrazolo[3,4-c]pyridine derivatives as potent, orally available, and brain-penetrating receptor interacting protein 1 (RIP1) kinase inhibitors: analysis of structure–kinetic relationships. J. Med. Chem. 61, 2384–2409 (2018).
Hamilton, G. L. et al. Potent and selective inhibitors of receptor-interacting protein kinase 1 that lack an aromatic back pocket group. Bioorg. Med. Chem. Lett. 29, 1497–1501 (2019).
Darwish, I., Yu, J., Chen, Y., Masuda, E. & Taylor, V. RIP1 inhibitory compounds and methods for making and using the same. https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019213445 (2019).
Weisel, K. et al. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 5, e00365 (2017). Together with Grievink et al. (2020) (ref. 183), this paper outlines pharmacokinetic and pharmacodynamic biomarkers used in clinical trials for RIPK1 inhibitors.
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04316585 (2020).
Weisel, K. et al. Response to inhibition of receptor-interacting protein kinase 1 (RIPK1) in active plaque psoriasis: a randomized placebo-controlled study. Clin. Pharmacol. Ther. https://doi.org/10.1002/cpt.1852 (2020).
Grievink, H. W. et al. DNL104, a centrally penetrant RIPK1 inhibitor, inhibits RIP1 kinase phosphorylation in a randomized phase I ascending dose study in healthy volunteers. Clin. Pharmacol. Ther. 107, 406–414 (2020).
Denali Therapeutics. Denali Therapeutics announces positive clinical results with its lead RIPK1 inhibitor molecule and intention to initiate patient studies in multiple indications in collaboration with Sanofi. https://denalitherapeutics.com/investors/press-release?id=6901 (2018).
Denali. Denali Therapeutics provides broad update on its RIPK1 program partnered with Sanofi. https://denalitherapeutics.com/investors/press-release?id=7626 (2020).
Denali Therapeutics. Denali Therapeutics announces that its partner Sanofi has commenced dosing of DNL758 in a phase 1 study https://www.globenewswire.com/news-release/2019/08/05/1896875/0/en/Denali-Therapeutics-Announces-That-Its-Partner-Sanofi-has-Commenced-Dosing-of-DNL758-in-a-Phase-1-Study.html (2019).
Bonanomi, G. et al. Isoxazolidine derived inhibitors of receptor interacting protein kinase 1 (ripk1). https://patentscope.wipo.int/search/en/detail.jsf?docId=US234728180 (2016).
Rigel Pharmaceuticals. Rigel Pharmaceuticals provides business update prior to investor & analyst call. https://ir.rigel.com/news-events/press-releases/detail/270/rigel-pharmaceuticals-provides-business-update-prior-to (2019).
Harris, P. A. et al. Identification of a RIP1 kinase inhibitor clinical candidate (GSK3145095) for the treatment of pancreatic cancer. ACS Med. Chem. Lett. 10, 857–862 (2019).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT03681951 (2018).
Zhu, K. et al. Necroptosis promotes cell-autonomous activation of proinflammatory cytokine gene expression. Cell Death Dis. 9, 500 (2018).
Denali. Discover, develop, defeat degeneration. https://denalitherapeutics.com/uploads/documents/events/Denali_GS_2018_06_12_rPDF.pdf (2018).
Aoki, T. et al. An association between asthma and TNF-308G/A polymorphism: meta-analysis. J. Hum. Genet. 51, 677–685 (2006).
McDermott, M. F. et al. Germline mutations in the extracellular domains of the 55kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97, 133–144 (1999).
Edilova, M. I., Abdul-Sater, A. A. & Watts, T. H. TRAF1 signaling in human health and disease. Front. Immunol. 9, 2969 (2018).
Speckmann, C. & Ehl, S. XIAP deficiency is a mendelian cause of late-onset IBD. Gut 63, 1031–1032 (2014).
Latour, S. & Aguilar, C. XIAP deficiency syndrome in humans. Semin. Cell Dev. Biol. 39, 115–123 (2015).
Chen, F. R. et al. Association of PELI1 polymorphisms in systemic lupus erythematosus susceptibility in a Chinese population. Lupus 24, 1037–1044 (2015).
Thienpont, B. et al. Haploinsufficiency of TAB2 causes congenital heart defects in humans. Am. J. Hum. Genet. 86, 839–849 (2010).
Pottier, C. et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92 (2015).
Zheng, C. et al. Infantile onset intractable inflammatory bowel disease due to novel heterozygous mutations in TNFAIP3 (A20). Inflamm. Bowel Dis. 24, 2613–2620 (2018).
Musone, S. L. et al. Multiple polymorphisms in the TNFAIP3 region are independently associated with systemic lupus erythematosus. Nat. Genet. 40, 1062–1064 (2008).
Shigemura, T. et al. Novel heterozygous C243Y A20/TNFAIP3 gene mutation is responsible for chronic inflammation in autosomal-dominant Behcet’s disease. RMD Open 2, e000223 (2016).
Vande Walle, L. et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512, 69–73 (2014).
Zhu, L. et al. Characteristics of A20 gene polymorphisms and clinical significance in patients with rheumatoid arthritis. J. Transl. Med. 13, 215 (2015).
Rizk, M. M., Elsayed, E. T., ElKeraie, A. F. & Ramzy, I. Association of tumor necrosis factor α-induced protein 3 interacting protein 1 (TNIP1) gene polymorphism (rs7708392) with lupus nephritis in Egyptian patients. Biochem. Genet. 56, 478–488 (2018).
Hovelmeyer, N. et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur. J. Immunol. 41, 595–601 (2011).
Matmati, M. et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 43, 908–912 (2011).
Adrianto, I. et al. Association of two independent functional risk haplotypes in TNIP1 with systemic lupus erythematosus. Arthritis Rheum. 64, 3695–3705 (2012).
Caster, D. J. et al. ABIN1 dysfunction as a genetic basis for lupus nephritis. J. Am. Soc. Nephrol. 24, 1743–1754 (2013).
McLaughlin, R. L. et al. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat. Commun. 8, 14774 (2017).
Patsopoulos, N. A. et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 70, 897–912 (2011).
Doffinger, R. et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat. Genet. 27, 277–285 (2001).
Smahi, A. et al. Genomic rearrangement in NEMO impairs NF-κB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 405, 466–472 (2000). Together with Doffinger et al. (2001) (ref. 210), this paper identifies mutations in NEMO as the cause of incontinentia pigmenti and anhidrotic ectodermal dysplasia with immunodeficiency.
Aradhya, S. et al. Atypical forms of incontinentia pigmenti in male individuals result from mutations of a cytosine tract in exon 10 of NEMO (IKKγ). Am. J. Hum. Genet. 68, 765–771 (2001).
Senegas, A., Gautheron, J., Maurin, A. G. & Courtois, G. IKK-related genetic diseases: probing NF-κB functions in humans and other matters. Cell Mol. Life Sci. 72, 1275–1287 (2015).
Niemela, J. et al. Caspase-8 deficiency presenting as late-onset multi-organ lymphocytic infiltration with granulomas in two adult siblings. J. Clin. Immunol. 35, 348–355 (2015).
Onizawa, M. et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627 (2015).
Guedes, R. P. et al. A20 deficiency causes spontaneous neuroinflammation in mice. J. Neuroinflammation 11, 122 (2014).
Lippens, S. et al. Keratinocyte-specific ablation of the NF-κB regulatory protein A20 (TNFAIP3) reveals a role in the control of epidermal homeostasis. Cell Death Differ. 18, 1845–1853 (2011).
Priem, D. et al. A20 protects cells from TNF-induced apoptosis through linear ubiquitin-dependent and -independent mechanisms. Cell Death Dis. 10, 692 (2019).
Harris, P. A. et al. Discovery of small molecule RIP1 kinase inhibitors for the treatment of pathologies associated with necroptosis. ACS Med. Chem. Lett. 4, 1238–1243 (2013).
Ren, Y. et al. Discovery of a highly potent, selective, and metabolically stable inhibitor of receptor-interacting protein 1 (RIP1) for the treatment of systemic inflammatory response syndrome. J. Med. Chem. 60, 972–986 (2017).
Delehouze, C. et al. 6E11, a highly selective inhibitor of receptor-interacting protein kinase 1, protects cells against cold hypoxia-reoxygenation injury. Sci. Rep. 7, 12931 (2017).
Hou, J. et al. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis. 10, 493 (2019).
Najjar, M. et al. Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 10, 1850–1860 (2015).
Wang, K. et al. Structure–activity relationship analysis of a novel necroptosis inhibitor, necrostatin-5. Bioorg. Med. Chem. Lett. 17, 1455–1465 (2007).
Zheng, W., Degterev, A., Hsu, E., Yuan, J. & Yuan, C. Structure–activity relationship study of a novel necroptosis inhibitor, necrostatin-7. Bioorg. Med. Chem. Lett. 18, 4932–4935 (2008).
Wu, Z., Li, Y., Cai, Y., Yuan, J. & Yuan, C. A novel necroptosis inhibitor-necrostatin-21 and its SAR study. Bioorg. Med. Chem. Lett. 23, 4903–4906 (2013).
US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/show/NCT03305419 (2017).
GlaxoSmithKline. Our pipeline: pipeline changes https://www.gsk.com/en-gb/research-and-development/our-pipeline/#pipeline-changes (2019).
Denali Therapeutics. Denali Therapeutics announces first-in-human dosing of its RIPK1 inhibitor clinical program. https://denalitherapeutics.com/investors/press-release?id=6601 (2018).
Oberst, A. et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011).
Feoktistova, M. et al. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43, 449–463 (2011).
Lipinski, M. M. et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl Acad. Sci. USA 107, 14164–14169 (2010).
Ali, M. & Mocarski, E. S. Proteasome inhibition blocks necroptosis by attenuating death complex aggregation. Cell Death Dis. 9, 346 (2018).
Liu, S. et al. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 9, 476 (2018).
Vieira, M. et al. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol. Dis. 68, 26–36 (2014).
Acknowledgements
Studies in the authors’ laboratories (J.Y.) are supported by NIH grants R21-AG059073, R01-AG047231 and RF1-AG055521. The authors thank Q. Zhou of Zhejiang University, J. Lewcock of Denali Therapeutics and D. Vucic of Genentech for helpful comments. The authors thank B. Toure of Nido Biosciences for assistance with figures.
Author information
Authors and Affiliations
Contributions
J.Y. conceived the review topic. L.M. and J.Y. wrote the manuscript with support from D.O.
Corresponding author
Ethics declarations
Competing interests
J.Y. is a consultant for Denali Therapeutics and Sanofi. D.O. is an employee of Sanofi.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- NF-κB
-
(Nuclear factor κ light chain enhancer of activated B cells). A protein complex whose pathway, which can be activated in response to cytokines, free radicals, viral or bacterial antigens and other stressors, mediates the transcription of genes required for pro-survival and pro-inflammatory signalling.
- TNFR1
-
(Tumour necrosis factor receptor 1). A type I membrane receptor that contains an intracellular death domain that, when activated, can recruit receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and TRADD to mediate inflammatory and pro-survival functions through the NF-κB pathway, as well as cell death mediated by RIPK1 kinase activity.
- TNF
-
(Tumour necrosis factor). A pro-inflammatory cytokine typically secreted by macrophages/monocytes and microglia. TNF signals through two receptors: TNFR1, which mediates cell death and NF-κB signalling; and TNFR2, which mediates non-canonical NF-κB signalling.
- Complex I
-
A transient complex associated with the intracellular domain of TNFR1 upon TNF stimulation that includes RIPK1 and many other regulators of NF-κB activation and cell death.
- RIPK1-dependent apoptosis
-
(RDA). A form of apoptosis enacted by activated RIPK1 kinase that forms a transient intermediate cell death complex comprising highly ubiquitylated, activated RIPK1, FADD and Casp8, known as iuRIPK1 complex, which then transitions to form complex IIa to mediate the activation of Casp8.
- RIPK1-independent apoptosis
-
A form of apoptosis enacted by activated (cleaved) caspase 8 that is independent of RIPK1 kinase activity.
- Loss of function
-
(LoF). Mutations or polymorphisms that lead to reduced functionality of a protein. This may include decreased transcription or translation, increased degradation or decreased functional activity relative to the wild-type protein.
- Gain of function
-
(GoF). Mutations or polymorphisms that lead to increased functionality of a protein. This may include increased transcription or translation, greater stability or decreased degradation, or increased or aberrant functional activity relative to the wild-type protein.
- Necrosome
-
(Also known as complex IIb). A complex comprising activated RIPK1, receptor-interacting serine/threonine-protein kinase 3 (RIPK3) and mixed-lineage kinase domain-like pseudokinase (MLKL) that enacts necroptosis, a regulated form of necrotic cell death.
Rights and permissions
About this article

Cite this article
Mifflin, L., Ofengeim, D. & Yuan, J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev Drug Discov 19, 553–571 (2020). https://doi.org/10.1038/s41573-020-0071-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41573-020-0071-y
This article is cited by
-
Immunomodulatory and clinical effects of receptor-interacting protein kinase 1 (RIPK1) inhibitor eclitasertib (SAR443122) in patients with severe COVID-19: a phase 1b, randomized, double-blinded, placebo-controlled study
Respiratory Research (2024)
-
Defective prelamin A processing promotes unconventional necroptosis driven by nuclear RIPK1
Nature Cell Biology (2024)
-
Mediators of necroptosis: from cell death to metabolic regulation
EMBO Molecular Medicine (2024)
-
Immunogenic cell death in cancer: targeting necroptosis to induce antitumour immunity
Nature Reviews Cancer (2024)
-
RIPK1 and necroptosis role in premature ageing
Nature Cell Biology (2024)
