
Abstract
The hawkmoth genus Rhodafra comprises two African species with unclear relationships, as their wing patterns are markedly different, with one species closely resembling species of a related genus, Hyles. The present paper aims to investigate the monophyly and phylogenetic position of Rhodafra in relation to Hyles and other genera of the subtribe Choerocampina (Sphingidae: Macroglossinae: Macroglossini) using mitochondrial and nuclear sequence data from more species and individuals than have hitherto been studied. As no fresh tissue of Rhodafra was available, ancient-DNA methodology was applied. All data corroborate the genus as monophyletic and that a similar wing pattern is not a good indicator of close phylogenetic relationship in this group of moths. Phylogenetic trees based on mitochondrial data agree in placing Rhodafra within Hyles. In contrast, analysis of nuclear EF1alpha sequences produces a topology in which Rhodafra is placed as the sister clade to Hyles. Although multispecies coalescent analyses suggest a polytomy between Rhodafra, Hyles lineata and the remaining Hyles, total evidence analyses corroborate Rhodafra as sister to Hyles. This relationship is interpreted as the favoured topology. For a more robust result, the question should be re-examined using genomic approaches.
Similar content being viewed by others

Introduction
The family Sphingidae (hawkmoths) comprises four subfamilies, Langiinae, Smerinthinae, Sphinginae and Macroglossinae (Kitching and Rougerie et al. 2018). Within the last of these, Choerocampina are a subtribe of largely tropical distribution in which the second segment of the labial palp and the labral pilifer have become modified into a unique bat-detecting ultrasonic hearing organ (Roeder et al. 1970; Kawahara et al. 2009 and references therein). Largely on the basis of modifications of this structure, Rothschild and Jordan (1903) divided the genera of Choerocampina into three subgroups: (1) Chaerocina Rothschild & Jordan, 1903 and Euchloron Boisduval, [1875]; (2) Deilephila [Laspeyres], 1809, Hyles Hübner, [1819], Phanoxyla, Rothschild & Jordan, 1903, Rhodafra Rothschild & Jordan, 1903, Xylophanes Hübner, [1819]; and (3) Basiothia Walker, 1856, Cechenena Rothschild & Jordan, 1903, Centroctena Rothschild & Jordan, 1903, Hippotion Hübner, [1819], Pergesa Walker, 1856, Rhagastis Rothschild & Jordan, 1903, and Theretra Hübner, [1819]. Subsequently, two further genera were described, Griseosphinx Kitching & Cadiou, 2000 and Cechetra Zolotuhin & Ryabov, 2012, to bring the total currently recognized number of genera in Choerocampina to 16. Although Rothschild and Jordan’s (1903) subdivision of the Choerocampina has been accepted for over a century without change, recent phylogenetic analyses using DNA sequence data (e.g. Kawahara et al. 2009) suggest that a different subdivision will be recognized once all the genera have been analysed together.
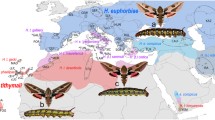
One of the genera whose phylogenetic placement is currently unclear is Rhodafra. It comprises two species, Rhodafra marshalli Rothschild & Jordan 1903, occurring from Kenya to Zambia and Zimbabwe, and R. opheltes (Cramer, 1780) from South Africa, which differ markedly in wing pattern (see Kitching 2019). Rhodafra opheltes superficially resembles moths of the genus Hyles (in particular Hyles euphorbiarum (Guérin-Méneville & Percheron, 1835) from South America), in having a broad oblique cream stripe across the forewing that is bordered distally by an olive-green triangular patch. This similarity has resulted in a close relationship being postulated between R. opheltes and Hyles. However, R. opheltes differs in having only a single pair of subdorsal black abdominal spots (as in some Theretra species) whereas all Hyles have two or more. Rhodafra marshalli also has only a single pair of black abdominal patches but the forewing pattern is very different, lacking the cream band and olive-green patch and having only a single curved postmedial line running from the posterior edge of the wing to the apex. On wing pattern alone, it would appear possible that the two species of Rhodafra are not that closely related; indeed, it is possible that the genus is not monophyletic. However, Rothschild and Jordan (1903) characterized the genus Rhodafra not on wing pattern but on a unique pattern of scaling on the outside on the second segment of the labial palp, in which long hair-like scales form a longitudinal crest that appears to be a continuation of the eyelashes. Some species of Hyles, e.g. H. gallii (von Rottemburg, 1775), have similar long hairs protruding through the general scaling of the labial palpus but these are not organized into a distinct crest, which appears to be unique not only in Choerocampina but also in all Sphingidae.
The maximum likelihood analysis by Kawahara et al. (2009) of DNA sequence data from five nuclear genes (131 ingroup Sphingidae taxa) included only two species of Hyles, Hyles lineata (Fabricius, 1775) and H. hippophaes (Esper, 1789), and one species of Rhodafra, R. marshalli. The results placed R. marshalli inside Hyles, as sister to H. hippophaes, although support for this pairing was weak (bootstrap support, BS = 66%). Support for the group comprising Hyles + Rhodafra was, however, high (BS = 100%). Adding sequences from the mitochondrial gene COI (but no further Hyles or Rhodafra specimens), Kawahara and Barber (2015) analysed a six-gene concatenated dataset (over 200 taxa) using both maximum likelihood and Bayesian inference, and a backbone constraint obtained from a tree based on transcriptomic data of over 900 genes with a well-supported branching pattern among five representative Sphingidae taxa. The results of that study yielded the same pattern of relationships as found by Kawahara et al. (2009): R. marshalli grouped with H. hippophaes (unfortunately mislabelled as “Hippotion hippophaes” in their Fig. S2) with Bayesian posterior probability (pp) of 0.93; and these plus H. lineata formed a fully supported group (pp 1). However, the maximum likelihood analysis (their Fig. S1) reversed the positions of the two Hyles species, such that H. lineata now was found sister to R. marshalli with high support (BS = 100%). Either way, albeit with very limited taxon sampling, DNA sequence data placed Rhodafra within the Hyles clade and thus called into question its validity as a separate genus.
Thus, given the currently ambiguous relationships of the genus, the present paper aims to investigate the monophyly and phylogenetic position of Rhodafra in relation to Hyles and within the Choerocampina using mitochondrial and nuclear sequence data of more species and individuals.
Material and methods
No fresh tissue of either Rhodafra species was available from multiple geographic localities to capture intra-specific variability. So, assuming degradation of historic DNA (i.e. a tissue that was not specifically preserved for later DNA analysis and is older than ~ 5 years) had occurred, DNA from 29 museum exemplars of Rhodafra (Table 1) was isolated using ancient-DNA (aDNA) techniques (protocols described by Hundsdoerfer et al. 2017; Mende and Hundsdoerfer 2013). Sequences of three mitochondrial (mt) genes, COI, t-RNA-Leu and COII, were obtained from 15 individuals (GenBank accession numbers LR700623-LR700637). The targeted 2284 base pairs (bp) were amplified in 13 fragments of ~ 110 to ~ 280 bp each. The targeted 774 bp of the nuclear EF1alpha (EF1a) gene fragment were obtained from 12 individuals (GenBank accession numbers LR721665-LR721676) in three fragments of ~ 280 bp using the primers Oscar-6143 and Bosie-6144 from Hundsdoerfer et al. (2009), together with newly developed primers (RhoEF356F: GTACTGGTGAGTTCGAAGCT, 46 °C; RhoEF739F: CGTCTTCCCCTGCAGGACG, 58 °C; Hyles_F6b: GCCAACATCACCACTGAAGTCAAG, 57 °C; RhoEF475R: CGGTGGAGTCCATTTTGTTG, 53 °C; RhoEF535R: TGATGTACGAGGACACTTCC, 47 °C; RhoEF979R: CAGCGACGTAGCCACGACG, 58°).
Datasets were compiled for three different approaches and all three were executed in two mt variants (COI + t-RNA-Leu + COII, and COI barcode only; overview in Table S2): (1) comparative gene trees not necessarily with the same taxonomic coverage, followed by two approaches with identical choices of OTUs for both EF1a and mt gene fragments; (2) concatenated alignments for total evidence (TE) analyses; and (3) multispecies coalescent (MSC) analyses with input from single gene trees.
Comparative gene trees
For the EF1a analyses, we selected sequences from the dataset of Hundsdoerfer et al. (2009; 918 bp published in GenBank, GB) of 21 samples of Hyles species and one to two individuals per outgroup genus. Sequences of R. marshalli from GenBank (EU479329.1; Kawahara et al. 2009) and one to two of all additionally available Choerocampina species not included in the data set of Hundsdoerfer et al. (2009) were included for comparison, resulting in a dataset of 61 OTUs (GenBank accession numbers are included in the sample names; see also Table S1). The choice was based on the additional availability of DNA barcode data for the OTUs (for TE analyses, see below). Two mitochondrial (mt) datasets were analysed for this approach. In the first (82 OTUs; mt1), long mt dataset (2279 bp COI, t-RNA-Leu, COII), two individuals of each Hyles (sub)species (four for H. lineata) were retained in addition to outgroups (data from Hundsdoerfer et al. 2009). A second mt dataset (118 OTUs; mt2), reduced to only the 658-bp DNA barcode sequence (“Folmer region” of COI) included only one sample per Hyles species (four for H. lineata) plus numerous publicly available barcode sequences of the tribe Choerocampina to allow for a broader taxon sampling (most were generated by the Barcode of Life (BOLD) team, see Ratnasingham and Hebert 2007; GenBank and BOLD accession numbers are again included in the sample names; see also Table S1).
Total evidence analyses
A third mt dataset (mt3) was compiled for TE analyses based on the second (mt2) barcode only data, but only includes those OTUs for which EF1a sequences were also available (61 OTUs), and the two alignments were concatenated using BioEdit (Hall 1999). In most OTUs, the barcode and EF1a data were from the same individual, but in some cases, this was not possible and so sequence data for the two genes were combined from different individuals. A fourth mt dataset (mt), also for TE analyses, was based on the third (mt3), but includes only those sequences for which mt data from all three-gene fragments (COI, t-RNA-Leu, COII) was available (46 OTUs).
Multispecies coalescent analyses
Mt3, mt4 and the corresponding EF1a datasets (with the same OTUs as mt3 and mt4) were each analysed separately to obtain the input gene trees for MSC analyses.
To compare the information content of the three datasets (the complete EF1a, mt1 and mt2), variability parameters (% variable versus constant bp; % parsimony-informative versus parsimony-uninformative bp) were determined using MEGA5 (Tamura et al. 2011). Maximum likelihood (ML) analyses were performed using RAxML (raxmlGUI1.5b2; Stamatakis 2014; GTRGAMMA, best of 10 replicate trees, 1000 thorough bootstrap replicates; no outgroup defined a priori, no per partition branch lengths). Partitioning followed the results of PartitionFinder analyses (Lanfear et al. 2012; settings: linked branch lengths, RAxML models, AICc model selection, greedy search, partition schemes in Table S2 and an AdditionalSupplements zip file, where the tree files can also be found). Trees are presented with mid-point rooting (using Figtree v. 1.4.2; Rambaut 2007), because this approach shows the best resolution of the ingroup in the presentation of trees. Multispecies coalescent analyses were performed using ASTRAL (v. 5.7.3; Zhang et al. 2018; default settings).
Results
Variability
To compare the information content of the different data sets, we report variability parameters. Of the 2279 bp in the mt three-gene (COI, t-RNA-Leu, COII) dataset mt1, 1612 were constant (c; 70.1%), 166 were variable but parsimony-uninformative (vpu; 7.2%) and 501 were variable and parsimony-informative (vpi, 22.0%). Within just the ingroup of Rhodafra and Hyles (81 sequences), 1824 bp were c (80.0%), 67 vpu (2.9%) and 388 vpi (17.0%). Reducing the mt dataset to just the 658 bp of the DNA barcode region (mt2; ~ 30% of the three-gene dataset) allowed an increased number of outgroups to be included, and 424 bp were c (64.4%), 31 vpu (4.7%) and 193 vpi (29.3%) (nearly half as many as in the three-gene dataset). Within the ingroup (86 sequences), 531 bp were c (80.7%), 42 vpu (6.4%) and only 85 vpi (12.9%). The 918-bp nuclear EF1a dataset was much less variable, with 793 c (86.4%), 45 vpu (4.9%) and 80 vpi bp (8.7%). Within the ingroup (33 sequences), 846 bp were c (92.2%), 34 vpu (3.7%) and 38 vpi (4.1%).
Comparative gene trees
The monophyly of the genus Rhodafra is corroborated by both mt and nuclear markers (Figs. 1 and 2) and also the monophyly of the two species, R. opheltes and R. marshalli. Analysing the entire available mt sequence data (2279 bp) increases BS for the genus from 80% in the barcode tree (Fig. 1) to 100% in the three-gene tree (Fig. S1). Support for a monophyletic Rhodafra based on analysis of the 918-bp nuclear EF1a data is also very high (BS = 96%; Fig. 2).

RAxML phylogram of Choerocampina with emphasis on Hyles, Rhodafra and Hippotion, based on 658 bp of sequence data from the barcode (first half of the mitochondrial gene COI, dataset mt2). Numbers on branches are bootstrap support values (only values > 25 shown)

RAxML phylogram of Hyles, Rhodafra and outgroups based on 918 bp of the nuclear EF1a gene. Numbers on branches are bootstrap support values (only values > 25 shown)
The two mt trees (from datasets mt1, mt2) agree in placing Rhodafra within Hyles (Figs. 1 and S1). However, in the tree derived from the analysis of DNA barcode sequences alone (mt2; Fig. 1), support for most groups is very poor. The group comprising Hyles + Rhodafra has a BS support of only 28% and support for placing Rhodafra as sister to all Hyles except H. lineata is only slightly higher at 38%. Furthermore, the nodes of the outgroup backbone all have very low support (< 25%), and thus, the relationships among the genera Basiothia, Cechenena, Centroctena, Chaerocina, Deilephila, Euchloron, Griseosphinx, Hippotion (part), Hyles, Pergesa, Phanoxyla, Rhagastis, Rhodafra, Theretra and Xylophanes are effectively unresolved. In addition, the DNA barcode tree suggests several genera, Cechenena, Hyles, Hippotion, Theretra and Rhagastis, are polyphyletic.
Analysis of the three mt genes (mt1) yields increased levels of support (Fig. S1), particularly within Hyles. The clade comprising Hyles and Rhodafra now receives near-full BS support (99%). Rhodafra is placed as sister to H. lineata (although with very low support, 36%), and these two together are sister to all remaining Hyles, which is well supported (BS = 95%). However, support for the former pairing is low (36%) and so the relationships among the three groups are best interpreted as an unresolved trichotomy. Hippotion (including Basiothia) (BS = 100%) and Deilephila (BS = 99%) are well supported, and the pairing of two of the three included Xylophanes species, X. falco (Walker, 1856) + X. porcus (Hübner, [1823]), receives moderate support (BS = 75%). All other groupings are unsupported.
In contrast, analysis of the nuclear EF1a sequences produced a topology in which Rhodafra is placed as the sister group of Hyles (Fig. 2; low BS support of 52%). These two genera are sister to Xylophanes (very low support of 25%) and these three genera are sister to Deilephila (also very low support of 26%). Most relationships among the outgroups are also poorly supported. Only two groupings comprising more than one genus have BS support above 80% (Fig. 2), one supporting Pergesa acteus (Cramer, 1779) plus Theretra silhetensis (Walker, 1856) (80%), rendering Theretra polyphyletic, and the other supporting Hippotion plus Basiothia (90%), in which Hippotion is paraphyletic.
Total evidence analyses
Combining mt barcode (mt3) and nuclear EF1a sequences in a TE analysis (Fig. S2) results in a tree that is similar to the EF1a gene tree (Fig. 2) but with much higher support for the clade Rhodafra plus Hyles (90%) than that of EF1a alone (52%). However, the support for the monophyly of Hyles in this tree (61%) is even lower than that of EF1a alone (77%). Combining the three mt gene fragments (mt4, COI, t-RNA-Leu, COII) with EF1a in a TE analysis (Fig. S3) reproduces this branching pattern for the relationship of Rhodafra and Hyles, but with full support for the clade Rhodafra plus Hyles, and lower support for Hyles (48% instead of 61%).
Multispecies coalescent analyses
The multispecies coalescent analysis using ASTRAL (EF1a and mt3, barcode only; Fig. S4) resulted in a topology with an ingroup trichotomy formed of Rhodafra, H. lineata, and a clade comprising all remaining Hyles. If more mt sequence data are included (mt4, COI, t-RNa-Leu, COII; Fig. S5), Rhodafra and all Hyles except H. lineata are grouped into a clade with a very short branch.
Discussion
By including the first DNA sequence data from R. opheltes, our study has provided the first molecular phylogenetic confirmation of the monophyly of the genus, as well as demonstrating the reciprocal monophyly of the two species of Rhodafra, despite their divergent wing patterns. Different authors favour either increasing taxon sampling or the number of characters to try to stabilize phylogenetic positioning during tree reconstruction (e.g. Mitchell et al. 2000; Heath et al. 2008; Hedtke et al. 2006; Rydin and Källersjö 2002; Sihvonen et al. 2011; Zwick et al. 2011). Unexpectedly, even though we sequenced only one nuclear gene, we also (slightly) increased the number of informative characters in the ingroup of Hyles and Rhodafra: our EF1a dataset contained 38 vpi ingroup characters, whereas the five gene datasets of Kawahara and Barber (2015, nuclear data only) contained only 22 vpi for Hyles, Rhodafra and Hippotion celerio (Linnaeus, 1758) (possibly due to the high amount of missing nuclear data: 46.5% in the sample R_marshalli_1; H. celerio had to be included to be able to have at least four taxa for the parsimony criterion, but all 22 of these sites were also variable within Hyles and Rhodafra). We thus consider the position of Rhodafra in our reconstruction based on nuclear genes to be slightly more reliable.
Our study corroborated the strong morphological synapomorphy of the labial palps first identified by Rothschild and Jordan (1903; see introduction) and removes any remaining doubts about the non-monophyly of the genus that may derive from the completely differing wing patterns of the two species of Rhodafra. Our study thus strengthens the conclusion of Hundsdoerfer et al. (2005) that a similar wing pattern is not a good indicator of close phylogenetic relationship in this group of moths (e.g. H. lineata, H. livornicoides (Lucas, 1892) and H. livornica (Esper, 1780); H. euphorbiae (Linnaeus, 1758) and H. nicaea (von Prunner, 1798); Fig. 1). Pioneering studies of the genes underlying Lepidoptera wing patterns in Heliconius (Van Belleghem et al. 2017) have revealed a modular architecture with narrow genomic intervals associated with specific differences in colour and pattern. One gene, cortex, has been suggested to regulate pattern switches across Lepidoptera (Nadeau et al. 2016). Thus, wing pattern and coloration can be very plastic, enabling very different patterning to occur in closely related species and similar patterning to occur in relatively distantly related taxa.
The placement of Rhodafra with respect to Hyles is more complex. Mitochondrial genes nest a monophyletic Rhodafra within Hyles (Figs. 1 and S1), whereas nuclear data place Rhodafra as sister to Hyles (Fig. 2). Relationships among all genera in our analyses are only weakly resolved and supported, but resolving these intergeneric relationships was not the aim of our study. Kawahara et al. (2009) and Kawahara and Barber (2015) included four additional nuclear genes (as well as EF1a) and resolved many intra-choerocampine relationships. The single Rhodafra sample they included (R. marshalli) was recovered as sister to H. hippophaes (Fig. 3 of Kawahara et al. 2009), whereas the second and only other Hyles sample they included, H. lineata, was placed as sister to the two, a relationship corresponding to our barcode tree result (Fig. 1). The same relationship was found by Kawahara and Barber (2015; which included nuclear and barcode sequence data) in their Bayesian reconstruction (their Fig. S2), whereas their RAxML tree showed a fully supported sister group of R. marshalli + H. lineata without H. hippophaes (their Fig. S1), corresponding to the result of our three-gene mitochondrial dataset (Fig. S1). Not having had the aim of clarifying the phylogenetic position of Rhodafra, these studies included only few Rhodafra and Hyles samples, but nevertheless consistently recovered Rhodafra within Hyles.
Despite this mt and nuclear incongruence, we performed a TE analysis (Fig. S2), which revealed strong support (90%) for a monophyletic Hyles plus Rhodafra, but only weak support (61%) for a monophyletic Hyles. Presumably, the support for Rhodafra plus Hyles, which is a feature of congruence between mt and nuclear data, is higher than in the single gene analyses (barcode, mt2 dataset only 28% and EF1a dataset 52%) because of the greater amount of total sequence data. With a trichotomy between Rhodafra, H. lineata and the remaining Hyles, the multispecies coalescent result (Fig. S3) reflects the mt and nuclear incongruence and thus most precisely the overall result.
Taken together, the mt-nuclear incongruence confirms that, as has been found in other studies (e.g. Cong et al. 2017; Dupont et al. 2016; Schniebs et al. 2016), DNA barcodes alone are a very poor data source for resolving phylogenetic relationships above the species level (as a result of such issues as maternal inheritance and rapid evolution) and all studies that purport to do so should be treated with extreme caution. Even the result based on the three times longer (double the number of ingroup vpi sites) mt fragment (COI, t-RNA-Leu, COII) is incongruent with that of the nuclear data, which contains only one-tenth of the barcode variability.
We have two explanations for the incongruence between mitochondrial and nuclear data. The first is that ancestral Rhodafra hybridized with and captured mitochondria from ancestral Hyles species after the split of H. lineata, followed by complete mitochondrial introgression. However, in the absence of a biogeographic reconstruction, which we have not undertaken as part of the present study, we are unable to determine where any such ancient introgression may have taken place.
The second explanation is ancient incomplete lineage sorting. The age estimate for Hyles/Rhodafra from the fossil-biogeographic dating by Kawahara and Barber (2015, their Fig. S3) is 6.5–8.5 Mya. During this time, the continents had already reached their current positions. If ancestral Rhodafra had evolved in Africa and ancestral Hyles in South America (c.f. Hundsdoerfer et al. 2005, 2009, 2017), then secondary contact for mitochondrial capture across continents is be expected to have been very rare. More likely, the mitochondria of the ancestors of Rhodafra and Hyles were incompletely sorted before the genera split, i.e. before the oldest age estimate of Hyles, 8.5 My (Kawahara and Barber 2015). This would have taken place in South America, given that this is both the geographic origin of Hyles and the current distribution of its postulated sister group Xylophanes (Hundsdoerfer et al. 2005, 2009).
Due to the topological incongruences in trees derived from different sets of OTUs, genes and analytical approaches, our study failed to recover a robust topology for the relationship between Rhodafra and Hyles. Although mt data can be misleading, it nevertheless represents part of the organisms’ evolutionary history that should not be completely ignored, even if it indicates unexpected results, such as the lack of monophyly for the genus Hyles in this case. For a more robust result, the question should be re-examined using genomic approaches.
Nevertheless, our study allows several observations, some of which may appear trivial, but are yet noteworthy: (1) Phylogenetic analyses based on mt data should be based on more than just the barcode (658 bp) region. BS increases greatly in all gene tree and TE analyses when mt data is augmented from 658 to 2279 bp (COI, t-RNA-Leu, COII), and analysis of full mitogenomes would be expected to improve support still further. (2) Despite mt-nuclear incongruence, TE analyses should nevertheless be performed as they need not necessarily reflect the result of the single dataset with the most vpi sites. Unexpectedly, in the present study, the TE trees (Figs. S3 and S4) reflected the branching pattern of the nuclear EF1a gene trees, whereas the variability, and thus the contribution of informative sites, was much higher from the mt data. Overall, therefore, we currently conclude that the preferred relationship between the two genera is that in which Rhodafra is the sister genus to a monophyletic Hyles.
References
Cong, Q., Shen, J., Borek, D., Robbins, R. K., Opler, P. A., Otwinowski, Z., & Grishin, N. V. (2017). When COI barcodes deceive: complete genomes reveal introgression in hairstreaks. Proceedings of the Royal Society B: Biological Sciences, 284(1848), 20161735.
Cramer, P. (1779). Uitlandsche Kapellen, voorkomende in de drie Waereld-Deelen Asia, Africa en America, by een verzameld en bescreeven. Vol. 3 (17–22). Amsteldam & Utrecht: S.J. Baalde & Barthelemy Wild.
Cramer, P. (1780). Uitlandsche Kapellen, voorkomende in de drie Waereld-Deelen Asia, Africa en America, by een verzameld en bescreeven. Vol. 3 (23–24). Amsteldam & Utrecht: S.J. Baalde & Barthelemy Wild.
Dupont, L., Porco, D., Symondson, W. O. C., & Roy, V. (2016). Hybridization relics complicate barcode-based identification of species in earthworms. Molecular Ecology Resources, 16(4), 883–894.
Esper, E. J. C. (1780). [(1778–1786)]. Die Schmetterlinge in Abbildungen nach der Natur mit Beschreibungen. Vol. II. Europäische Gattungen. Erlangen: Verlage Wolfgang Walthers.
Esper, E. J. C. (1789). [(1789-[1803–1804])]. Die Schmetterlinge in Abbildungen nach der Natur mit Beschreibungen. Fortsetzung der europäischer Schmetterlinge. Vol. II. Zweiter Abschnitt. Zu dem Geschlecht der Abendschmetterlinge. Erlangen: Verlage Wolfgang Walthers.
Fabricius, J. C. (1775). Systema entomologiae, sistens insectorum classes, ordines, genera, species, adiectis synonymis, locis, descriptionibus, observationibus. Libraria Kortii: Flensburgi et Lipsiae.
Guérin-Méneville, F. E. & Percheron, A. (1835). [(1835-1838)]. Genera des insectes ou exposition détailée de tous les caractères propres a chacun des genres de cette class d’animaux. Paris: J.B. Baillière.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Heath, T. A., Hedtke, S. M., & Hillis, D. M. (2008). Taxon sampling and the accuracy of phylogenetic analyses. Journal of Systematics and Evolution, 46(3), 239–257.
Hedtke, S. M., Townsend, T. M., & Hillis, D. M. (2006). Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Systematic Biology, 55(3), 522–529.
Hübner, J. (1823). [([1819]-[1827)]. Sammlung exotischer Schmetterlinge. Vol. 2. Augsburg: J. Hübner.
Hundsdoerfer, A. K., Kitching, I. J., & Wink, M. (2005). A molecular phylogeny of the hawkmoth genus Hyles (Lepidoptera: Sphingidae, Macroglossinae). Molecular Phylogenetics and Evolution, 35(2), 442–458.
Hundsdoerfer, A. K., Rubinoff, D., Attié, M., Wink, M., & Kitching, I. J. (2009). A revised molecular phylogeny of the globally distributed hawkmoth genus Hyles (Lepidoptera: Sphingidae), based on mitochondrial and nuclear DNA sequences. Molecular Phylogenetics and Evolution, 52(3), 852–865.
Hundsdoerfer, A. K., Päckert, M., Kehlmaier, C., Strutzenberger, P., & Kitching, I. J. (2017). Museum archives revisited: Central Asiatic hawkmoths reveal exceptionally high late Pliocene species diversification (Lepidoptera, Sphingidae). Zoologica Scripta, 46(5), 552–570.
Kawahara, A. Y., & Barber, J. R. (2015). Tempo and mode of antibat ultrasound production and sonar jamming in the diverse hawkmoth radiation. Proceedings of the National Academy of Sciences, 112(20), 6407–6412.
Kawahara, A. Y., Mignault, A. A., Regier, J. C., Kitching, I. J., & Mitter, C. (2009). Phylogeny and biogeography of hawkmoths (Lepidoptera: Sphingidae): evidence from five nuclear genes. PLoS One, 4(5), e5719.
Kitching, I. J. (2019). Sphingidae taxonomic inventory. Creating a Taxonomic e-Science. Resource document. Scratchpads biodiversity online. http://sphingidae.myspecies.info/. Accessed 13 May 2019.
Kitching, I. J., & Cadiou, J. M. (2000). Hawkmoths of the world: an annotated and illustrated revisionary checklist (p. 227). London & Ithaca: The Natural History Museum & Cornell University Press.
Kitching, I. J. & Rougerie, R., Zwick, A., Hamilton, C. A., St Laurent, R. A., Naumann, S., Ballesteros Mejia, L., & Kawahara, A. (2018). A global checklist of the Bombycoidea (Insecta: Lepidoptera). Biodiversity Data Journal, 6, e22236.
Lanfear, R., Calcott, B., Ho, S. Y., & Guindon, S. (2012). PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution, 29(6), 1695–1701.
Linnaeus, C. (1758). Systema naturae per regna tri naturae, secundum classes, ordines, genera, species, cum characteribus, differintiis, synonymis, locis. 1. Animalia. 10th ed., Holmiae: Laurentii Salvii.
Lucas, T. P. (1892). On 34 new species of Australian Lepidoptera, with additional localities, &c. Proceedings of the Royal Society of Queensland, 8, 68-94.
Mende, M. B., & Hundsdoerfer, A. K. (2013). Mitochondrial lineage sorting in action–historical biogeography of the Hyles euphorbiae complex (Sphingidae, Lepidoptera) in Italy. BMC Evolutionary Biology, 13(1), 83.
Mitchell, A., Mitter, C., & Regier, J. C. (2000). More taxa or more characters revisited: combining data from nuclear protein-encoding genes for phylogenetic analyses of Noctuoidea (Insecta: Lepidoptera). Systematic Biology, 49(2), 202–224.
Nadeau, N. J., Pardo-Diaz, C., Whibley, A., Supple, M. A., Saenko, S. V., Wallbank, R. W., et al. (2016). The gene cortex controls mimicry and crypsis in butterflies and moths. Nature, 534(7605), 106–110.
Rambaut, A. (2007) FigTree, a graphical viewer of phylogenetic trees. Resource document. http://tree.bio.ed.ac.uk/software/figtree/ Accessed 01 July 2016.
Ratnasingham, S., & Hebert, P. D. (2007). BOLD: the barcode of life data system (http://www.barcodinglife.org). Molecular Ecology Resources, 7, 355–364.
Roeder, K. D., Treat, A. E., & Vande Berg, J. S. (1970). Distal lobe of the pilifer: an ultrasonic receptor in choerocampine hawkmoths. Science, N.Y., 170, 1098–1099.
Rothschild, LW, Jordan, K (1903). A revision of the lepidopterous family Sphingidae. Novitates Zoologicae (Supplement) 9, i-cxxxv + 1–972.
Rydin, C., & Källersjö, M. (2002). Taxon sampling and seed plant phylogeny. Cladistics, 18(5), 485–513.
Schniebs, K., Gloeer, P., Vinarski, M. V., & Hundsdoerfer, A. K. (2016). A barcode pitfall in Palaearctic Stagnicola specimens (Mollusca: Lymnaeidae): incongruence of mitochondrial genes, a nuclear marker and morphology. North-Western Journal of Zoology, 12(2).
Sihvonen, P., Mutanen, M., Kaila, L., Brehm, G., Hausmann, A., & Staude, H. S. (2011). Comprehensive molecular sampling yields a robust phylogeny for geometrid moths (Lepidoptera: Geometridae). PLoS One, 6(6), e20356.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Van Belleghem, S. M., Rastas, P., Papanicolaou, A., Martin, S. H., Arias, C. F., Supple, M. A., et al. (2017). Complex modular architecture around a simple toolkit of wing pattern genes. Nature Ecology & Evolution, 1(3), 0052.
von Prunner, L. J. (1798). Lepidoptera Pedemontana illustrata. Augusta Taurinorum: Mathaeus Guaita.
von Rottemburg, S. A. (1775). Anmerkungen zu den Hufnagelischen Tabellen der Schmetterlinge. Erste Abtheilung. Naturforscher, Halle, 6, 1–34.
Walker, F. (1856). Sphingidae. List of specimens of Lepidopterous insects in the collection of the British Museum, 8, 1–271.
Zhang, C., Rabiee, M., Sayyari, E., & Mirarab, S. (2018). ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics, 19(S6), 153.
Zwick, A., Regier, J. C., Mitter, C., & Cummings, M. P. (2011). Increased gene sampling yields robust support for higher-level clades within Bombycoidea (Lepidoptera). Systematic Entomology, 36(1), 31–43.
Acknowledgements
Collecting of specimens and DNA sequencing of our previously published sequences was funded by the Deutsche Forschungsgemeinschaft [grant numbers DFG HU1561/1-1, 1-2] and Michael Wink, Institute of Pharmacy and Molecular Biotechnology, University of Heidelberg. The European Union’s Improving Human Potential program SYNTHESYS [grant numbers GB-TAF-3410, GB-TAF-5177] enabled A.K. Hundsdörfer to undertake a collaborative study of the morphology of Hyles at the Natural History Museum, London, UK. We would like to thank Geoff Martin and Alessandro Giusti, Insects Division, Department of Life Sciences, Natural History Museum, London, UK, for facilitating access to the sphingid collections, arranging loans of material and permitting sequencing of specimens. Similarly, we thank Ulf Eitschberger (Entomological Museum Eitschberger Marktleuthen, Germany) and Tomáš Melichar (Sphingidae Museum, Czech Republic) for access to their collections, Martin Husemann for access to the collection of the Zoological Museum Hamburg, Germany, and Simon van Noort, Aisha Mayekiso (Iziko Museum, Cape Town, South Africa) and the late Martin Kruger (Ditsong Museum, Pretoria, South Africa) for sending Rhodafra legs (that unfortunately did not yield sequence data). All new sequences were generated by Anke Mueller, Molecular Laboratory Senckenberg Dresden (SGN-SNSD-Mol-Lab). We thank Alberto Zilli and two anonymous reviewers for their helpful suggestions on an earlier version of this paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Table S1
GenBank accession numbers of the sequences analysed (XLSX 14.4 kb)
Table S2
Overview of alignments, number of OTU’s, number of bp, analyses and RAxML partitions (determined by PartitionFinder) (XLSX 10.4 kb)
Fig. S1
RAxML phylogram of Hyles, Rhodafra and outgroups based on the three mt gene dataset (mt1, 2279 bp COI, t-RNA-Leu, COII). Numbers on branches are bootstrap support values (only values >25 shown) (PDF 365 kb)
Fig. S2
Total evidence RAxML phylogram of Hyles, Rhodafra and outgroups tree based on the mt barcode (mt3, 658 bp COI) and EF1a (918 bp). GenBank accession numbers of publicly available sequences are given in the names of samples (and also in Table S1). Numbers on branches are bootstrap support values (only values >25 shown) (PDF 278 kb)
Fig. S3
Total evidence RAxML phylogram of Hyles, Rhodafra and outgroups tree based on the three mt genes (mt4, 2279 bp COI, t-RNA-Leu, COII) and EF1a (918 bp). GenBank accession numbers of publicly available sequences are given in the names of samples (and also in Table S1). Numbers on branches are bootstrap support values (only values >25 shown) (PDF 272 kb)
Fig. S4
Multispecies coalescent tree from the ASTRAL analysis based on the two single gene ML trees (mt3, COI and EF1a). GenBank accession numbers of publicly available sequences are given in the names of samples (and also in Table S1). Numbers on branches are bootstrap support values (only values >25 shown) (PDF 251 kb)
Fig. S5
Multispecies coalescent tree from the ASTRAL analysis based on two ML trees from mt and nuclear data (mt4, 2279 bp COI, t-RNA-Leu, COII and EF1a). GenBank accession numbers of publicly available sequences are given in the names of samples (and also in Table S1). Numbers on branches are bootstrap support values (only values >25 shown) (PDF 222 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hundsdoerfer, A.K., Kitching, I.J. Ancient incomplete lineage sorting of Hyles and Rhodafra (Lepidoptera: Sphingidae). Org Divers Evol 20, 527–536 (2020). https://doi.org/10.1007/s13127-020-00445-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-020-00445-0