
Abstract
Identifying the genes causing male infertility is important to increase our biological understanding as well as the diagnostic yield and clinical relevance of genetic testing in this disorder. While significant progress has been made in some areas, mainly in our knowledge of the genes underlying rare qualitative sperm defects, the same cannot be said for the genetics of quantitative sperm defects. Technological advances and approaches in genomics are critical for the process of disease gene identification. In this review we highlight the impact of various technological developments on male infertility gene discovery as well as functional validation, going from the past to the present and the future. In particular, we draw attention to the use of unbiased genomics approaches, the development of increasingly relevant functional assays and the importance of large-scale international collaboration to advance disease gene identification in male infertility.
Similar content being viewed by others
Introduction
Infertility is a complex pathological condition that affects close to 7% of the global male human population and presents as a wide range of heterogeneous phenotypes, from congenital or acquired urogenital abnormalities, endocrine disturbances and immunological factors to spermatogenic quantitative and qualitative defects (Krausz 2011; Tournaye et al. 2017). While severe forms of infertility cannot be directly inherited—by default an affected man is incapable of naturally passing on his genetic information (unless facilitated by assisted reproductive technologies)—genetics plays a major role in this disorder. Unaffected parents can for example pass on genetic mutations that result in autosomal recessive or X-linked forms of male infertility (Chillón et al. 1995; Yatsenko et al. 2015). In addition, many mutational events take place during normal gametogenesis that can result in germline de novo mutations (DNMs), de novo copy number variations (CNVs) or de novo chromosomal abnormalities, some of which can result in male infertility in the offspring (Jacobs and Strong 1959; Reijo et al. 1995; Sun et al. 1999).
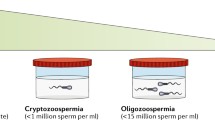
In approximately 15% of infertile men a genetic defect is most likely the underlying cause of the pathology (Tournaye et al. 2017; Krausz and Riera-Escamilla 2018). However, despite widespread usage of karyotyping, azoospermia factor (AZF) deletion screening and cystic fibrosis transmembrane conductance regulator (CFTR) mutation analysis, a recent study in a large unselected patient cohort revealed a causal genetic diagnosis in only 4% of infertile men (Tüttelmann et al. 2018). Similar to what has been found for other disorders, genetics is found to play a more prominent role in the most severe forms of spermatogenic impairment such as severe oligozoospermia (< 5 million sperm cells per ml) or azoospermia (no sperm in ejaculate) (Lopes et al. 2013; Krausz and Riera-Escamilla 2018).
The identification of genetic causes of male infertility, which began in the middle of the twentieth century, continues to this day aided by the development of novel molecular techniques and technological advancements that have allowed for the discovery and characterisation of key genes responsible for the various subtypes of human male infertility (Fig. 1). Although evidence for a genetic cause of male infertility was first reported in the 1950s, when an extra X chromosome was found in patients diagnosed with Klinefelter Syndrome, it was not until the late 1990s that research efforts were intensified to search for additional genetic factors, initially focusing on specific deletions and mutations in the androgen receptor and the CFTR gene (Dumur et al. 1990; Akin et al. 1991; McPhaul et al. 1992; Patrizio et al. 1993) and Y chromosomal abnormalities (Vogt et al. 1992; Reijo et al. 1995). In recent years, researchers have increasingly applied array-based genome-wide approaches and, more recently, next-generation sequencing (NGS) technologies to perform more unbiased genomic studies in male infertility and identify numerous male infertility genes (Aston and Carrell 2009; Talkowski et al. 2011; Tüttelmann et al. 2011; Ayhan et al. 2014).

Timeline of the discovery of key genes involved in male infertility. The development of novel molecular techniques and technological advancements introduced since 1956 have allowed for the identification of key genes responsible for the various types of male infertility in humans. Although the first male infertility genes were first identified in the late 1980s, the widespread application of microarray and NGS approaches has resulted in an increase in the detection of male infertility genes
In this review, we contextualise how different technological advances have contributed to our understanding of the genetics of male infertility, detailing current shortcomings and challenges. We conclude by providing some perspective on future directions and a call-to-arms among researchers, clinicians and patients for stronger collaboration and sharing of information for the benefit of all.
The past: chromosome studies and specific gene analysis revealed the first male infertility genes
Chromosomal abnormalities causing male infertility
Karyotyping was the first test employed to investigate the presence of genetic abnormalities in infertile men and to this day remains the most widely used diagnostic test in male infertility. This cytogenetic technique revealed several very important chromosomal abnormalities associated to male infertility, with the most common being the presence of an additional X chromosome (47, XXY) which characterises Klinefelter syndrome (Jacobs and Strong 1959), present in 15% of non-obstructive azoospermic patients (Ferlin et al. 2006a; Jungwirth et al. 2012; Punab et al. 2016; Vockel et al. 2019). The combination of karyotype analysis with fluorescence in situ hybridisation (FISH) revealed additional chromosomal abnormalities to underlie primary infertility and sex development disorders, specifically 46, XX male; Robertsonian and reciprocal translocations (Therkelsen 1964; Chapelle et al. 1964; Hamerton 1968; Koulischer and Schoysman 1974; Jacobs et al. 1975).
Karyotype analysis was also essential to identify the location of genetic factors controlling normal spermatogenesis. It was through the usage of this technique that in 1976 a deletion at the distal portion of band q11 of the Y chromosome was found in 6 men with azoospermia and the region was identified as essential for spermatogenesis (Tiepolo and Zuffardi 1976). Although this pointed to the presence of a gene or genes controlling human spermatogenesis in this deleted portion of the Y chromosome, it was not until the 1990s that the first gene directly involved in spermatogenic failure was identified by novel molecular technologies.
The search for the azoospermia factor (AZF) within the deleted regions of the Y chromosome involved the use of polymerase chain reaction (PCR) analysis and Y-specific sequence-tagged sites (STSs) to identify potential candidate genes (Ma et al. 1992, 1993; Kobayashi et al. 1994). Appropriately, the strongest candidate gene found to be absent in the azoospermic men with Y microdeletions was named Deleted in Azoospermia or DAZ (Reijo et al. 1995, 1996; Vogt 1998; Ferlin et al. 1999), of which four paralogs (DAZ1-4) have now been identified on the Y chromosome (Saxena et al. 2000). Further application of these PCR-based techniques revealed three genomic regions on the Y chromosome frequently deleted in men with spermatogenic failure, named AZF regions a, b, and c (Vogt 1998). Each of these regions contains candidate genes highly or exclusively expressed in the testis and essential in spermatogenesis, including BPY2, CDY, DAZ, HSFY, RBMY, PRY, TSPY; VCY and XKRY (Reijo et al. 1995; Elliott et al. 1997; Lahn and Page 1997; Yen 1998; Sun et al. 1999; Skaletsky et al. 2003; Krausz and Casamonti 2017). Microdeletions affecting AZF regions result in a variable phenotype, ranging from oligozoospermia to azoospermia in 2–10% in infertile men (Krausz and Riera-Escamilla 2018). However, the genetic causes for the infertility of most men remained unknown, requiring scientists to look elsewhere in the genome.
Identification of specific gene mutations resulting in male infertility
The first gene linked to male infertility outside of the Y chromosome was identified in 1988 on chromosome X. At the time, mutations in the murine androgen receptor gene, mapped via linkage analysis to the X chromosome, had already been well established to cause testicular feminisation in mice (Lyon and Hawkes 1970). Based on this information, Brown et al. used a PCR-based approach coupled with southern blotting to reveal that deletion of the human Androgen Receptor (AR, also known as NR3C4) was responsible for infertility in patients with mild or partial androgen insensitivity syndrome, as well as sex reversal in patients with complete androgen insensitivity syndrome (Brown et al. 1988). Initially, PCR-amplified exons of the AR gene were screened for mutations using techniques such as denaturing gradient gel electrophoresis (DGGE) and single-strand conformational polymorphism analysis (SSCP) and by denaturing high performance liquid chromatography (DHPLC) screening (Quigley et al. 1995; Ferlin et al. 2006b). Since the gene was first linked to male infertility, mutations in AR have been identified in 2% of infertile men causing either mild or partial androgen insensitivity syndrome (McPhaul et al. 1992; Ferlin et al. 2006b; Gottlieb et al. 2012; Vockel et al. 2019).
In 1989, mutations in the CFTR gene (Cystic Fibrosis Transmembrane Conductance Regulator) on chromosome 7 were discovered to underlie Cystic Fibrosis, using restriction fragment length polymorphism (RFLP) analysis to pinpoint the genomic locus of interest followed by PCR-based sequencing to identify mutations (Kerem et al. 1989; Riordan et al. 1989). Since the original reporting, further studies have identified specific mutations in this gene responsible for isolated infertility. In particular, mutations in CFTR have been found to cause obstructive azoospermia as a result of Congenital Bilateral Absence of the Vas Deferens using DGGE and SSCP techniques (Dumur et al. 1990; Anguiano 1992; Culard et al. 1994). The high prevalence of CFTR mutations in the global population, particularly in individuals of European descent where 1 in 25 are carriers of pathogenic variants, explains why CFTR gene mutations cause 60–70% of congenital absence of the vas deferens (CAVD) cases (Weiske et al. 2000). Overall, however, CAVDs is a rare condition found in only 1–2% of all infertile men (Bieth et al. 2020).
Optimization and upscaling of Sanger sequencing on automated DNA sequencers resulted in wide-scale adoption of DNA sequencing in human disease research and diagnostics in the early 2000s (Metzker 2005; Hutchison 2007). It was particularly successful in testing for mutations in candidate disease genes identified by either positional cloning or by evidence from orthologous genes studied in model organisms. A good example of this was shown by the identification of mutations in SYCP3 gene related to meiotic arrest during spermatogenesis (Miyamoto et al. 2003). The role of this gene in male infertility had been previously established in mice where male homozygous null mutants for the Sycp3 gene were found to be infertile due to massive apoptotic cell loss during spermatogenesis (Yuan et al. 2000). After isolation of the human SYCP3 gene it was found to have an identical function and similar negative consequences to human fertility if disrupted (Martinez-Garay et al. 2002; Miyamoto et al. 2003). Of note, while in mice bi-allelic mutations are required to reveal an infertility phenotype, in humans a single heterozygous mutation in SYCP3 is sufficient to compromise spermatogenesis.
The present: technological advances allowing unbiased analysis of the infertile genome
Genome-wide homozygosity screening using microsatellite scans and SNP microarrays
Adaptations made to existing sequencing technology as well as the introduction of single nucleotide polymorphism (SNP) microarrays in the 1990s permitted once more a shift in the research approaches available to investigate the genomes of infertile men and identify novel genes associated with male infertility. Initially, this work was focused on developing and applying large sets of polymorphic markers to screen genomes of infertile men from consanguineous descent for regions of homozygosity. In 2007, this new positional cloning approach resulted in the identification of two novel male infertility genes, AURKC and SPATA16, causing multiple morphological sperm abnormalities (Dieterich et al. 2007; Dam et al. 2007). Similarly, homozygosity mapping was used to identify an homozygous variant in DNAH1 in a small cohort of infertile men with morphological abnormalities in their sperm flagella (Ben Khelifa et al. 2014).
Microarray-based detection of genomic copy number variation
While structural variants such as CNVs present on the Y chromosome have been clearly shown to have a negative effect on spermatogenesis (Nathanson et al. 2005; Visser et al. 2009; Krausz and Casamonti 2017), only few other structural genomic variations have been robustly linked to male infertility. Microarray-based comparative genomic hybridization (array CGH) and SNP arrays have dramatically increased the detection resolution of genomic deletions and duplications since the late 90 s (Solinas-Toldo et al. 1997; Pinkel et al. 1998).
The application of SNP microarray technology revealed one important CNV associated with male infertility in 2011, a 200 kb homozygous deletion affecting the DPY19L2 gene (Harbuz et al. 2011; Koscinski et al. 2011). Specific amplification and sequencing of DPY19L2 was initially hampered by the presence of a pseudogene. In 2012, Coutton et al. optimized the conditions to specifically amplify and sequence this gene and identified globozoospermia patients with a combination of a deletion and a nonsense or missense mutation, as well as a patient with homozygous missense mutations (Coutton et al. 2012). Together, deletions and point mutations in DPY19L2 are now known to explain most cases of globozoospermia.
In 2011, Tüttelmann et al. employed microarray technology to discover an excess of rare CNVs on the sex chromosomes of azoospermic and severe oligozoospermic German men (Tüttelmann et al. 2011), a signature that was later replicated in other populations (Stouffs et al. 2012; Krausz et al. 2012; Lopes et al. 2013; Lo Giacco et al. 2014). More recently, high-resolution array-CGH was used by Yatsenko et al. to identify an identical deletion of three exons of the TEX11 gene on chromosome X in two patients with azoospermia. Sanger sequencing of this gene revealed additional pathogenic (truncating and splice) mutations in azoospermia patients (Yatsenko et al. 2015). Mutations in TEX11 were also reported in azoospermia patients by another group in the same year (Yang et al. 2015). Interestingly, this group decided to study TEX11 because they previously showed that male mice lacking this gene show meiotic arrest, resulting in azoospermia (Yang et al. 2008).
Using a similar microarray approach, recurrent deletions affecting the DMRT1 gene located on chromosomes 9 have been associated with azoospermia (Lopes et al. 2013; Lima et al. 2015). While putative pathogenic mutations in azoospermic men have also been described in this gene (Tewes et al. 2014), it’s role in infertility is still not completely clear. The clinical relevance of other rare CNVs, reported in a limited number or even a single infertile man, remains uncertain. As an example, an approximately 1 Mb deletion found in a single azoospermic man was reported in 2014. The CNV on chromosome 11 spans nine genes, of which the WT1 gene was hypothesized as the potential cause for spermatogenic failure (Seabra et al. 2014). Another example involves an 11–15 kb deletion on chromosome X observed so far exclusively in azoospermic men from the Mediterranean, causing a partial deletion of the proximal copy of the MAGEA9B gene (Lo Giacco et al. 2014; Shen et al. 2017).
While SNP and CGH microarrays have highlighted the important role of CNVs in male infertility, the widespread application of next-generation sequencing (NGS) now offers the opportunity to combine the detection of CNVs and other more complex structural variations with the simultaneous detection of SNVs (Zhou et al. 2018; Ho et al. 2019). Recently, a balanced reciprocal translocation affecting the SYCP2 gene was reported in a patient with severe oligozoospermia, initially discovered by karyotyping but further characterized using both microarrays as well as NGS (Schilit et al. 2020). Interestingly, through international collaboration, loss-of-function mutations affecting this gene were identified in an additional three infertile patients from Germany, highlighting the importance of combining structural genomic variation and single nucleotide variation analysis as well as multi-institutional collaboration.
Next-generation sequencing to detect new disease genes
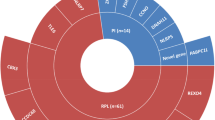
The development of high-throughput NGS platforms in the past decade has resulted in a dramatic drop in sequencing costs and an equally dramatic increase in sequencing throughput. Rather than relying on the need to select and sequence individual candidate genes or evaluate specific polymorphisms or large structural defects in a small number of patients and controls, NGS allows for unbiased sequencing of large numbers of genes, all coding exons (whole exome sequencing) or sequencing of the entire human genome, and increasingly allows this to be done in very large cohorts of patients and controls. Accordingly, NGS has provided an inexpensive and rapid genetic screening approach to discover novel disease-associated genes (Boycott et al. 2013; Fernandez-Marmiesse et al. 2018). Figure 2 shows how in recent years exome sequencing has become the predominant technology used for disease gene studies in male infertility.

Breakdown of genetic tools used in published male infertility studies over the past decade. Based on an analysis of all published literature related to monogenic forms of male infertility, Sanger sequencing was the predominant tool used (82%) in 2010 but has since then seen a reduced usage down to 26% in 2019. In the same time period, NGS-based techniques gained ground to become currently the most common tool (61%)
In Multiple Morphological Abnormalities of the Sperm Flagella (MMAF), exome sequencing of relatively small cohorts of patients has been particularly successful, resulting in the identification of many important new recessive disease genes (Touré et al. 2020). Exome sequencing has also been successfully applied to identify new disease genes for patients with acephalic spermatozoa. In particular, homozygous and compound heterozygous mutations in testis-specific genes BRDT, SUN5 and PMFBP1 (Zhu et al. 2016, 2018; Li et al. 2017; Sha et al. 2019) have been found to disrupt the head-flagella junction of the spermatozoa of these infertile men. In addition, NGS has also been instrumental in the study of congenital isolated hypogonadotropic hypogonadism, characterised by incomplete or absent puberty and infertility. A large number of pathogenic mutations have been identified in genes and genetic loci that result in neurodevelopmental defects of gonadotropic hormone-releasing hormone (GnRH) neuron migration or disrupt neuroendocrine GnRH secretion and action (Cangiano et al. 2020; Butz et al. 2020).
The investigation of the genetic basis for the much more common forms of non-obstructive azoospermia has proven to be more difficult, likely because of the enormous genetic heterogeneity resulting in a wide diversity of phenotypes encompassed in NOA, from the complete lack of germ cells (Sertoli cell only syndrome) to various forms of maturation arrest. Nevertheless, significant progress has been made on this front, where exome sequencing of azoospermic men in both consanguineous and non-consanguineous families has revealed pathogenic mutations in genes such as TEX15 (Okutman et al. 2015), FANCM (Kasak et al. 2018), XRCC2 (Yang et al. 2018), MEIOB (Gershoni et al. 2019) and FANCA (Krausz et al. 2019). In addition, exome sequencing revealed mutations in ADGRG2 in patients with congenital obstructive azoospermia (Patat et al. 2016). In conclusion, the widespread application of exome sequencing in azoospermia and severe oligozoospermia is uncovering mutations in many new candidate disease genes (see also Kasak and Laan 2020). At this stage, however, many of these new candidate genes have not been independently replicated and their role in quantitative sperm defects therefore remains to be established.
Clinical validity assessment of candidate infertility genes
Up to this point we have described the different approaches used to identify genetic variants associated with male infertility. However, in order to convincingly link variants in a certain gene to disease, one needs to consider various levels of evidence. Incorrect and misleading conclusions about the role of genes in causing quantitative or qualitative spermatogenic defects could lead to inappropriate diagnoses and even mismanagement and counselling of couples. Furthermore, incorrectly characterized genes may impede follow-up research by contaminating candidate disease gene lists and pathway analyses. It is therefore important to identify genes with insufficient, inconclusive and low-quality evidence for involvement in the aetiology of male infertility. Recently, a systematic clinical validity assessment of all evidence available for gene-disease relationships in male infertility was performed (Oud et al. 2019). Based on a previously published method (Smith et al. 2017), 92 genes were classified as at least moderately linked to a human male infertility phenotype. This included only 18% of all 521 gene-disease relationships described in male infertility at that time, demonstrating that the quality and extent of evidence varies greatly.
In the years to come, the number of novel genes described for male infertility is expected to increase rapidly as NGS methods become even more widely and frequently used. It is therefore important to continue adding and regularly re-assessing the genetic and functional evidence for all genes described (Soraggi et al. 2020; Houston et al. 2020).
Recent developments in functional validation
The proper design of functional validation experiments, as well as reliance on previously developed functional models with male infertility phenotypes, are critical for the validation of newly identified variants. The emergence of new techniques for gene editing and the implementation of novel model organism and in vitro systems for the study of spermatogenesis has and will continue to expand our ability to perform the critical step of functional validation (Fig. 3).

Mutagenesis techniques for functional validation and examples in male infertility research. Outline of the current mutagenesis techniques for functional validation, and the approximate year of application. Comparison of the main characteristics of the described techniques and some published examples of the application of these technologies in male infertility research
In many diseases, in vitro modelling systems are widely used and highly effective in screening large numbers of variants across multiple genes. Additionally, in many cases in vitro systems can be established from individuals carrying a specific variant, enabling the direct assessment of the functional impact of the variant. However, despite significant efforts, the development of in vitro spermatogenesis systems in humans has proven elusive to date (Komeya et al. 2018). The ability to culture and maintain the function of human testicular tissue would enable the functional validation of novel variants, and perhaps the modification of variants that cause azoospermia, potentially enabling the restoration of spermatogenic capacity in the not-too-distant future (Sato et al. 2011; Ibtisham et al. 2017).
While in vitro systems are not well established for the study of male infertility, the application of gene editing tools in a variety of model organisms has proven effective in the functional validation of suspected infertility-causing variants. For example, in the mouse, N-ethyl-N-nitrosourea (ENU), an alkylating agent that induces genome-wide mutagenesis at random loci (Lewis et al. 1992; Stainier et al. 2017), has been successfully used for the identification of novel genes required for male fertility. Following random mutagenesis by ENU, mice displaying an infertility phenotype are subsequently screened by a variety of approaches, including selective breeding and linkage analysis, and more recently whole genome sequencing, to identify the mutations underlying reproductive defects (Kennedy and O’Bryan 2006; Jamsai and O’Bryan 2010; Geister et al. 2018).
In cases where a candidate variant has been identified and requires functional validation, a variety of targeted approaches can nowadays be applied. These approaches include the use of morpholino oligonucleotides for relatively short-term suppression of gene activity, zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALEN) and Clustered regularly interspaced short palindromic repeats (CRISPR) (Summerton 1999; Gaj et al. 2013; Rumi et al. 2014; Ramanagoudr-Bhojappa et al. 2018; Anuar et al. 2019), which enable genomic modification to inactivate a gene through the introduction of a premature stop codon or frameshift mutation. Alternatively, these approaches can be used to recapitulate specific mutations, for example a missense mutation that is predicted to be pathogenic (Gaj et al. 2013; Anzalone et al. 2019). While all of these tools have been applied successfully in male infertility research, the advantages of CRISPR in terms of cost, efficiency and simplicity have made this gene editing technology the tool of choice for most contemporary studies, and its use in male infertility research will certainly expand rapidly in the coming years.
The enormous value of mouse knock-out experiments for the identification of human male infertility genes has already been highlighted in early studies (Lyon and Hawkes 1970; Yuan et al. 2000). For example, studies of the murine Klhl10 gene, shown to be essential for spermiogenesis in mice, motivated male infertility studies in humans and pointed to one of the few known examples of autosomal dominant male infertility (Yan et al. 2004). After employing reverse-transcription PCR on RNA isolated from sperm of infertile and healthy men, two pathogenic-predicted KLHL10 missense mutations and one splicing mutation were identified by Sanger sequencing in severely oligozoospermic patients. Of note one fertile man was found to harbour one of these missense mutations, questioning the penetrance of this variant (Yatsenko et al. 2006). Another example is hsf2, which was initially found to regulate normal spermatogenesis in mice (He et al. 2003) and it was only later that dominant negative heterozygous mutations in the human orthologue HSF2 were found in infertile men by targeted NGS sequencing of this and ~ 600 other infertility candidate genes (Mou et al. 2013).
Zebrafish have been used increasingly to model human diseases including infertility. Lin et al. showed using a zebrafish model and Crispr/Cas9 that the amh gene is essential to control the balance between proliferation and differentiation of male germ cells (Lin et al. 2017). More recently CRISPR technology was used to systematically characterize the function of 17 Fanconi Anemia (FA) genes, revealing their essential role in growth, sexual development and fertility (Ramanagoudr-Bhojappa et al. 2018). Studies like these are essential for the clinical validity assessment of genes related to male infertility and provide additional evidence for the role of recently identified mutations of these genes in azoospermic men (Kasak et al. 2018; Krausz et al. 2019).
High throughput screens in Drosophila using ethyl-methanesulfonate-induced mutations, similar to the ENU approach in mice (Wakimoto et al. 2004) or more recently RNA interference (RNAi) knockdown (Yu et al. 2015) enabled the efficient identification of candidate infertility genes and downstream functional assessment of multiple candidate genes in a single experiment. For example, discovery of the role of boule (homologous to human DAZ) in meiosis was first discovered in a large-scale mutation screen using drosophila (Castrillon et al. 1993; Eberhart et al. 1996).
Lastly, flagellated and ciliated unicellular organisms including Chlamydomonas, Trypanosoma and Tetrahymena have been used to study the functional consequences of mutations in genes involved in sperm flagellar formation and function (e.g. CFAP43, CFAP44, ODA7, SPAG16L, and WDR66) in patients displaying MMAF (Zhang et al. 2006; Duquesnoy et al. 2009; Coutton et al. 2018; Kherraf et al. 2018). Yeast have likewise been an important model organism for the study of meiosis, enabling the identification of novel meiotic genes associated with male infertility, for example gda1 -orthologous to human ENTPD6 (Wang et al. 2015).
The use of gene editing tools in these and other model organisms will continue to be important in identifying and characterizing genetic variants associated with male infertility.
The future: overcoming existing limitations to improve biological understanding, patient diagnostics and prognostics
The introduction of unbiased NGS approaches have revolutionized the identification of genetic causes for many diseases (Payne et al. 2018; Bean et al. 2020). Although these methods are frequently used in research laboratories to study the genetics of male infertility, they have yet to identify the majority of genes underlying this disorder. For several types of male infertility such as DFS-MMAF, acephalic sperm syndrome and globozoospermia, the current diagnostic yield based on known genetic causes explains approximately 50% of cases. In contrast, the diagnostic yield for the most common forms of male infertility, azoospermia and oligozoospermia, remains much more limited (Krausz 2011; Tüttelmann et al. 2018; Krausz and Riera-Escamilla 2018). Given the enormous potential of new genomic technologies in both research and diagnostics, how can we most efficiently improve our understanding of the genetics of male infertility? To conclude this review, we will discuss the limitations and pitfalls of current approaches and highlight some important new developments in genomic technologies and improvements in the organization of our research that should help to accelerate disease gene identification.
Challenges to gene discovery in a relatively common heterogeneous disorder
Exome and especially genome sequencing now allow us to perform unbiased genetic studies that can help to identify novel disease genes or genomic regions. The power to discover novel infertility genes is, however, still heavily influenced by a number of study design considerations. As with other disease models, careful phenotyping and cohort selection are critical. In addition, an understanding of the genetic architecture of the disease is important in designing appropriately powered studies. As previously discussed, discrete sperm morphological abnormalities are expected to involve a relatively modest number of genes, whereas a quantitative spermatogenic failure phenotype may arise from variants in any one of hundreds or even thousands of genes. In the case of extremely rare disorders with discrete phenotypes, a small case/control design may be appropriate, as it is likely that variants will be localized to a small number of genes. Genetic studies in consanguineous families have proven to be an effective means of identifying novel male infertility genes accounting for rare qualitative sperm defects. In cases of consanguinity, analyses can be focussed on regions of the genome that are homozygous by descent, as it is likely that rare recessive disease-causing mutations will exist in a homozygous state more frequently in these families (Okutman et al. 2015; Kasak et al. 2018).
In the investigation of NOA in outbred populations, it is likely that individual genes will harbour disease-causing variants at extremely low frequency in the patient population, given the effect of the disease on overall fitness. In this case, large patient and control cohorts and robust statistical methods are essential to identify genes with an increased mutational load in the genome of affected patients, as was shown for other genetically heterogeneous disorders (Fitzgerald et al. 2015; Wilfert et al. 2016). This calls for large-scale collaborative studies and widespread data sharing to reliably and robustly identify and functionally validate new male infertility genes.
International consortia have recently been established in the field of male infertility genetics to promote this, including the GEMINI consortium (https://gemini.conradlab.org/) and the IMIGC consortium (https://www.imigc.org/). Collaboration, both within and beyond these consortia, will be essential to identify recurrently mutated genes, study detailed clinical presentations, perform relevant functional studies to confirm the pathogenicity of certain mutations and unravel the underlying biological mechanisms. Moreover, these consortia can help to replicate single case observations and confirm or question the role of new candidate genes in infertility, establish clinical guidelines and help to improve genetic diagnostics. Increased research collaboration can be a driver that benefits both our biological understanding as well as patient diagnostics in the field of male infertility.
Establishing the role of inherited and de novo mutations in infertility
So far, most research has focused on studying either autosomal recessive or X/Y-linked forms of male infertility. Severe male infertility cannot be dominantly inherited from fathers, but it can be maternally inherited or caused by de novo germline and post-zygotic mutations or CNVs. Exome and genome sequencing of patient-parent trios has revealed the importance of de novo germline mutations in many sporadic genetic diseases, in particular in intellectual disability and related disorders (Vissers et al. 2010, 2016; Veltman and Brunner 2012; McRae et al. 2017). This approach can be an immensely powerful means of identifying novel disease genes, but one of the main challenges for applications in male infertility research is that parental DNA samples are often unavailable. Recently, however, several groups have started to assemble cohorts of patient-parent trios for male infertility genetic research, aiming to identify de novo mutations causing male infertility as well as providing insight into dominant maternal inheritance.
Towards a comprehensive overview of all genomic variation in male infertility
Most NGS approaches use short-read sequencing which limits the detection of repeat structures (Schatz et al. 2010), highly homologous sequences and structural variation (Zhou et al. 2018; Ho et al. 2019). This affects male infertility research significantly, given the importance of chromosome microdeletions in highly homologous and repetitive regions on the Y chromosome as well as causal structural variation reported in other genomic regions. Recently, new sequencing platforms have emerged that have increased sequencing read lengths from a few hundred nucleotides to 10 kb or even longer (Rhoads and Au 2015; Gordon et al. 2016; Jiao et al. 2017). Because of their increased read length these platforms are much better able to detect repeat expansions, homologous sequences and structural genomic variation (Pendleton et al. 2015; Cretu Stancu et al. 2017). Unfortunately, the current per-base accuracy and cost of these technologies does not yet compare favourably to short-read sequencing methods (Rhoads and Au 2015; Rang et al. 2018). Therefore, it is not yet possible to reliably and affordably ascertain all genetic variation present in the genome of an infertile patient in a single experiment. However, complete, accurate and affordable human genome sequencing will likely be available in the next decade, and the major challenge in male infertility will become variant interpretation, not detection.
Conclusion
Significant progress has been made in our understanding of the genetics of male infertility. Using a variety of genomic technologies, the genes involved in rare qualitative sperm defects have now been largely identified, resulting in the development and application of NGS gene panels with a high diagnostic yield. Progress in our understanding of the genetics underlying the more common quantitative sperm defects has not been as rapid as we have seen in qualitative sperm defects or in other genetic disorders. The organization of large well-funded consortia studying thousands of patients with unbiased genomics approaches and the application of relevant functional validation assays should result in a much-needed breakthrough in this field in the coming years.
References
Akin JW, Behzadian A, Tho SPT, McDonough PG (1991) Evidence for a partial deletion in the androgen receptor gene in a phenotypic male with azoospermia. Am J Obstet Gynecol 165:1891–1894. https://doi.org/10.1016/0002-9378(91)90052-S
Anguiano A (1992) Congenital bilateral absence of the vas deferens. JAMA 267:1794. https://doi.org/10.1001/jama.1992.03480130110034
Anuar ND, Kurscheid S, Field M et al (2019) Gene editing of the multi-copy H2A.B gene and its importance for fertility. Genome Biol 20:23. https://doi.org/10.1186/s13059-019-1633-3
Anzalone AV, Randolph PB, Davis JR et al (2019) Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576:149–157. https://doi.org/10.1038/s41586-019-1711-4
Aston KI, Carrell DT (2009) Genome-wide study of single-nucleotide polymorphisms associated with azoospermia and severe oligozoospermia. J Androl 30:711–725. https://doi.org/10.2164/jandrol.109.007971
Ayhan Ö, Balkan M, Guven A et al (2014) Truncating mutations in TAF4B and ZMYND15 causing recessive azoospermia. J Med Genet 51:239–244. https://doi.org/10.1136/jmedgenet-2013-102102
Bean LJH, Funke B, Carlston CM et al (2020) Diagnostic gene sequencing panels: from design to report—a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 22:453–461. https://doi.org/10.1038/s41436-019-0666-z
Ben Khelifa M, Coutton C, Zouari R et al (2014) Mutations in DNAH1, which encodes an inner arm heavy chain dynein, lead to male infertility from multiple morphological abnormalities of the sperm flagella. Am J Hum Genet 94:95–104. https://doi.org/10.1016/j.ajhg.2013.11.017
Bieth E, Hamdi SM, Mieusset R (2020) Genetics of the congenital absence of the vas deferens. Hum Genet. https://doi.org/10.1007/s00439-020-02122-w
Boycott KM, Vanstone MR, Bulman DE, MacKenzie AE (2013) Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat Rev Genet 14:681–691. https://doi.org/10.1038/nrg3555
Brown TR, Lubahn DB, Wilson EM et al (1988) Deletion of the steroid-binding domain of the human androgen receptor gene in one family with complete androgen insensitivity syndrome: evidence for further genetic heterogeneity in this syndrome. Proc Natl Acad Sci 85:8151–8155. https://doi.org/10.1073/pnas.85.21.8151
Butz H, Nyírő G, Kurucz PA et al (2020) Molecular genetic diagnostics of hypogonadotropic hypogonadism: from panel design towards result interpretation in clinical practice. Hum Genet. https://doi.org/10.1007/s00439-020-02148-0
Cangiano B, Swee DS, Quinton R, Bonomi M (2020) Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. https://doi.org/10.1007/s00439-020-02147-1
Castrillon DH, Gonczy P, Alexander S et al (1993) Toward a molecular genetic analysis of spermatogenesis in Drosophila melanogaster: characterization of male-sterile mutants generated by single P element mutagenesis. Genetics 135:489–505
Chapelle A, Hortling H, Niemi M, Wennström J (1964) XX sex chromosomes in a human male. Acta Med Scand 175:25–38. https://doi.org/10.1111/j.0954-6820.1964.tb04630.x
Chillón M, Casals T, Mercier B et al (1995) Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med 332:1475–1480. https://doi.org/10.1056/NEJM199506013322204
Coutton C, Vargas AS, Amiri-Yekta A et al (2018) Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in Trypanosoma and human. Nat Commun https://doi.org/10.1038/s41467-017-02792-7
Coutton C, Zouari R, Abada F et al (2012) MLPA and sequence analysis of DPY19L2 reveals point mutations causing globozoospermia. Hum Reprod 27:2549–2558. https://doi.org/10.1093/humrep/des160
Cretu Stancu M, van Roosmalen MJ, Renkens I et al (2017) Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun 8:1326. https://doi.org/10.1038/s41467-017-01343-4
Culard J-F, Desgeorges M, Costa P et al (1994) Analysis of the whole CFTR coding regions and splice junctions in azoospermic men with congenital bilateral aplasia of epididymis or vas deferens. Hum Genet 93:467–470. https://doi.org/10.1007/BF00201678
Dam AHDM, Koscinski I, Kremer JAM et al (2007) Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet 81:813–820. https://doi.org/10.1086/521314
Dieterich K, Soto Rifo R, Faure AK et al (2007) Homozygous mutation of AURKC yields large-headed polyploid spermatozoa and causes male infertility. Nat Genet 39:661–665. https://doi.org/10.1038/ng2027
Dumur V, Gervais R, Rigot J-M et al (1990) Abnormal distribution of CF ΔF508 allele in azoospermic men with congenital aplasia of epididymis and vas deferens. Lancet 336:512. https://doi.org/10.1016/0140-6736(90)92066-Q
Duquesnoy P, Escudier E, Vincensini L et al (2009) Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am J Hum Genet 85:890–896. https://doi.org/10.1016/j.ajhg.2009.11.008
Eberhart CG, Maines JZ, Wasserman SA (1996) Meiotic cell cycle requirement for a fly homologue of human deleted in Azoospermia. Nature 381:783–785
Elliott DJ, Millar MR, Oghene K et al (1997) Expression of RBM in the nuclei of human germ cells is dependent on a critical region of the Y chromosome long arm. Proc Natl Acad Sci 94:3848–3853. https://doi.org/10.1073/pnas.94.8.3848
Ferlin A, Arredi B, Foresta C (2006a) Genetic causes of male infertility. Reprod Toxicol 22:133–141. https://doi.org/10.1016/j.reprotox.2006.04.016
Ferlin A, Moro E, Garolla A, Foresta C (1999) Human male infertility and Y chromosome deletions: role of the AZF-candidate genes DAZ, RBM and DFFRY. Hum Reprod 14:1710–1716. https://doi.org/10.1093/humrep/14.7.1710
Ferlin A, Vinanzi C, Garolla A et al (2006b) Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations. Clin Endocrinol (Oxf) 65:606–610. https://doi.org/10.1111/j.1365-2265.2006.02635.x
Fernandez-Marmiesse A, Gouveia S, Couce ML (2018) NGS technologies as a turning point in rare disease research. Diagnosis and Treatment. Curr Med Chem https://doi.org/10.2174/0929867324666170718101946
Fitzgerald TW, Gerety SS, Jones WD et al (2015) Large-scale discovery of novel genetic causes of developmental disorders. Nature 519:223–228. https://doi.org/10.1038/nature14135
Gaj T, Gersbach CA, Barbas CF (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405. https://doi.org/10.1016/j.tibtech.2013.04.004
Geister KA, Timms AE, Beier DR (2018) Optimizing genomic methods for mapping and identification of candidate variants in ENU mutagenesis screens using inbred mice. G3 Genes Genomes Genet 8:401–409. https://doi.org/10.1534/g3.117.300292
Gershoni M, Hauser R, Barda S et al (2019) A new MEIOB mutation is a recurrent cause for azoospermia and testicular meiotic arrest. Hum Reprod 34:666–671. https://doi.org/10.1093/humrep/dez016
Gordon D, Huddleston J, Chaisson MJP et al (2016) Long-read sequence assembly of the gorilla genome. Science 352:aae0344–aae0344. https://doi.org/10.1126/science.aae0344
Gottlieb B, Beitel LK, Nadarajah A et al (2012) The androgen receptor gene mutations database: 2012 update. Hum Mutat 33:887–894. https://doi.org/10.1002/humu.22046
Hamerton JL (1968) Robertsonian translocations in man: evidence for prezygotic selection. Cytogenet Genome Res 7:260–276. https://doi.org/10.1159/000129990
Harbuz R, Zouari R, Pierre V et al (2011) A recurrent deletion of DPY19L2 causes infertility in man by blocking sperm head elongation and acrosome formation. Am J Hum Genet 88:351–361. https://doi.org/10.1016/j.ajhg.2011.02.007
He H, Soncin F, Grammatikakis N et al (2003) Elevated expression of heat shock factor (HSF) 2A stimulates HSF1-induced transcription during stress. J Biol Chem 278:35465–35475. https://doi.org/10.1074/jbc.M304663200
Ho SS, Urban AE, Mills RE (2019) Structural variation in the sequencing era. Nat Rev Genet. https://doi.org/10.1038/s41576-019-0180-9
Houston BJ, Conrad DF, O’Bryan MK (2020) A framework for high-resolution phenotyping of candidate male infertility mutants: from human to mouse. Hum Genet. https://doi.org/10.1007/s00439-020-02159-x
Hutchison CA (2007) DNA sequencing: bench to bedside and beyond. Nucleic Acids Res 35:6227–6237. https://doi.org/10.1093/nar/gkm688
Ibtisham F, Wu J, Xiao M et al (2017) Progress and future prospect of in vitro spermatogenesis. Oncotarget 8:66709–66727. https://doi.org/10.18632/oncotarget.19640
Jacobs PA, Frackiewicz A, Law P et al (1975) The effect of structural aberrations of the chromosomes on reproductive fitness in man: II. Results. Clin Genet 8:169–178. https://doi.org/10.1111/j.1399-0004.1975.tb01490.x
Jacobs PA, Strong JA (1959) A case of human intersexuality having a possible XXY sex-determining mechanism. Nature 183:302–303. https://doi.org/10.1038/183302a0
Jamsai D, O’Bryan MK (2010) Genome-wide ENU mutagenesis for the discovery of novel male fertility regulators. Syst Biol Reprod Med 56:246–259. https://doi.org/10.3109/19396361003706424
Jiao Y, Peluso P, Shi J et al (2017) Improved maize reference genome with single-molecule technologies. Nature 546:524–527. https://doi.org/10.1038/nature22971
Jungwirth A, Giwercman A, Tournaye H et al (2012) European association of urology guidelines on male infertility: the 2012 update. Eur Urol 62:324–332. https://doi.org/10.1016/j.eururo.2012.04.048
Kasak L, Laan M (2020) Monogenic causes of non-obstructive azoospermia: challenges, established knowledge, limitations and perspectives. Hum Genet. https://doi.org/10.1007/s00439-020-02112-y
Kasak L, Punab M, Nagirnaja L et al (2018) Bi-allelic recessive loss-of-function variants in FANCM cause non-obstructive azoospermia. Am J Hum Genet 103:200–212. https://doi.org/10.1016/j.ajhg.2018.07.005
Kennedy CL, O’Bryan MK (2006) N-ethyl-N-nitrosourea (ENU) mutagenesis and male fertility research. Hum Reprod Update 12:293–301. https://doi.org/10.1093/humupd/dmk004
Kerem B, Rommens J, Buchanan J et al (1989) Identification of the cystic fibrosis gene: genetic analysis. Science 245:1073–1080. https://doi.org/10.1126/science.2570460
Kherraf Z-E, Amiri-Yekta A, Dacheux D et al (2018) A homozygous ancestral SVA-insertion-mediated deletion in WDR66 induces multiple morphological abnormalities of the sperm flagellum and male infertility. Am J Hum Genet 103:400–412. https://doi.org/10.1016/j.ajhg.2018.07.014
Kobayashi K, Mlzuno K, Hida A et al (1994) PCR analysis of the Y chromosome long arm in azoospermic patients: evidence for a second locus required for spermatogenesis. Hum Mol Genet 3:1965–1967. https://doi.org/10.1093/hmg/3.11.1965
Komeya M, Sato T, Ogawa T (2018) In vitro spermatogenesis: a century-long research journey, still half way around. Reprod Med Biol 17:407–420. https://doi.org/10.1002/rmb2.12225
Koscinski I, ElInati E, Fossard C et al (2011) DPY19L2 deletion as a major cause of Globozoospermia. Am J Hum Genet 88:344–350. https://doi.org/10.1016/j.ajhg.2011.01.018
Koulischer L, Schoysman R (1974) Chromosomes and human infertility. I. Mitotic and meiotic chromosome studies in 202 consecutive male patients. Clin Genet 5:116–126. https://doi.org/10.1111/j.1399-0004.1974.tb01673.x
Krausz C (2011) Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab 25:271–285. https://doi.org/10.1016/j.beem.2010.08.006
Krausz C, Casamonti E (2017) Spermatogenic failure and the Y chromosome. Hum Genet 136:637–655. https://doi.org/10.1007/s00439-017-1793-8
Krausz C, Giachini C, Lo Giacco D et al (2012) High resolution X chromosome-SPECIFIC Array-CGH detects new CNVs in infertile males. PLoS ONE 7:e44887. https://doi.org/10.1371/journal.pone.0044887
Krausz C, Riera-Escamilla A (2018) Genetics of male infertility. Nat Rev Urol 15:369–384. https://doi.org/10.1038/s41585-018-0003-3
Krausz C, Riera-Escamilla A, Chianese C et al (2019) From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet Med 21:189–194. https://doi.org/10.1038/s41436-018-0037-1
Lahn BT, Page DC (1997) Functional coherence of the human Y chromosome. Science 278:675–680. https://doi.org/10.1126/science.278.5338.675
Lewis SE, Barnett LB, Shelby MD (1992) ENU mutagenesis in the mouse electrophoretic specific-locus test 2. Mutational studies of mature oocytes. Mutat Res Genet Toxicol 296:129–133. https://doi.org/10.1016/0165-1110(92)90036-9
Li L, Sha Y, Wang X et al (2017) Whole-exome sequencing identified a homozygous BRDT mutation in a patient with acephalic spermatozoa. Oncotarget 8:19914–19922. https://doi.org/10.18632/oncotarget.15251
Lima AC, Carvalho F, Gonçalves J et al (2015) Rare double sex and mab-3-related transcription factor 1 regulatory variants in severe spermatogenic failure. Andrology 3:825–833. https://doi.org/10.1111/andr.12063
Lin Q, Mei J, Li Z et al (2017) Distinct and cooperative roles of amh and dmrt1 in self-renewal and differentiation of male germ cells in Zebrafis. Genetics 207:1007–1022. https://doi.org/10.1534/genetics.117.300274/-/DC1.1
Lo Giacco D, Chianese C, Ars E et al (2014) Recurrent X chromosome-linked deletions: discovery of new genetic factors in male infertility. J Med Genet 51:340–344. https://doi.org/10.1136/jmedgenet-2013-101988
Lopes AM, Aston KI, Thompson E et al (2013) Human spermatogenic failure purges deleterious mutation load from the autosomes and both sex chromosomes, including the gene DMRT1. PLoS Genet 9:e1003349. https://doi.org/10.1371/journal.pgen.1003349
Lyon MF, Hawkes SG (1970) X-linked gene for testicular feminization in the mouse. Nature 227:1217–1219. https://doi.org/10.1038/2271217a0
Ma K, Inglis JD, Sharkey A et al (1993) A Y chromosome gene family with RNA-binding protein homology: candidates for the azoospermia factor AZF controlling human spermatogenesis. Cell 75:1287–1295. https://doi.org/10.1016/0092-8674(93)90616-X
Ma K, Sharkey A, Kirsch S et al (1992) Towards the molecular localisation of the AZF locus: mapping of microdeletions in azoospermic men within 14 subintervals of interval 6 of the human Y chromosome. Hum Mol Genet 1:29–33. https://doi.org/10.1093/hmg/1.1.29
Martinez-Garay I, Jablonka S, Sutajova M et al (2002) A new gene family (FAM9) of low-copy repeats in Xp22.3 expressed exclusively in testis: implications for recombinations in this region. Genomics 80:259–267. https://doi.org/10.1006/geno.2002.6834
McPhaul MJ, Marcelli M, Zoppi S et al (1992) Mutations in the ligand-binding domain of the androgen receptor gene cluster in two regions of the gene. J Clin Invest 90:2097–2101. https://doi.org/10.1172/JCI116093
McRae JF, Clayton S, Fitzgerald TW et al (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature 542:433–438. https://doi.org/10.1038/nature21062
Metzker ML (2005) Emerging technologies in DNA sequencing. Genome Res 15:1767–1776. https://doi.org/10.1101/gr.3770505
Miyamoto T, Hasuike S, Yogev L et al (2003) Azoospermia in patients heterozygous for a mutation in SYCP3. Lancet 362:1714–1719. https://doi.org/10.1016/S0140-6736(03)14845-3
Mou L, Wang Y, Li H et al (2013) A dominant-negative mutation of HSF2 associated with idiopathic azoospermia. Hum Genet 132:159–165. https://doi.org/10.1007/s00439-012-1234-7
Nathanson KL, Kanetsky PA, Hawes R et al (2005) The Y deletion gr/gr and susceptibility to testicular germ cell tumor. Am J Hum Genet 77:1034–1043. https://doi.org/10.1086/498455
Okutman O, Muller J, Baert Y et al (2015) Exome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish family. Hum Mol Genet 24:5581–5588. https://doi.org/10.1093/hmg/ddv290
Oud MS, Volozonoka L, Smits RM et al (2019) A systematic review and standardized clinical validity assessment of male infertility genes. Hum Reprod 34:932–941. https://doi.org/10.1093/humrep/dez022
Patat O, Pagin A, Siegfried A et al (2016) Truncating mutations in the adhesion g protein-coupled receptor G2 Gene ADGRG2 cause an X-linked congenital bilateral absence of vas deferens. Am J Hum Genet 99:437–442. https://doi.org/10.1016/j.ajhg.2016.06.012
Patrizio P, Ord T, Silber SJ, Asch RH (1993) Andrology: cystic fibrosis mutations impair the fertilization rate of epididymal sperm from men with congenital absence of the vas deferens. Hum Reprod 8:1259–1263. https://doi.org/10.1093/oxfordjournals.humrep.a138237
Payne K, Gavan SP, Wright SJ, Thompson AJ (2018) Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat Rev Genet 19:235–246. https://doi.org/10.1038/nrg.2017.108
Pendleton M, Sebra R, Pang AWC et al (2015) Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat Methods 12:780–786. https://doi.org/10.1038/nmeth.3454
Pinkel D, Segraves R, Sudar D et al (1998) High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat Genet 20:207–211. https://doi.org/10.1038/2524
Punab M, Poolamets O, Paju P et al (2016) Causes of male infertility: a 9-year prospective monocentre study on 1737 patients with reduced total sperm counts. Hum Reprod 32:18–31. https://doi.org/10.1093/humrep/dew284
Quigley CA, De BA, Marschke KB et al (1995) Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev 16:271–321. https://doi.org/10.1210/edrv-16-3-271
Ramanagoudr-Bhojappa R, Carrington B, Ramaswami M et al (2018) Multiplexed CRISPR/Cas9-mediated knockout of 19 Fanconi anemia pathway genes in zebrafish revealed their roles in growth, sexual development and fertility. PLOS Genet 14:e1007821. https://doi.org/10.1371/journal.pgen.1007821
Rang FJ, Kloosterman WP, de Ridder J (2018) From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol 19:90. https://doi.org/10.1186/s13059-018-1462-9
Reijo R, Alagappan RK, Page DC, Patrizio P (1996) Severe oligozoospermia resulting from deletions of azoospermia factor gene on Y chromosome. Lancet 347:1290–1293. https://doi.org/10.1016/S0140-6736(96)90938-1
Reijo R, Lee T, Salo P et al (1995) Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat Genet 10:383–393. https://doi.org/10.1038/ng0895-383
Rhoads A, Au KF (2015) PacBio sequencing and its applications. Genomics Proteomics Bioinf 13:278–289. https://doi.org/10.1016/j.gpb.2015.08.002
Riordan RJ, Kerem B et al (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 245:1066–1073. https://doi.org/10.1126/science.2475911
Rumi MAK, Dhakal P, Kubota K et al (2014) Generation of Esr1-knockout rats using zinc finger nuclease-mediated genome editing. Endocrinology 155:1991–1999. https://doi.org/10.1210/en.2013-2150
Sato T, Katagiri K, Gohbara A et al (2011) In vitro production of functional sperm in cultured neonatal mouse testes. Nature 471:504–507. https://doi.org/10.1038/nature09850
Saxena R, de Vries JWA, Repping S et al (2000) Four DAZ genes in two clusters found in the AZFc region of the human Y chromosome. Genomics 67:256–267. https://doi.org/10.1006/geno.2000.6260
Schatz MC, Delcher AL, Salzberg SL (2010) Assembly of large genomes using second-generation sequencing. Genome Res 20:1165–1173. https://doi.org/10.1101/gr.101360.109
Schilit SLP, Menon S, Friedrich C et al (2020) SYCP2 translocation-mediated dysregulation and frameshift variants cause human male infertility. Am J Hum Genet 106:41–57. https://doi.org/10.1016/j.ajhg.2019.11.013
Seabra CM, Quental S, Neto AP et al (2014) A novel Alu-mediated microdeletion at 11p13 removes WT1 in a patient with cryptorchidism and azoospermia. Reprod Biomed Online 29:388–391. https://doi.org/10.1016/j.rbmo.2014.04.017
Sha Y-W, Wang X, Xu X et al (2019) Biallelic mutations in PMFBP1 cause acephalic spermatozoa. Clin Genet 95:277–286. https://doi.org/10.1111/cge.13461
Shen Y, Xu J, Yang X et al (2017) Evidence for the involvement of the proximal copy of the MAGEA9 gene in Xq28-linked CNV67 specific to spermatogenic failure†. Biol Reprod 96:610–616. https://doi.org/10.1093/biolre/iox006
Skaletsky H, Kuroda-Kawaguchi T, Minx PJ et al (2003) The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837. https://doi.org/10.1038/nature01722
Smith ED, Radtke K, Rossi M et al (2017) Classification of genes: standardized clinical validity assessment of gene-disease associations aids diagnostic exome analysis and reclassifications. Hum Mutat 38:600–608. https://doi.org/10.1002/humu.23183
Solinas-Toldo S, Lampel S, Stilgenbauer S et al (1997) Matrix-based comparative genomic hybridization: Biochips to screen for genomic imbalances. Genes Chromosom Cancer 20:399–407. https://doi.org/10.1002/(SICI)1098-2264(199712)20:4<399:AID-GCC12>3.0.CO;2-I
Soraggi S, Riera M, Rajpert-De Meyts E et al (2020) Evaluating genetic causes of azoospermia: what can we learn from a complex cellular structure and single-cell transcriptomics of the human testis? Hum Genet. https://doi.org/10.1007/s00439-020-02116-8
Stainier DYR, Raz E, Lawson ND et al (2017) Guidelines for morpholino use in zebrafish. PLOS Genet 13:e1007000. https://doi.org/10.1371/journal.pgen.1007000
Stouffs K, Vandermaelen D, Massart A et al (2012) Array comparative genomic hybridization in male infertility. Hum Reprod 27:921–929. https://doi.org/10.1093/humrep/der440
Summerton J (1999) Morpholino antisense oligomers: the case for an RNase H-independent structural type. Biochim Biophys Acta Gene Struct Expr 1489:141–158. https://doi.org/10.1016/S0167-4781(99)00150-5
Sun C, Skaletsky H, Birren B et al (1999) An azoospermic man with a de novo point mutation in the Y-chromosomal gene USP9Y. Nat Genet 23:429–432. https://doi.org/10.1038/70539
Talkowski ME, Ernst C, Heilbut A et al (2011) Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet 88:469–481. https://doi.org/10.1016/j.ajhg.2011.03.013
Tewes A-C, Ledig S, Tüttelmann F et al (2014) DMRT1 mutations are rarely associated with male infertility. Fertil Steril 102:816–820.e3. https://doi.org/10.1016/j.fertnstert.2014.05.022
Therkelsen A (1964) Sterile male with the chromosome constitution 46 XX. Cytogenet Genome Res 3:207–218. https://doi.org/10.1159/000129812
Tiepolo L, Zuffardi O (1976) Localization of factors controlling spermatogenesis in the nonfluorescent portion of the human y chromosome long arm. Hum Genet 34:119–124. https://doi.org/10.1007/BF00278879
Touré A, Martinez G, Kherraf Z-E et al (2020) The genetic architecture of morphological abnormalities of the sperm tail. Hum Genet. https://doi.org/10.1007/s00439-020-02113-x
Tournaye H, Krausz C, Oates RD (2017) Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol 5:544–553. https://doi.org/10.1016/S2213-8587(16)30040-7
Tüttelmann F, Ruckert C, Röpke A (2018) Disorders of spermatogenesis. Medizinische Genet 30:12–20. https://doi.org/10.1007/s11825-018-0181-7
Tüttelmann F, Simoni M, Kliesch S et al (2011) Copy number variants in patients with severe oligozoospermia and sertoli-cell-only syndrome. PLoS ONE 6:e19426. https://doi.org/10.1371/journal.pone.0019426
Veltman JA, Brunner HG (2012) De novo mutations in human genetic disease. Nat Rev Genet 13:565–575. https://doi.org/10.1038/nrg3241
Visser L, Westerveld GH, Korver CM et al (2009) Y chromosome gr/gr deletions are a risk factor for low semen quality. Hum Reprod 24:2667–2673. https://doi.org/10.1093/humrep/dep243
Vissers LELM, de Ligt J, Gilissen C et al (2010) A de novo paradigm for mental retardation. Nat Genet 42:1109–1112. https://doi.org/10.1038/ng.712
Vissers LELM, Gilissen C, Veltman JA (2016) Genetic studies in intellectual disability and related disorders. Nat Rev Genet 17:9–18. https://doi.org/10.1038/nrg3999
Vockel M, Riera-Escamilla A, Tüttelmann F, Krausz C (2019) The X chromosome and male infertility. Hum Genet. https://doi.org/10.1007/s00439-019-02101-w
Vogt P, Chandley AC, Hargreave TB et al (1992) Microdeletions in interval 6 of the Y chromosome of males with idiopathic sterility point to disruption of AZF, a human spermatogenesis gene. Hum Genet 89:491–496. https://doi.org/10.1007/BF00219172
Vogt PH (1998) Human chromosome deletions in Yq11, AZF candidate genes and male infertility: history and update. Mol Hum Reprod 4:739–744. https://doi.org/10.1093/molehr/4.8.739
Wakimoto BT, Lindsley DL, Herrera C (2004) Toward a comprehensive genetic analysis of male fertility in Drosophila melanogaster. Genetics 167:207–216. https://doi.org/10.1534/genetics.167.1.207
Wang Q, Liu C, Tang C et al (2015) Yeast model identifies ENTPD6 as a potential non-obstructive azoospermia pathogenic gene. Sci Rep 5:1–13. https://doi.org/10.1038/srep11762
Weiske W-H, Sälzler N, Schroeder-Printzen I, Weidner W (2000) Clinical findings in congenital absence of the vasa deferentia. Andrologia 32:13–18. https://doi.org/10.1111/j.1439-0272.2000.tb02859.x
Wilfert AB, Chao KR, Kaushal M et al (2016) Genome-wide significance testing of variation from single case exomes. Nat Genet 48:1455–1461. https://doi.org/10.1038/ng.3697
Yan W, Ma L, Burns KH, Matzuk MM (2004) Haploinsufficiency of kelch-like protein homolog 10 causes infertility in male mice. Proc Natl Acad Sci 101:7793–7798. https://doi.org/10.1073/pnas.0308025101
Yang F, Gell K, van der Heijden GW et al (2008) Meiotic failure in male mice lacking an X-linked factor. Genes Dev 22:682–691. https://doi.org/10.1101/gad.1613608
Yang F, Silber S, Leu NA et al (2015) TEX 11 is mutated in infertile men with azoospermia and regulates genome-wide recombination rates in mouse. EMBO Mol Med 7:1198–1210. https://doi.org/10.15252/emmm.201404967
Yang Y, Guo J, Dai L et al (2018) XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J Med Genet 55:628–636. https://doi.org/10.1136/jmedgenet-2017-105145
Yatsenko AN, Georgiadis AP, Röpke A et al (2015) X-linked TEX11 mutations, meiotic arrest, and azoospermia in infertile men. N Engl J Med 372:2097–2107. https://doi.org/10.1056/NEJMoa1406192
Yatsenko AN, Roy A, Chen R et al (2006) Non-invasive genetic diagnosis of male infertility using spermatozoal RNA: KLHL10mutations in oligozoospermic patients impair homodimerization. Hum Mol Genet 15:3411–3419. https://doi.org/10.1093/hmg/ddl417
Yen PH (1998) A long-range restriction map of deletion interval 6 of the human Y chromosome: a region frequently deleted in azoospermic males. Genomics 54:5–12. https://doi.org/10.1006/geno.1998.5526
Yu J, Wu H, Wen Y et al (2015) Identification of seven genes essential for male fertility through a genome-wide association study of non-obstructive azoospermia and RNA interference mediated large-scale functional screening in Drosophila. Hum Mol Genet 24:1493–1503. https://doi.org/10.1093/hmg/ddu557
Yuan L, Liu J-G, Zhao J et al (2000) The murine SCP3 gene is required for synaptonemal complex assembly, chromosome synapsis, and male fertility. Mol Cell 5:73–83. https://doi.org/10.1016/S1097-2765(00)80404-9
Zhang Z, Kostetskii I, Tang W et al (2006) Deficiency of SPAG16L causes male infertility associated with impaired sperm motility. Biol Reprod 74:751–759. https://doi.org/10.1095/biolreprod.105.049254
Zhou B, Ho SS, Zhang X et al (2018) Whole-genome sequencing analysis of CNV using low-coverage and paired-end strategies is efficient and outperforms array-based CNV analysis. J Med Genet 55:735–743. https://doi.org/10.1136/jmedgenet-2018-105272
Zhu F, Wang F, Yang X et al (2016) Biallelic SUN5 mutations cause autosomal-recessive acephalic spermatozoa syndrome. Am J Hum Genet 99:942–949. https://doi.org/10.1016/j.ajhg.2016.08.004
Zhu F, Liu C, Wang F et al (2018) Mutations in PMFBP1 cause acephalic spermatozoa syndrome. Am J Hum Genet 103:188–199. https://doi.org/10.1016/j.ajhg.2018.06.010
Acknowledgements
This work was supported in part by the National Institutes of Health funding to K.I.A. (R01HD078641). This work was supported by a VICI grant from The Netherlands Organization for Scientific Research (918-15-667 to J.A.V.), the Royal Society and Wolfson Foundation (WM160091 to J.A.V.) as well as an Investigator Award in Science from the Wellcome Trust (209451 to J.A.V.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xavier, M.J., Salas-Huetos, A., Oud, M.S. et al. Disease gene discovery in male infertility: past, present and future. Hum Genet 140, 7–19 (2021). https://doi.org/10.1007/s00439-020-02202-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02202-x