
Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review

 , ,
, , 
Abstract
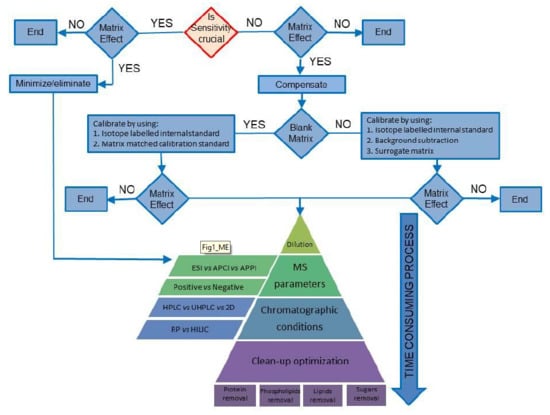
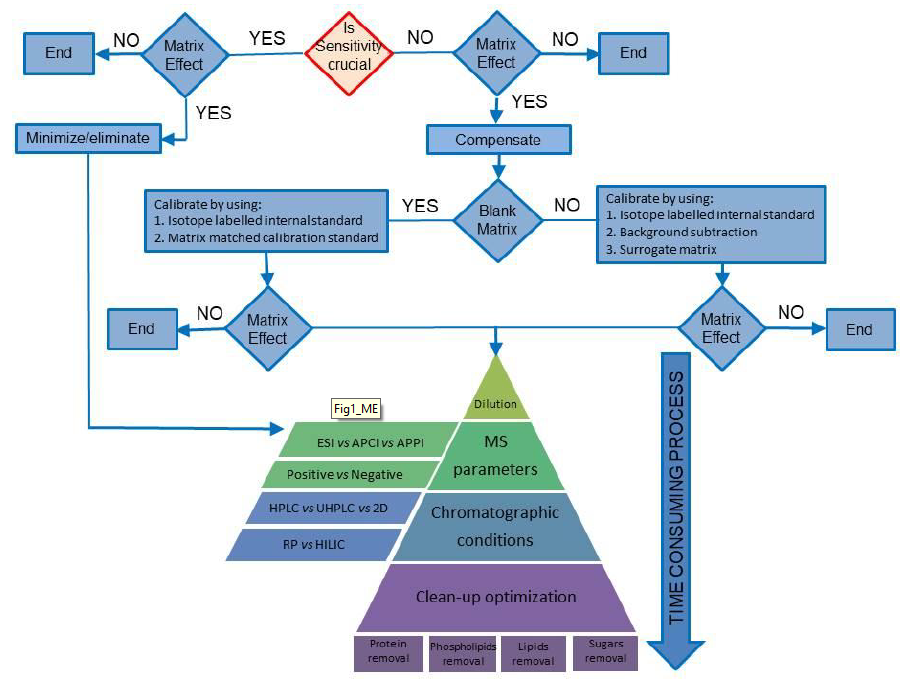
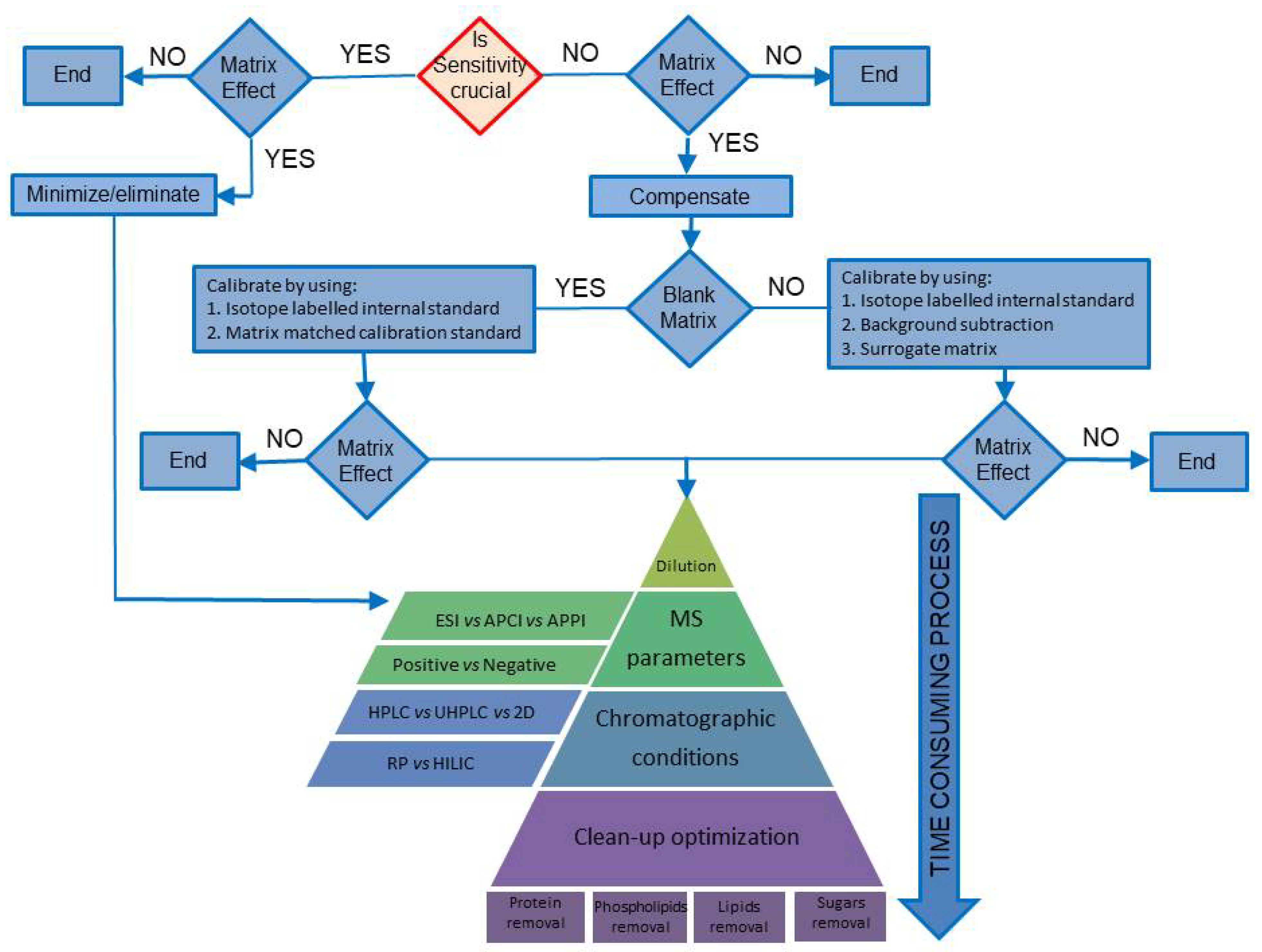
:
1. Introduction
2. Evaluation of MEs
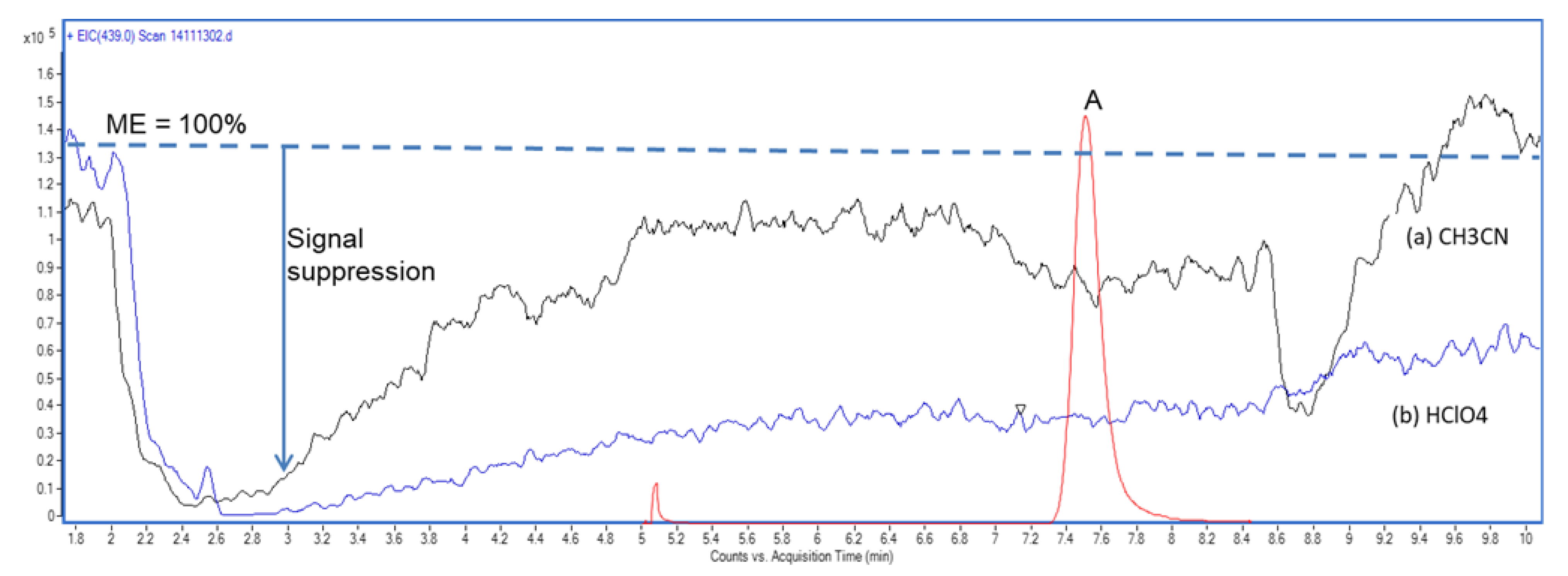
2.1. Post-Column Infusion
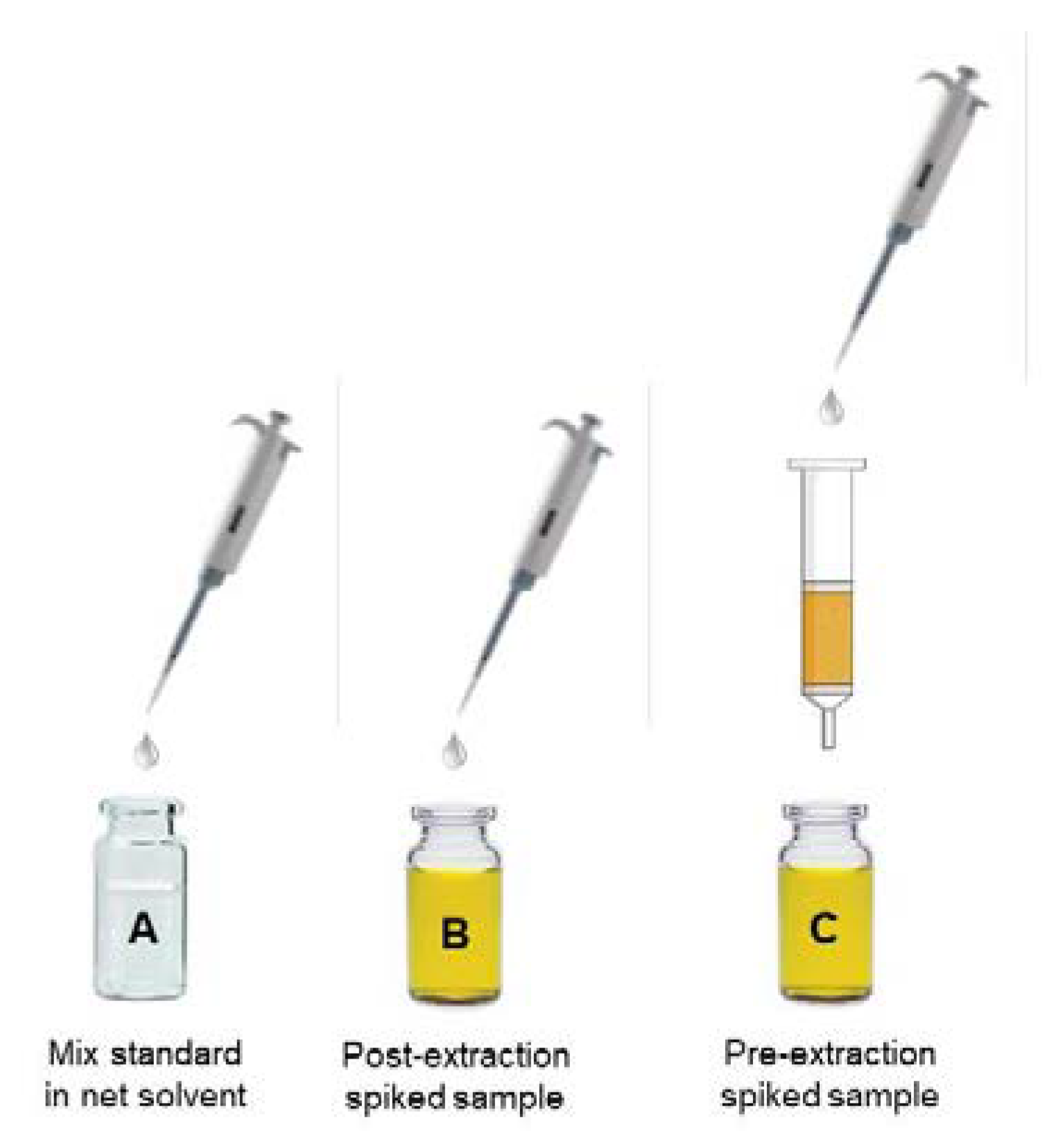
2.2. The Matuszewski Post-Extraction Spike Method
2.3. Slope Ratio Analysis
2.4. Evaluation of Relative MEs
3. Compensating for MEs
3.1. Case Study I: The Blank Matrix Is Available
3.2. Case Study II: The Blank Matrix Is Not Available
4. Minimizing Matrix Effects
4.1. Sample Dilution
4.2. Mass Spectrometric Conditions
4.3. Chromatographic Conditions
4.4. Clean-Up Optimization
4.4.1. Removal of Proteins
4.4.2. Phospholipid Removal
4.4.3. Removal of Lipids
4.4.4. Removal of Sugars
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kloepfer, A.; Quintana, J.B.; Reemtsma, T. Operational options to reduce matrix effects in liquid chromatography–electrospray ionisation-mass spectrometry analysis of aqueous environmental samples. J. Chromatogr. A 2005, 1067, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Künnapas, A.; Herodes, K.; Leito, I. Matrix effects in pesticide multi-residue analysis by liquid chromatography–mass spectrometry. J. Chromatogr. A 2008, 1187, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Dams, R.; Huestis, M.A.; Lambert, W.E.; Murphy, C.M. Matrix effect in bio-analysis of illicit drugs with LC-MS/MS: Influence of ionization type, sample preparation, and biofluid. J. Am. Soc. Mass Spectrom. 2003, 14, 1290–1294. [Google Scholar] [CrossRef] [Green Version]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Matrix effect in quantitative LC/MS/MS analyses of biological fluids: A method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 1998, 70, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Breaux, J.; Jones, K.; Boulas, P. Analytical methods development and validation. Pharm. Technol. 2003, 5, 6–13. [Google Scholar]
- Kaza, M.; Karaźniewicz-Łada, M.; Kosicka, K.; Siemiątkowska, A.; Rudzki, P.J. Bioanalytical method validation: New FDA guidance vs. EMA guideline. Better or worse? J. Pharm. Biomed. Anal. 2019, 165, 381–385. [Google Scholar] [CrossRef]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part II. Anal. Chim. Acta 2015, 870, 8–28. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, R.; IUPAC. Compendium of Chemical Terminology, 2nd ed.; (the “Gold Book”); Blackwell Scientific Publications: Oxford, UK, 1997; Available online: https://doi.org/10.1351/goldbook (accessed on 25 June 2019).
- Kermit, K.M.; Robert, K.B.; Marcos, N.E.; Langley, G.J.; Liang, L.; Yasuhide, N. Definitions of terms relating to mass spectrometry (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1515–1609. [Google Scholar]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An overview of matrix effects in liquid chromatography–mass spectrometry. Mass Spectrom. Rev. 2011, 30, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Cech, N.B.; Enke, C.G. Relating Electrospray Ionization Response to Nonpolar Character of Small Peptides. Anal. Chem. 2000, 72, 2717–2723. [Google Scholar] [CrossRef] [PubMed]
- King, R.; Bonfiglio, R.; Fernandez-Metzler, C.; Miller-Stein, C.; Olah, T. Mechanistic investigation of ionization suppression in electrospray ionization. J. Am. Soc. Mass Spectrom. 2000, 11, 942–950. [Google Scholar] [CrossRef] [Green Version]
- Annesley, T.M. Ion Suppression in Mass Spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, W. Pitfalls in LC-MS(-MS) Analysis. Toxichem. Krimtech. 2004, 71, 64–68. [Google Scholar]
- Yuan, C.; Chen, D.; Wang, S. Drug confirmation by mass spectrometry: Identification criteria and complicating factors. Clin. Chim. Acta 2015, 438, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wan, S.; Chen, L.; Wang, X.; Wang, Z.; Huang, Y. Determination of cycloserine in microdialysis samples using liquid chromatography-tandem mass spectrometry with benzoyl chloride derivatization. Biomed. Chromatogr. 2018, 32, e4187. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.; Patel, N.; Patel, B.; Patel, H.; Bahekar, R.; Srinivas, N.R.; Patel, P.R.; Desai, R. Sensitive and specific LC-ESI-MS/MS method for determination of ZYDPLA1, a novel long-acting dipeptidyl peptidase 4 inhibitor in rat plasma: An application for toxicokinetic study in rats. Biomed. Chromatogr. 2017, 31. [Google Scholar] [CrossRef]
- Tang, J.C.Y.; Dutton, J.J.; Piec, I.; Green, D.; Fisher, E.; Washbourne, C.J.; Fraser, W.D. LC–MS/MS application for urine free pyridinoline and free deoxypyridinoline: Urine markers of collagen and bone degradation. Clin. Mass Spectrom. 2016, 1, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, K.; Kataoka, H. Analysis of urinary 8-isoprostane as an oxidative stress biomarker by stable isotope dilution using automated online in-tube solid-phase microextraction coupled with liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2015, 112, 36–42. [Google Scholar] [CrossRef]
- Kataoka, H. Recent developments and applications of microextraction techniques in drug analysis. Anal. Bioanal. Chem. 2010, 396, 339–364. [Google Scholar] [CrossRef]
- Chen, L.; Xu, S.; Li, J. Recent advances in molecular imprinting technology: Current status, challenges and highlighted applications. Chem. Soc. Rev. 2011, 2292. [Google Scholar] [CrossRef]
- Yang, Z.; Cai, Q.; Chen, N.; Zhou, X.; Hong, J. Selective separation and identification of metabolite groups of Polygonum cuspidatum extract in rat plasma using dispersion solid-phase extraction by magnetic molecularly imprinted polymers coupled with LC/Q-TOF-MS. RSC Adv. 2016, 6, 12193–12204. [Google Scholar] [CrossRef]
- Thakare, R.; Chhonker, Y.S.; Gautam, N.; Alamoudi, J.A.; Alnouti, Y. Quantitative analysis of endogenous compounds. J. Pharm. Biomed. Anal. 2016, 128, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Nováková, L. Challenges in the development of bioanalytical liquid chromatography-mass spectrometry method with emphasis on fast analysis. J. Chromatogr. A 2013, 1292, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Bonfiglio, R.; King, R.C.; Olah, T.V.; Merkle, K. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun. Mass Spectrom. 1999, 13, 1175–1185. [Google Scholar] [CrossRef]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Romero-González, R.; Garrido Frenich, A.; Martínez Vidal, J.L.; Prestes, O.D.; Grio, S.L. Simultaneous determination of pesticides, biopesticides and mycotoxins in organic products applying a quick, easy, cheap, effective, rugged and safe extraction procedure and ultra-high performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Krska, R.; Schuhmacher, R. A liquid chromatography/tandem mass spectrometric multi-mycotoxin method for the quantification of 87 analytes and its application to semi-quantitative screening of moldy food samples. Anal. Bioanal. Chem. 2007, 389, 1505–1523. [Google Scholar] [CrossRef]
- González, O.; Van Vliet, M.; Damen, C.W.; Van der Kloet, F.M.; Vreeken, R.J.; Hankemeier, T. Matrix Effect Compensation in Small-Molecule Profiling for an LC-TOF Platform Using Multicomponent Postcolumn Infusion. Anal. Chem. 2015, 87, 5921–5929. [Google Scholar] [CrossRef]
- Kamiguchi, H.; Yamaguchi, M.; Murabayashi, M.; Mori, I.; Horinouchi, A. Method development and validation for simultaneous quantitation of endogenous hippuric acid and phenylacetylglycine in rat urine using liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1035, 76–83. [Google Scholar] [CrossRef]
- Stahnke, H.; Reemtsma, T.; Alder, L. Compensation of matrix effects by postcolumn infusion of a monitor substance in multiresidue analysis with LC-MS/MS. Anal. Chem. 2009, 81, 2185–2192. [Google Scholar] [CrossRef]
- Rossmann, J.; Gurke, R.; Renner, L.D.; Oertel, R.; Kirch, W. Evaluation of the matrix effect of different sample matrices for 33 pharmaceuticals by post-column infusion. J. Chromatogr. B 2015, 1000, 84–94. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, S.; Lee, J.; Kim, B. Accurate determination of ochratoxin A in Korean fermented soybean paste by isotope dilution-liquid chromatography tandem mass spectrometry. Food Chem. 2016, 190, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Hoff, R.B.; Rübensam, G.; Jank, L.; Barreto, F.; Ruaro Peralba, M.d.C.; Pizzolato, T.M.; Silvia Díaz-Cruz, M.; Barceló, D. Analytical quality assurance in veterinary drug residue analysis methods: Matrix effects determination and monitoring for sulfonamides analysis. Talanta 2015, 132, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antignac, J.-P.; De Wasch, K.; Monteau, F.; De Brabander, H.; Andre, F.; Le Bizec, B. The ion suppression phenomenon in liquid chromatography–mass spectrometry and its consequences in the field of residue analysis. Anal. Chim. Acta 2005, 529, 129–136. [Google Scholar] [CrossRef]
- Kittlaus, S.; Schimanke, J.; Kempe, G.; Speer, K. Assessment of sample cleanup and matrix effects in the pesticide residue analysis of foods using postcolumn infusion in liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 8399–8410. [Google Scholar] [CrossRef]
- Ghosh, C.; Shinde, C.P.; Chakraborty, B.S. Influence of ionization source design on matrix effects during LC-ESI-MS/MS analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 893, 193–200. [Google Scholar] [CrossRef]
- Rentsch, K.M. Knowing the unknown – State of the art of LCMS in toxicology. Trac. Trends Anal. Chem. 2016, 84, 88–93. [Google Scholar] [CrossRef]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef]
- Van Eeckhaut, A.; Lanckmans, K.; Sarre, S.; Smolders, I.; Michotte, Y. Validation of bioanalytical LC-MS/MS assays: Evaluation of matrix effects. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 2198–2207. [Google Scholar] [CrossRef]
- Nurit, E.; Lyan, B.; Piquet, A.; Branlard, G.; Pujos-Guillot, E. Development of a LC-MS/MS method for the simultaneous screening of seven water-soluble vitamins in processing semi-coarse wheat flour products. Anal. Bioanal. Chem. 2015, 407, 3471–3479. [Google Scholar] [CrossRef]
- Lasaosa, M.; Patel, P.; Givler, S.; De León, D.D.; Seeholzer, S.H. A liquid chromatography-mass spectrometry assay for quantification of Exendin [9-39] in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 947, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Qian, X.; Yang, Z.; Zhang, L. Study on the matrix effect in the determination of selected pharmaceutical residues in seawater by solid-phase extraction and ultra-high-performance liquid chromatography-electrospray ionization low-energy collision-induced dissociation tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 1471–1475. [Google Scholar] [PubMed]
- Caban, M.; Migowska, N.; Stepnowski, P.; Kwiatkowski, M.; Kumirska, J. Matrix effects and recovery calculations in analyses of pharmaceuticals based on the determination of β-blockers and β-agonists in environmental samples. J. Chromatogr. A 2012, 1258, 117–127. [Google Scholar] [CrossRef]
- Food and Drug Administration. Food and Drug Administration, Guidance for Industry: Bioanalytical Method Validation. Available online: http://www.labcompliance.de/FDA/FDA-Others/Laboratory/f-507-bioanalytical-4252fnl.pdf (accessed on 25 May 2020).
- Matuszewski, B.K. Standard line slopes as a measure of a relative matrix effect in quantitative HPLC–MS bioanalysis. J. Chromatogr. B 2006, 830, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ismaiel, O.A.; Halquist, M.S.; Elmamly, M.Y.; Shalaby, A.; Thomas Karnes, H. Monitoring phospholipids for assessment of ion enhancement and ion suppression in ESI and APCI LC/MS/MS for chlorpheniramine in human plasma and the importance of multiple source matrix effect evaluations. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 875, 333–343. [Google Scholar] [CrossRef]
- Niessen, W.M.; Manini, P.; Andreoli, R. Matrix effects in quantitative pesticide analysis using liquid chromatography-mass spectrometry. Mass Spectrom. Rev. 2006, 25, 881–899. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Bioanalytical Method Validation; Amsterdam, The Netherlands, 2012; Volume 44, pp. 1–23. [Google Scholar]
- Franco, P.; Porru, E.; Fiori, J.; Gioiello, A.; Cerra, B.; Roda, G.; Caliceti, C.; Simoni, P.; Roda, A. Identification and quantification of oxo-bile acids in human faeces with liquid chromatography–mass spectrometry: A potent tool for human gut acidic sterolbiome studies. J. Chromatogr. A 2019, 1585, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, D.L.; Price, P.I.; Rudewicz, P.J. Quantitation of SR 27417 in human plasma using electrospray liquid chromatography-tandem mass spectrometry: A study of ion suppression. J. Am. Soc. Mass Spectrom. 1996, 7, 1099–1105. [Google Scholar] [CrossRef] [Green Version]
- Heller, D.N. Ruggedness testing of quantitative atmospheric pressure ionization mass spectrometry methods: The effect of co-injected matrix on matrix effects. Rapid Commun. Mass Spectrom. 2007, 21, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Guidelines for the Validation of Chemical Methods for the FDA FVM Program. Available online: https://cannabis.ca.gov/wp-content/uploads/sites/13/2018/07/Documents-Incorporated-by-Reference-%E2%80%93-US-FDA-Guidelines.pdf (accessed on 25 May 2020).
- González, O.; Blanco, M.E.; Iriarte, G.; Bartolomé, L.; Maguregui, M.I.; Alonso, R.M. Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect. J. Chromatogr. A 2014, 1353, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Genangeli, M.; Caprioli, G.; Cortese, M.; Laus, F.; Matteucci, M.; Petrelli, R.; Ricciutelli, M.; Sagratini, G.; Sartori, S.; Vittori, S. Development and application of a UHPLC-MS/MS method for the simultaneous determination of 17 steroidal hormones in equine serum. J. Mass Spectrom. 2017, 52, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Hényková, E.; Vránová, H.P.; Amakorová, P.; Pospíšil, T.; Žukauskaitė, A.; Vlčková, M.; Urbánek, L.; Novák, O.; Mareš, J.; Kaňovský, P.; et al. Stable isotope dilution ultra-high performance liquid chromatography-tandem mass spectrometry quantitative profiling of tryptophan-related neuroactive substances in human serum and cerebrospinal fluid. J. Chromatogr. A 2016, 1437, 145–157. [Google Scholar]
- Ares, A.M.; Ayuso, I.; Bernal, J.L.; Nozal, M.J.; Bernal, J. Trace analysis of sulforaphane in bee pollen and royal jelly by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1012, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Yarita, T.; Aoyagi, Y.; Otake, T. Evaluation of the impact of matrix effect on quantification of pesticides in foods by gas chromatography-mass spectrometry using isotope-labeled internal standards. J. Chromatogr. A 2015, 1396, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Brockbals, L.; Karlsen, M.; Ramsey, J.; Miserez, B. Single injection quantification of cocaine using multiple isotopically labeled internal standards. Forensic Toxicol. 2017, 35, 153–161. [Google Scholar] [CrossRef]
- Mathias, P.C.; Hayden, J.A.; Laha, T.J.; Hoofnagle, A.N. Evaluation of matrix effects using a spike recovery approach in a dilute-and-inject liquid chromatography-tandem mass spectrometry opioid monitoring assay. Clin. Chim. Acta 2014, 437, 38–42. [Google Scholar] [CrossRef]
- Kang, J.; Hick, L.A.; Price, W.E. Using calibration approaches to compensate for remaining matrix effects in quantitative liquid chromatography/electrospray ionization multistage mass spectrometric analysis of phytoestrogens in aqueous environmental samples. Rapid Commun. Mass Spectrom. 2007, 21, 4065–4072. [Google Scholar] [CrossRef] [Green Version]
- Berg, T.; Strand, D.H. 13C labelled internal standards—A solution to minimize ion suppression effects in liquid chromatography–tandem mass spectrometry analyses of drugs in biological samples? J. Chromatogr. A 2011, 1218, 9366–9374. [Google Scholar] [CrossRef]
- Topics, R. The Fitness for Pupose of Analytical Methods. 2014. Available online: https://www.diva-portal.org/smash/record.jsf?pid=diva2%3A948751&dswid=2303 (accessed on 19 May 2020).
- Hewavitharana, A.K. Matrix matching in liquid chromatography–mass spectrometry with stable isotope labelled internal standards—Is it necessary? J. Chromatogr. A 2011, 1218, 359–361. [Google Scholar] [CrossRef]
- Gosetti, F.; Mazzucco, E.; Zampieri, D.; Gennaro, M.C. Signal suppression/enhancement in high-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 3929–3937. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, L.; Zhang, J.; Yang, Y.; Wu, Y.; Shao, B. Simultaneous determination of seven bisphenols in environmental water and solid samples by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. A 2014, 1328, 26–34. [Google Scholar] [CrossRef]
- Ding, J.; Jin, G.; Jin, G.; Shen, A.; Guo, Z.; Yu, B.; Jiao, Y.; Yan, J.; Liang, X. Determination of Underivatized Glyphosate Residues in Plant-Derived Food with Low Matrix Effect by Solid Phase Extraction-Liquid Chromatography-Tandem Mass Spectrometry. Food Anal. Methods 2016, 9, 2856–2863. [Google Scholar] [CrossRef]
- Redruello, B.; Ladero, V.; Del Rio, B.; Fernández, M.; Martin, M.C.; Alvarez, M.A. A UHPLC method for the simultaneous analysis of biogenic amines, amino acids and ammonium ions in beer. Food Chem. 2017, 217, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Gachet, M.S.; Rhyn, P.; Bosch, O.G.; Quednow, B.B.; Gertsch, J. A quantitiative LC-MS/MS method for the measurement of arachidonic acid, prostanoids, endocannabinoids, N-acylethanolamines and steroids in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 976, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Kline, J.A.; Wang, M. Development, validation, and comparison of four methods to simultaneously quantify l-arginine, citrulline, and ornithine in human plasma using hydrophilic interaction liquid chromatography and electrospray tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1005, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Khamis, M.M.; Adamko, D.J.; El-Aneed, A. Development of a validated LC- MS/MS method for the quantification of 19 endogenous asthma/COPD potential urinary biomarkers. Anal. Chim. Acta 2017, 989, 45–58. [Google Scholar] [CrossRef]
- Ongay, S.; Hendriks, G.; Hermans, J.; Van den Berge, M.; Ten Hacken, N.H.; Van de Merbel, N.C.; Bischoff, R. Quantification of free and total desmosine and isodesmosine in human urine by liquid chromatography tandem mass spectrometry: A comparison of the surrogate-analyte and the surrogate-matrix approach for quantitation. J. Chromatogr. A 2014, 1326, 13–19. [Google Scholar] [CrossRef]
- Cequier, E.; Marcé, R.M.; Becher, G.; Thomsen, C. A high-throughput method for determination of metabolites of organophosphate flame retardants in urine by ultra performance liquid chromatography-high resolution mass spectrometry. Anal. Chim. Acta 2014, 845, 98–104. [Google Scholar] [CrossRef]
- Busardò, F.P.; Kyriakou, C.; Marchei, E.; Pacifici, R.; Pedersen, D.S.; Pichini, S. Ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS) for determination of GHB, precursors and metabolites in different specimens: Application to clinical and forensic cases. J. Pharm. Biomed. Anal. 2017, 137, 123–131. [Google Scholar] [CrossRef]
- Zhang, J.; Yasuda, M.; Desnick, R.J.; Balwani, M.; Bishop, D.; Yu, C. A LC-MS/MS method for the specific, sensitive, and simultaneous quantification of 5-aminolevulinic acid and porphobilinogen. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 2389–2396. [Google Scholar] [CrossRef] [Green Version]
- Keski-Rahkonen, P.; Desai, R.; Jimenez, M.; Harwood, D.T.; Handelsman, D.J. Measurement of Estradiol in Human Serum by LC-MS/MS Using a Novel Estrogen-Specific Derivatization Reagent. Anal. Chem. 2015, 87, 7180–7186. [Google Scholar] [CrossRef]
- Steiner, C.; Von Eckardstein, A.; Rentsch, K.M. Quantification of the 15 major human bile acids and their precursor 7α-hydroxy-4-cholesten-3-one in serum by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 2870–2880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, T.; Myint, K.T.; Sato, K.; Wada, O.; Kakiyama, G.; Iida, T.; Hishinuma, T.; Mano, N.; Goto, J. LC/ESI-tandem mass spectrometric determination of bile acid 3-sulfates in human urine 3beta-Sulfooxy-12alpha-hydroxy-5beta-cholanoic acid is an abundant nonamidated sulfate. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 846, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Han, Y.; Neuvonen, M.; Laitila, J.; Neuvonen, P.J.; Niemi, M. High performance liquid chromatography-tandem mass spectrometry for the determination of bile acid concentrations in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 51–60. [Google Scholar] [CrossRef]
- Minato, K.; Suzuki, M.; Nagao, H.; Suzuki, R.; Ochiai, H. Development of analytical method for simultaneous determination of five rodent unique bile acids in rat plasma using ultra-performance liquid chromatography coupled with time-of-flight mass spectrometry. J. Chromatogr. B 2015, 1002, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yeh, C.Y.; Li, K.; Chung-Davidson, Y.W.; Li, W. An UPLC-MS/MS method for quantitative profiling of bile acids in sea lamprey plasma and tissues. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 980, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Bathena, S.P.; Huang, J.; Nunn, M.E.; Miyamoto, T.; Parrish, L.C.; Lang, M.S.; McVaney, T.P.; Toews, M.L.; Cerutis, D.R.; Alnouti, Y. Quantitative determination of lysophosphatidic acids (LPAs) in human saliva and gingival crevicular fluid (GCF) by LC-MS/MS. J. Pharm. Biomed. Anal. 2011, 56, 402–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palandra, J.; Prusakiewicz, J.; Ozer, J.S.; Zhang, Y.; Heath, T.G. Endogenous ethanolamide analysis in human plasma using HPLC tandem MS with electrospray ionization. J. Chromatogr. B 2009, 877, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Liu, F.; Li, X.; Wang, Y.; Gu, X.; Dai, J.; Wang, G.; Cheng, Y.; Yan, C. Fast quantification of endogenous carbohydrates in plasma using hydrophilic interaction liquid chromatography coupled with tandem mass spectrometry. J. Sep. Sci. 2015, 38, 34–41. [Google Scholar] [CrossRef]
- Seegmiller, J.C.; Barnidge, D.R.; Burns, B.E.; Larson, T.S.; Lieske, J.C.; Kumar, R. Quantification of urinary albumin by using protein cleavage and LC-MS/MS. Clin. Chem. 2009, 55, 1100–1107. [Google Scholar] [CrossRef]
- Kamčeva, T.; Bjånes, T.; Svardal, A.; Riedel, B.; Schjøtt, J.; Eide, T. Liquid chromatography/tandem mass spectrometry method for simultaneous quantification of eight endogenous nucleotides and the intracellular gemcitabine metabolite dFdCTP in human peripheral blood mononuclear cells. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1001, 212–220. [Google Scholar] [CrossRef]
- Owen, D.H.; Katz, D.F. A vaginal fluid simulant. Contraception 1999, 59, 91–95. [Google Scholar] [CrossRef]
- Tanaka, T.; Hayashi, Y. Determination of silicon, calcium, magnesium and phosphorus in urine using inductively-coupled plasma emission spectrometry and a matrix-matching technique. Clin. Chim. Acta 1986, 156, 109–113. [Google Scholar] [CrossRef]
- Mirejovsky, D.; Patel, A.S.; Rodriguez, D.D.; Hunt, T.J. Lipid adsorption onto hydrogel contact lens materials. Advantages of Nile red over oil red O in visualization of lipids. Optom. Vis. Sci. 1991, 68, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Oka, K.; Yamamoto, M.; Nonaka, T.; Tomonaga, M. The significance of artificial cerebrospinal fluid as perfusate and endoneurosurgery. Neurosurgery 1996, 38, 733–736. [Google Scholar]
- Mashru, R.C.; Sutariya, V.B.; Sankalia, M.G.; Parikh, P.P. Development and Evaluation of Fast-Dissolving Film of Salbutamol Sulphate. Drug Dev. Ind. Pharm. 2005, 31, 25–34. [Google Scholar] [CrossRef]
- Shama, N.; Bai, S.W.; Chung, B.C.; Jung, B.H. Quantitative analysis of 17 amino acids in the connective tissue of patients with pelvic organ prolapse using capillary electrophoresis-tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 865, 18–24. [Google Scholar] [CrossRef]
- Le, A.; Ng, A.; Kwan, T.; Cusmano-Ozog, K.; Cowan, T.M. A rapid, sensitive method for quantitative analysis of underivatized amino acids by liquid chromatography-tandem mass spectrometry (LC-MS/MS). J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 944, 166–174. [Google Scholar] [CrossRef]
- Zhao, X.E.; Zhu, S.; Yang, H.; You, J.; Song, F.; Liu, Z.; Liu, S. Simultaneous determination of amino acid and monoamine neurotransmitters in PC12 cells and rats models of Parkinson’s disease using a sensitizing derivatization reagent by UHPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 995, 15–23. [Google Scholar] [CrossRef]
- Han, J.; Liu, Y.; Wang, R.; Yang, J.; Ling, V.; Borchers, C.H. Metabolic profiling of bile acids in human and mouse blood by LC-MS/MS in combination with phospholipid-depletion solid-phase extraction. Anal. Chem. 2015, 87, 1127–1136. [Google Scholar] [CrossRef]
- Bobeldijk, I.; Hekman, M.; De Vries-van der Weij, J.; Coulier, L.; Ramaker, R.; Kleemann, R.; Kooistra, T.; Rubingh, C.; Freidig, A.; Verheij, E. Quantitative profiling of bile acids in biofluids and tissues based on accurate mass high resolution LC-FT-MS: Compound class targeting in a metabolomics workflow. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 306–313. [Google Scholar] [CrossRef]
- Montuschi, P.; Martello, S.; Felli, M.; Mondino, C.; Chiarotti, M. Ion trap liquid chromatography/tandem mass spectrometry analysis of leukotriene B4 in exhaled breath condensate. Rapid Commun. Mass Spectrom. Rcm 2004, 18, 2723–2729. [Google Scholar] [CrossRef] [PubMed]
- Squellerio, I.; Porro, B.; Songia, P.; Veglia, F.; Caruso, D.; Tremoli, E.; Cavalca, V. Liquid chromatography-tandem mass spectrometry for simultaneous measurement of thromboxane B2 and 12(S)-hydroxyeicosatetraenoic acid in serum. J. Pharm. Biomed. Anal. 2014, 96, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Sriboonvorakul, N.; Leepipatpiboon, N.; Dondorp, A.M.; Pouplin, T.; White, N.J.; Tarning, J.; Lindegardh, N. Liquid chromatographic-mass spectrometric method for simultaneous determination of small organic acids potentially contributing to acidosis in severe malaria. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 941, 116–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.; Bogan, K.L.; Song, P.; Burant, C.F.; Kennedy, R.T.; Brenner, C. NAD+ metabolite levels as a function of vitamins and calorie restriction: Evidence for different mechanisms of longevity. BMC Chem. Biol. 2010, 10. [Google Scholar] [CrossRef] [Green Version]
- Ohlenbusch, G.; Zwiener, C.; Meckenstock, R.U.; Frimmel, F.H. Identification and quantification of polar naphthalene derivatives in contaminated groundwater of a former gas plant site by liquid chromatography-electrospray ionization tandem mass spectrometry. J. Chromatogr. A 2002, 967, 201–207. [Google Scholar] [CrossRef]
- Sagawa, N.; Takino, T.; Kurogochi, S. A simple method with liquid chromatography/tandem mass spectrometry for the determination of the six trichothecene mycotoxins in rice medium. Biosci. Biotechnol. Biochem. 2006, 70, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Stahnke, H.; Kittlaus, S.; Kempe, G.; Alder, L. Reduction of Matrix Effects in Liquid Chromatography–Electrospray Ionization–Mass Spectrometry by Dilution of the Sample Extracts: How Much Dilution is Needed? Anal. Chem. 2012, 84, 1474–1482. [Google Scholar] [CrossRef]
- Ferrer, C.; Lozano, A.; Agüera, A.; Girón, A.J.; Fernández-Alba, A.R. Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables. J. Chromatogr. A 2011, 1218, 7634–7639. [Google Scholar] [CrossRef]
- Kruve, A.; Leito, I.; Herodes, K. Combating matrix effects in LC/ESI/MS: The extrapolative dilution approach. Anal. Chim. Acta 2009, 651, 75–80. [Google Scholar] [CrossRef]
- Malachová, A.; Sulyok, M.; Beltrán, E.; Berthiller, F.; Krska, R. Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J. Chromatogr. A 2014, 1362, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Deventer, K.; Pozo, O.J.; Verstraete, A.G.; Van Eenoo, P. Dilute-and-shoot-liquid chromatography-mass spectrometry for urine analysis in doping control and analytical toxicology. Trac Trends Anal. Chem. 2014, 55, 1–13. [Google Scholar] [CrossRef]
- Thiry, J.; Evrard, B.; Nys, G.; Fillet, M.; Kok, M.G.M. Sampling only ten microliters of whole blood for the quantification of poorly soluble drugs: Itraconazole as case study. J. Chromatogr. A 2017, 1479, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Cappiello, A.; Famiglini, G.; Pierini, E.; Palma, P.; Trufelli, H. Advanced Liquid Chromatography−Mass Spectrometry Interface Based on Electron Ionization. Anal. Chem. 2007, 79, 5364–5372. [Google Scholar] [CrossRef] [PubMed]
- Cody, R.B.; Laramée, J.A.; Durst, H.D. Versatile New Ion Source for the Analysis of Materials in Open Air under Ambient Conditions. Anal. Chem. 2005, 77, 2297–2302. [Google Scholar] [CrossRef]
- Stahnke, H.; Kittlaus, S.; Kempe, G.; Hemmerling, C.; Alder, L. The influence of electrospray ion source design on matrix effects. J. Mass Spectrom. 2012, 47, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Periat, A.; Kohler, I.; Bugey, A.; Bieri, S.; Versace, F.; Staub, C.; Guillarme, D. Hydrophilic interaction chromatography versus reversed phase liquid chromatography coupled to mass spectrometry: Effect of electrospray ionization source geometry on sensitivity. J. Chromatogr. A 2014, 1356, 211–220. [Google Scholar] [CrossRef]
- Sunner, J.; Nicol, G.; Kebarle, P. Factors determining relative sensitivity of analytes in positive mode atmospheric pressure ionization mass spectrometry. Anal. Chem. 1988, 60, 1300–1307. [Google Scholar] [CrossRef]
- Souverain, S.; Rudaz, S.; Veuthey, J.-L. Matrix effect in LC-ESI-MS and LC-APCI-MS with off-line and on-line extraction procedures. J. Chromatogr. A 2004, 1058, 61–66. [Google Scholar] [CrossRef]
- Barnes, K.A.; Fussell, R.J.; Startin, J.R.; Thorpe, S.A.; Reynolds, S.L. Determination of the pesticides diflubenzuron and clofentezine in plums, strawberries and blackcurrant-based fruit drinks by high performance liquid chromatographic/atmospheric pressure chemical ionization-mass spectrometry. Rapid Commun. Mass Spectrom. 1995, 9, 1441–1445. [Google Scholar] [CrossRef]
- Schuhmacher, J.; Zimmer, D.; Tesche, F.; Pickard, V. Matrix effects during analysis of plasma samples by electrospray and atmospheric pressure chemical ionization mass spectrometry: Practical approaches to their elimination. Rapid Commun. Mass Spectrom. 2003, 17, 1950–1957. [Google Scholar] [CrossRef]
- Vanderford, B.J.; Pearson, R.A.; Rexing, D.J.; Snyder, S.A. Analysis of endocrine disruptors, pharmaceuticals, and personal care products in water using liquid chromatography/tandem mass spectrometry. Anal. Chem. 2003, 75, 6265–6274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, H.; Hsieh, Y.; Nardo, C.; Xu, X.; Wang, S.; Ng, K.; Korfmacher, W.A. Investigation of matrix effects in bioanalytical high-performance liquid chromatography/tandem mass spectrometric assays: Application to drug discovery. Rapid Commun. Mass Spectrom. 2003, 17, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.R.; Foltz, R.L.; Meng, M.; Bennett, P. Ionization enhancement in atmospheric pressure chemical ionization and suppression in electrospray ionization between target drugs and stable-isotope-labeled internal standards in quantitative liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2815–2821. [Google Scholar] [CrossRef]
- Sangster, T.; Spence, M.; Sinclair, P.; Payne, R.; Smith, C. Unexpected observation of ion suppression in a liquid chromatography/atmospheric pressure chemical ionization mass spectrometric bioanalytical method. Rapid Commun. Mass Spectrom. 2004, 18, 1361–1664. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, L.; Tarcomnicu, I.; Rizea, S. Matrix Effect in Mass Spectrometry Combined with Separation Methods. Comparison HPLC, GC and Discussion on Methods to Control These Effects. In Tandem Mass Spectrom: Molecular Characterization; Intech: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]
- Hanold, K.A.; Fischer, S.M.; Cormia, P.H.; Miller, C.E.; Syage, J.A. Atmospheric pressure photoionization. 1. General properties for LC/MS. Anal. Chem. 2004, 76, 2842–2851. [Google Scholar] [CrossRef]
- Hajšlová, J.; Zrostlíková, J. Matrix effects in (ultra)trace analysis of pesticide residues in food and biotic matrices. J. Chromatogr. A 2003, 1000, 181–197. [Google Scholar] [CrossRef]
- Wang, Y.-Q.; Ye, D.-Q.; Zhu, B.-Q.; Wu, G.-F.; Duan, C.-Q. Rapid HPLC analysis of amino acids and biogenic amines in wines during fermentation and evaluation of matrix effect. Food Chem. 2014, 163, 6–15. [Google Scholar] [CrossRef]
- Chambers, E.; Wagrowski-Diehl, D.M.; Lu, Z.; Mazzeo, J.R. Systematic and comprehensive strategy for reducing matrix effects in LC/MS/MS analyses. J. Chromatogr. B 2007, 852, 22–34. [Google Scholar] [CrossRef]
- Marchi, I.; Viette, V.; Badoud, F.; Fathi, M.; Saugy, M.; Rudaz, S.; Veuthey, J.L. Characterization and classification of matrix effects in biological samples analyses. J. Chromatogr. A 2010, 1217, 4071–4078. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.K.; Hercules, D.M.; Gusev, A.I. Effect of liquid chromatography separation of complex matrices on liquid chromatography-tandem mass spectrometry signal suppression. J. Chromatogr. A 2001, 907, 337–342. [Google Scholar] [CrossRef]
- Benijts, T.; Dams, R.; Lambert, W.; De Leenheer, A. Countering matrix effects in environmental liquid chromatography–electrospray ionization tandem mass spectrometry water analysis for endocrine disrupting chemicals. J. Chromatogr. A 2004, 1029, 153–159. [Google Scholar] [CrossRef]
- Van Damme, T.; Lachová, M.; Lynen, F.; Szucs, R.; Sandra, P. Solid-phase extraction based on hydrophilic interaction liquid chromatography with acetone as eluent for eliminating matrix effects in the analysis of biological fluids by LC-MS. Anal. Bioanal. Chem. 2014, 406, 401–407. [Google Scholar] [CrossRef]
- Rezk, M.R.; Bendas, E.R.; Basalious, E.B.; Karim, I.A. Development and validation of sensitive and rapid UPLC-MS/MS method for quantitative determination of daclatasvir in human plasma: Application to a bioequivalence study. J. Pharm. Biomed. Anal. 2016, 128, 61–66. [Google Scholar] [CrossRef] [PubMed]
- González-Antuña, A.; Domínguez-Romero, J.C.; García-Reyes, J.F.; Rodríguez-González, P.; Centineo, G.; García Alonso, J.I.; Molina-Díaz, A. Overcoming matrix effects in electrospray: Quantitation of β-agonists in complex matrices by isotope dilution liquid chromatography-mass spectrometry using singly (13)C-labeled analogues. J. Chromatogr. A 2013, 1288, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Liu, Q.; Shi, J.-b.; Wei, F.-s.; Jiang, G.-b. Direct analysis of eight chlorophenols in urine by large volume injection online turbulent flow solid-phase extraction liquid chromatography with multiple wavelength ultraviolet detection. Talanta 2014, 119, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Naidong, W. Bioanalytical liquid chromatography tandem mass spectrometry methods on underivatized silica columns with aqueous/organic mobile phases. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 796, 209–224. [Google Scholar] [CrossRef]
- Xu, R.N.; Fan, L.; Rieser, M.J.; El-Shourbagy, T.A. Recent advances in high-throughput quantitative bioanalysis by LC–MS/MS. J. Pharm. Biomed. Anal. 2007, 44, 342–355. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Schug, K.A. The advantages of ESI-MS detection in conjunction with HILIC mode separations: Fundamentals and applications. J. Sep. Sci. 2008, 31, 1465–1480. [Google Scholar] [CrossRef]
- Olesti, E.; Rodríguez-Morató, J.; Gomez-Gomez, A.; Ramaekers, J.G.; De la Torre, R.; Pozo, O.J. Quantification of endogenous neurotransmitters and related compounds by liquid chromatography coupled to tandem mass spectrometry. Talanta 2019, 192, 93–102. [Google Scholar] [CrossRef]
- Nemoto, T.; Lee, X.P.; Kumazawa, T.; Hasegawa, C.; Fujishiro, M.; Marumo, A.; Shouji, Y.; Inagaki, K.; Sato, K. High-throughput determination of nonsteroidal anti-inflammatory drugs in human plasma by HILIC-MS/MS. J. Pharm. Biomed. Anal. 2014, 88, 71–80. [Google Scholar] [CrossRef]
- Vilhena Rde, O.; Pontes, F.L.; Marson, B.M.; Ribeiro, R.P.; De Carvalho, K.A.; Cardoso, M.A.; Pontarolo, R. A new HILIC-MS/MS method for the simultaneous analysis of carbidopa, levodopa, and its metabolites in human plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 967, 41–49. [Google Scholar] [CrossRef]
- Isokawa, M.; Kanamori, T.; Funatsu, T.; Tsunoda, M. Recent advances in hydrophilic interaction chromatography for quantitative analysis of endogenous and pharmaceutical compounds in plasma samples. Bioanalysis 2014, 6, 2421–2439. [Google Scholar] [CrossRef] [PubMed]
- Pontarolo, R.; Gimenez, A.C.; De Francisco, T.M.G.; Ribeiro, R.P.; Pontes, F.L.D.; Gasparetto, J.C. Simultaneous determination of metformin and vildagliptin in human plasma by a HILIC-MS/MS method. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 965, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.L.; Lipman, J.; Roberts, J.A.; Wallis, S.C. A simple LC-MS/MS method using HILIC chromatography for the determination of fosfomycin in plasma and urine: Application to a pilot pharmacokinetic study in humans. J. Pharm. Biomed. Anal. 2015, 105, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meulder, M.; Waldron, M.P.; Li, L.; Peay, M.G.; Tingler, M.J.; Hidy, B.J.; Verhaeghe, T.; Jenkins, R.G. Development and validation of HILIC-ESI/MS/MS methods for simultaneous quantitation of several antipsychotics in human plasma and blood. Bioanalysis 2016, 8, 765–794. [Google Scholar] [CrossRef] [PubMed]
- Periat, A.; Kohler, I.; Thomas, A.; Nicoli, R.; Boccard, J.; Veuthey, J.L.; Schappler, J.; Guillarme, D. Systematic evaluation of matrix effects in hydrophilic interaction chromatography versus reversed phase liquid chromatography coupled to mass spectrometry. J. Chromatogr. A 2016, 1439, 42–53. [Google Scholar] [CrossRef]
- Mess, J.N.; Côté, C.; Bergeron, A.; Furtado, M.; Garofolo, F. Selection of HILIC columns to handle matrix effect due to phospholipids. Bioanalysis 2009, 1, 57–62. [Google Scholar] [CrossRef]
- Ha, J.; Seo, H.-Y.; Shim, Y.-S.; Seo, D.-W.; Seog, H.; Ito, M.; Nakagawa, H. Determination of capsaicinoids in foods using ultra high performance liquid chromatography. Food Sci. Biotechnol. 2010, 19, 1005–1009. [Google Scholar] [CrossRef]
- Vierikova, M.; Germuska, R.; Lehotay, J. Determination of Coumarin in Food Using Ultra-Performance Liquid Chromatography–Electrospray-Tandem Mass Spectrometry. J. Liq. Chromatogr. Relat. Technol. 2008, 32, 95–105. [Google Scholar] [CrossRef]
- Huang, L.C.; Zheng, N.; Zheng, B.Q.; Wen, F.; Cheng, J.B.; Han, R.W.; Xu, X.M.; Li, S.L.; Wang, J.Q. Simultaneous determination of aflatoxin M1, ochratoxin A, zearalenone and α-zearalenol in milk by UHPLC-MS/MS. Food Chem. 2014, 146, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, E.; Ibáñez, M.; Sancho, J.V.; Hernández, F. Determination of patulin in apple and derived products by UHPLC-MS/MS. Study of matrix effects with atmospheric pressure ionisation sources. Food Chem. 2014, 142, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, P. Rapid screening of mycotoxins in liquid milk and milk powder by automated size-exclusion SPE-UPLC-MS/MS and quantification of matrix effects over the whole chromatographic run. Food Chem. 2015, 173, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Tian, Y.; Mi, J.; Wang, J.; Xu, Y. Simultaneous determination and pharmacokinetic study of eight components in rat plasma by UHPLC-MS/MS after oral administration of Hypericum japonicum Thunb extract. J. Pharm. Biomed. Anal. 2016, 118, 228–234. [Google Scholar] [CrossRef]
- Bedani, F.; Schoenmakers, P.J.; Janssen, H.G. Theories to support method development in comprehensive two-dimensional liquid chromatography--a review. J. Sep. Sci. 2012, 35, 1697–1711. [Google Scholar] [CrossRef] [PubMed]
- Guiochon, G.; Beaver, L.A.; Gonnord, M.F.; Siouffi, A.M.; Zakaria, M. Theoretical investigation of the potentialities of the use of a multidimensional column in chromatography. J. Chromatogr. A 1983, 255, 415–437. [Google Scholar] [CrossRef]
- Leonhardt, J.; Teutenberg, T.; Buschmann, G.; Gassner, O.; Schmidt, T.C. A new method for the determination of peak distribution across a two-dimensional separation space for the identification of optimal column combinations. Anal. Bioanal. Chem. 2016, 408, 8079–8088. [Google Scholar] [CrossRef]
- Česla, P.; Fischer, J.; Jandera, P. Separation of phenolic acids and flavone natural antioxidants by two-dimensional method combining liquid chromatography and micellar electrokinetic capillary chromatography. Electrophoresis 2010, 31, 2200–2210. [Google Scholar] [CrossRef] [Green Version]
- Stoll, D.R.; Talus, E.S.; Harmes, D.C.; Zhang, K. Evaluation of detection sensitivity in comprehensive two-dimensional liquid chromatography separations of an active pharmaceutical ingredient and its degradants. Anal. Bioanal. Chem. 2015, 407, 265–277. [Google Scholar] [CrossRef]
- Stoll, D.R.; Sajulga, R.W.; Voigt, B.N.; Larson, E.J.; Jeong, L.N.; Rutan, S.C. Simulation of elution profiles in liquid chromatography—II: Investigation of injection volume overload under gradient elution conditions applied to second dimension separations in two-dimensional liquid chromatography. J. Chromatogr. A 2017, 1523, 162–172. [Google Scholar] [CrossRef]
- Raterink, R.-J.; Lindenburg, P.W.; Vreeken, R.J.; Ramautar, R.; Hankemeier, T. Recent developments in sample-pretreatment techniques for mass spectrometry-based metabolomics. Trac Trends Anal. Chem. 2014, 61, 157–167. [Google Scholar] [CrossRef]
- Moein, M.M.; El Beqqali, A.; Abdel-Rehim, M. Bioanalytical method development and validation: Critical concepts and strategies. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1043, 3–11. [Google Scholar] [CrossRef]
- Guo, J.; Zhao, Y.; Zhao, L.; Zhang, W.; Zhang, A.; Xu, B. Simultaneous quantification of CTN986 and its deglycosylation products in rat serum using liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. Rcm 2006, 20, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Chen, X.; Zhong, D. Stability of baicalin in biological fluids in vitro. J. Pharm. Biomed. Anal. 2005, 39, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.Y.; Lee, H.W.; Kim, H.K.; Kim, H.H.; Chang, S.G.; Sohn, D.H.; Kim, J.; Lee, H.S. Simultaneous determination of ginsenoside Rb(1) and Rg(1) in human plasma by liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2004, 35, 207–212. [Google Scholar] [CrossRef]
- Moreno-González, D.; Rodríguez-Ramírez, R.; Del Olmo-Iruela, M.; García-Campaña, A.M. Validation of a new method based on salting-out assisted liquid-liquid extraction and UHPLC-MS/MS for the determination of betalactam antibiotics in infant dairy products. Talanta 2017, 167, 493–498. [Google Scholar] [CrossRef]
- Li, T.; Zhang, L.; Tong, L.; Liao, Q. High-throughput salting-out-assisted homogeneous liquid-liquid extraction with acetonitrile for determination of baicalin in rat plasma with high-performance liquid chromatography. Biomed. Chromatogr. 2014, 28, 648–653. [Google Scholar] [CrossRef]
- Bakhtiar, R.; Majumdar, T.K. Tracking problems and possible solutions in the quantitative determination of small molecule drugs and metabolites in biological fluids using liquid chromatography-mass spectrometry. J. Pharmacol. Toxicol. Methods 2007, 55, 262–278. [Google Scholar] [CrossRef]
- Jiang, H.; Cao, H.; Zhang, Y.; Fast, D.M. Systematic evaluation of supported liquid extraction in reducing matrix effect and improving extraction efficiency in LC-MS/MS based bioanalysis for 10 model pharmaceutical compounds. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 891, 71–80. [Google Scholar] [CrossRef]
- Ismaiel, O.A.; Zhang, T.; Jenkins, R.G.; Karnes, H.T. Investigation of endogenous blood plasma phospholipids, cholesterol and glycerides that contribute to matrix effects in bioanalysis by liquid chromatography/mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2010, 878, 3303–3316. [Google Scholar] [CrossRef]
- Xing, J.; Xie, C.; Lou, H. Recent applications of liquid chromatography–mass spectrometry in natural products bioanalysis. J. Pharm. Biomed. Anal. 2007, 44, 368–378. [Google Scholar] [CrossRef]
- Tan, Z.-R.; Ouyang, D.-S.; Zhou, G.; Wang, L.-S.; Li, Z.; Wang, D.; Zhou, H.-H. Sensitive bioassay for the simultaneous determination of pseudoephedrine and cetirizine in human plasma by liquid-chromatography–ion trap spectrometry. J. Pharm. Biomed. Anal. 2006, 42, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Little, J.L.; Wempe, M.F.; Buchanan, C.M. Liquid chromatography-mass spectrometry/mass spectrometry method development for drug metabolism studies: Examining lipid matrix ionization effects in plasma. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 833, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Bylda, C.; Thiele, R.; Kobold, U.; Volmer, D.A. Simultaneous quantification of digoxin, digitoxin, and their metabolites in serum using high performance liquid chromatography-tandem mass spectrometry. Drug Test. Anal. 2015, 7, 937–946. [Google Scholar] [CrossRef]
- Salatti-Dorado, J.Á.; Caballero-Casero, N.; Sicilia, M.D.; Lunar, M.L.; Rubio, S. The use of a restricted access volatile supramolecular solvent for the LC/MS-MS assay of bisphenol A in urine with a significant reduction of phospholipid-based matrix effects. Anal. Chim. Acta 2017, 950, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Baduel, C.; Mueller, J.F.; Tsai, H.; Gomez Ramos, M.J. Development of sample extraction and clean-up strategies for target and non-target analysis of environmental contaminants in biological matrices. J. Chromatogr. A 2015, 1426, 33–47. [Google Scholar] [CrossRef]
- Carmical, J.; Brown, S. The impact of phospholipids and phospholipid removal on bioanalytical method performance. Biomed. Chromatogr. 2016, 30, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Teo, C.C.; Chong, W.P.K.; Tan, E.; Basri, N.B.; Low, Z.J.; Ho, Y.S. Advances in sample preparation and analytical techniques for lipidomics study of clinical samples. Trac Trends Anal. Chem. 2015, 66, 1–18. [Google Scholar] [CrossRef]
- Murphy, R.C.; Fiedler, J.; Hevko, J. Analysis of nonvolatile lipids by mass spectrometry. Chem. Rev. 2001, 101, 479–526. [Google Scholar] [CrossRef]
- Pulfer, M.; Murphy, R.C. Electrospray mass spectrometry of phospholipids. Mass Spectrom. Rev. 2003, 22, 332–364. [Google Scholar] [CrossRef]
- Xia, Y.Q.; Jemal, M. Phospholipids in liquid chromatography/mass spectrometry bioanalysis: Comparison of three tandem mass spectrometric techniques for monitoring plasma phospholipids, the effect of mobile phase composition on phospholipids elution and the association of phospholipids with matrix effects. Rapid Commun. Mass Spectrom. Rcm 2009, 23, 2125–2138. [Google Scholar] [CrossRef]
- Pucci, V.; Di Palma, S.; Alfieri, A.; Bonelli, F.; Monteagudo, E. A novel strategy for reducing phospholipids-based matrix effect in LC-ESI-MS bioanalysis by means of HybridSPE. J. Pharm. Biomed. Anal. 2009, 50, 867–871. [Google Scholar] [CrossRef]
- Ahmad, S.; Kalra, H.; Gupta, A.; Raut, B.; Hussain, A.; Rahman, M.A. HybridSPE: A novel technique to reduce phospholipid-based matrix effect in LC-ESI-MS Bioanalysis. J. Pharm. Bioallied Sci. 2012, 4, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Danaceau, J.; Yu, H.; Chambers, E.; Fountain, K.J. Matrix effects in metabolite quantification for MIST assessment: The impact of phospholipid removal and HPLC column particle size. Bioanalysis 2014, 6, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Tulipani, S.; Mora-Cubillos, X.; Jáuregui, O.; Llorach, R.; García-Fuentes, E.; Tinahones, F.J.; Andres-Lacueva, C. New and vintage solutions to enhance the plasma metabolome coverage by LC-ESI-MS untargeted metabolomics: The not-so-simple process of method performance evaluation. Anal. Chem. 2015, 87, 2639–2647. [Google Scholar] [CrossRef] [PubMed]
- Ask, K.S.; Bardakci, T.; Parmer, M.P.; Halvorsen, T.G.; Øiestad, E.L.; Pedersen-Bjergaard, S.; Gjelstad, A. Parallel artificial liquid membrane extraction as an efficient tool for removal of phospholipids from human plasma. J. Pharm. Biomed. Anal. 2016, 129, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Neville, D.; Houghton, R.; Garrett, S. Efficacy of plasma phospholipid removal during sample preparation and subsequent retention under typical UHPLC conditions. Bioanalysis 2012, 4, 795–807. [Google Scholar] [CrossRef]
- Zhang, D.; Park, J.-A.; Kim, S.-K.; Cho, S.-H.; Jeong, D.; Cho, S.-M.; Yi, H.; Shim, J.-H.; Kim, J.-S.; Abd El-Aty, A.M.; et al. Simultaneous detection of flumethasone, dl-methylephedrine, and 2-hydroxy-4,6-dimethylpyrimidine in porcine muscle and pasteurized cow milk using liquid chromatography coupled with triple-quadrupole mass spectrometry. J. Chromatogr. B 2016, 1012, 8–16. [Google Scholar] [CrossRef]
- Chiesa, L.M.; Nobile, M.; Biolatti, B.; Pavlovic, R.; Panseri, S.; Cannizzo, F.T.; Arioli, F. Detection of selected corticosteroids and anabolic steroids in calf milk replacers by liquid chromatography–electrospray ionisation – Tandem mass spectrometry. Food Control 2016, 61, 196–203. [Google Scholar] [CrossRef]
- Yoon, S.H.; Kim, M.S.; Kim, S.H.; Park, H.M.; Pyo, H.; Lee, Y.M.; Lee, K.T.; Hong, J. Effective application of freezing lipid precipitation and SCX-SPE for determination of pyrrolizidine alkaloids in high lipid foodstuffs by LC-ESI-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 992, 56–66. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, R.; Jiménez-Díaz, I.; Zafra-Gómez, A.; Ballesteros, O.; Navalón, A. A multiresidue method for the determination of selected endocrine disrupting chemicals in human breast milk based on a simple extraction procedure. Talanta 2014, 130, 561–570. [Google Scholar] [CrossRef] [PubMed]
- ISO. Heat Treated Milk-Determination of Lactulose Content—Method Using High-Performance Liquid Chromatography. Available online: https://www.iso.org/standard/44510.html (accessed on 21 April 2020).
- Rodríguez-Gómez, R.; Dorival-García, N.; Zafra-Gómez, A.; Camino-Sánchez, F.J.; Ballesteros, O.; Navalón, A. New method for the determination of parabens and bisphenol A in human milk samples using ultrasound-assisted extraction and clean-up with dispersive sorbents prior to UHPLC–MS/MS analysis. J. Chromatogr. B 2015, 992, 47–55. [Google Scholar] [CrossRef]
- Alcántara-Durán, J.; Moreno-González, D.; García-Reyes, J.F.; Molina-Díaz, A. Use of a modified QuEChERS method for the determination of mycotoxin residues in edible nuts by nano flow liquid chromatography high resolution mass spectrometry. Food Chem. 2019, 279, 144–149. [Google Scholar] [CrossRef]
- Rejczak, T.; Tuzimski, T. QuEChERS-based extraction with dispersive solid phase extraction clean-up using PSA and ZrO2-based sorbents for determination of pesticides in bovine milk samples by HPLC-DAD. Food Chem. 2017, 217, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Tomasini, D.; Sampaio, M.R.F.; Caldas, S.S.; Buffon, J.G.; Duarte, F.A.; Primel, E.G. Simultaneous determination of pesticides and 5-hydroxymethylfurfural in honey by the modified QuEChERS method and liquid chromatography coupled to tandem mass spectrometry. Talanta 2012, 99, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Irungu, J.; Raina, S.; Torto, B. Determination of pesticide residues in honey: A preliminary study from two of Africa’s largest honey producers. Int. J. Food Contam. 2016, 3, 14. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zhang, J.; Zhao, W.; Zhang, W.; Wang, L.; Zhou, J.; Li, Y. Development and validation of a multiclass method for the quantification of veterinary drug residues in honey and royal jelly by liquid chromatography–tandem mass spectrometry. Food Chem. 2017, 221, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Schlittenbauer, L.; Seiwert, B.; Reemtsma, T. Matrix effects in human urine analysis using multi-targeted liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2015, 1415, 91–99. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Method | Description of the Method | Limits | References |
|---|---|---|---|
| Post-column infusion method | The post-column infusion method provides a qualitative assessment of matrix effects. It permits the identification of the retention time zones in a chromatographic plot most likely to experience phenomena of ion enhancement or suppression. It consists in a constant flow through the LC-MS column of the mobile phase or blank, and the post column injection through a T-piece of the analyte standard. ME can be assessed as suppression or enhancement of the analyte signal in specific regions of the chromatogram. |
| [7,10,12,25,29,30,31,32,33,34,35,36,37,38,39] |
| Post-extraction spike method | In the post-extraction spike method, the response of the analyte in a standard solution is compared to that of the analyte spiked into a blank matrix sample at the same concentration. Deviations from the responses of the two solutions are identified as ion enhancement or suppression. This method is able to provide a quantitative assessment of matrix effect. |
| [26,40,41,42,43,44] |
| Slope Ratio Analysis | It allows a semi-quantitative screening of matrix effect. It exploits spiked samples and matrix-matched calibration standards at different calibration levels. This modified approach evaluates the same parameters obtained by post-extracion addition method in an entire selected range of concentrations instead of a single level. |
| [28] |
| Relative MEs evaluation | It permits the evaluation of the variability of MEs lot by lot |
| [3,26,45,46,47,48,49] |
| Method | Theory or Mechanism | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Matrix-matched calibration | External calibration presupposes the preparation of several samples from blank matrix spiked at different analyte concentrations before injection with linear calibrations calculated for each analyte. |
|
| [46,57] |
| Isotope labeled internal standard | The use of internal standard implies the use of a substance with identical or similar ionization properties and very close retention time to that of the analyte. According to literature 13C-labelled IS mimes better than 2H-labelled IS the target analyte. |
|
| [58,59,60,61,62,63,64,65] |
| Method | Theory or Mechanism | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Standard addition | It requires that the analyte be spiked in same sample extract at different concentration levels. |
|
| [23,67,68] |
| Background subtraction | The calibration curve is built by subtracting the background. |
|
| [23,66,69,70] |
| Surrogate matrix | It exploits surrogate matrixes such as neat solvent, stripped and artificial matrixes, that act as a blank-like matrix. |
|
| [23,55,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99] |
| Surrogate analyte method | It requires stable-isotope-labeled standard as a surrogate analyte to allow calibration. |
|
| [23,72,100] |
| Matrix | Technique | Effectiveness | References |
|---|---|---|---|
| Infant food, plasma | Salting out | Residual ion suppression in ESI | [162,163] |
| Plasma, Urine | Precipitation with organic solvents | Not effective for other interference removal like phospholipids, lipids, aminoacids | [3,12,125,129,164,165,166] |
| Plasma, serum | ZnSO4 solution | Effective for PPT and decrease the phospholipids amount | [169,170] |
| Urine | Restricted access, volatile supramolecular solvents (RAM-VOL-SUPRAS) | Avoids or dramatically reduces the ME. | [171] |
| Fish muscle- Breast Milk | Protein-lipid removal filter cartridges (Captiva ND Lipids) | The best option to reduce ME in LC-MS applications | [172] |
| Matrix | Technique | Effectiveness | References |
|---|---|---|---|
| Plasma | PPT | No extensive removal of PLs—ion suppression for many analytes | [125] |
| Plasma | Liquid-liquid extraction | Clean final extract but unacceptable analyte recovery especially for polar analytes | [125] |
| Plasma | Pure cationic exchange solid-phase extraction (SPE) | Cleaner extracts and reduced matrix effects compared to PPT | [125] |
| Plasma | Reversed-phase SPE | Cleaner extracts and reduced matrix effects compared to PPT | [125] |
| Plasma | Mixed-mode SPE | Best effectiveness because it combines the retention mechanisms of reverse-phase and ion exchange | [125] |
| Plasma | Solid supported liquid extraction (SLE) | Able to remove the majority of PLs through appropriate loading of buffers and eluting solvents | [165] |
| Plasma | Hybrid SPE-PPT(HybridSPE™, Ostro™, Phenomenex Phree™, Captiva™) | Effectively reduces the ME in biological samples | [130,178,179,180,181] |
| Urine | Restricted access, volatile supramolecular solvents (RAM-VOL-SUPRAS) | Avoids or dramatically reduces the ME. | [171] |
| Human plasma | Parallel Artificial Liquid Membrane Extraction (PALME) | Complete absence of ion suppression and high recovery percentage | [182] |
| Matrix | Technique | Effectiveness | References |
|---|---|---|---|
| Porcine muscle-milk | LLE with hexane | [184,185] | |
| High lipid foodstuff | Freezing lipid precipitation (LFP) | Effective sample clean-up | [186] |
| Milk | Lipid precipitation by zinc and tungsten salt solutions in acidic media | Good sensitivity | [187,188] |
| Milk | Dispersive solid phase extraction (d-SPE) by using a combination of PSA-C18 solid sorbent or PSA- Zirconium-dioxide | Very effective ME reduction | [189,191] |
| Fish muscle- Breast Milk | Protein-lipid removal filter cartridges (Captiva ND Lipids) | The best option to reduce ME in LC-MS applications. Effective also in protein removal | [172] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortese, M.; Gigliobianco, M.R.; Magnoni, F.; Censi, R.; Di Martino, P. Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review. Molecules 2020, 25, 3047. https://doi.org/10.3390/molecules25133047
Cortese M, Gigliobianco MR, Magnoni F, Censi R, Di Martino P. Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review. Molecules. 2020; 25(13):3047. https://doi.org/10.3390/molecules25133047
Chicago/Turabian StyleCortese, Manuela, Maria Rosa Gigliobianco, Federico Magnoni, Roberta Censi, and Piera Di Martino. 2020. "Compensate for or Minimize Matrix Effects? Strategies for Overcoming Matrix Effects in Liquid Chromatography-Mass Spectrometry Technique: A Tutorial Review" Molecules 25, no. 13: 3047. https://doi.org/10.3390/molecules25133047