Proteomic Profiling of Emiliania huxleyi Using a Three-Dimensional Separation Method Combined with Tandem Mass Spectrometry

 ,
,  ,
,
Abstract
:
1. Introduction
2. Results
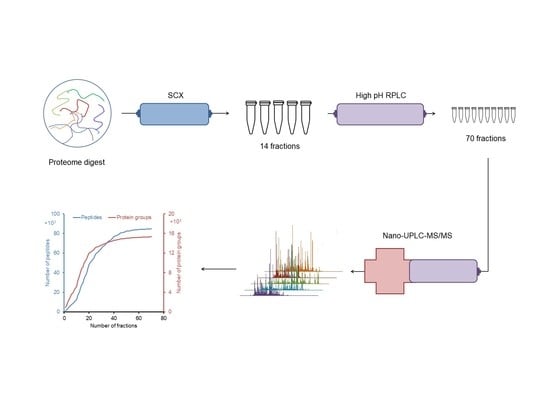
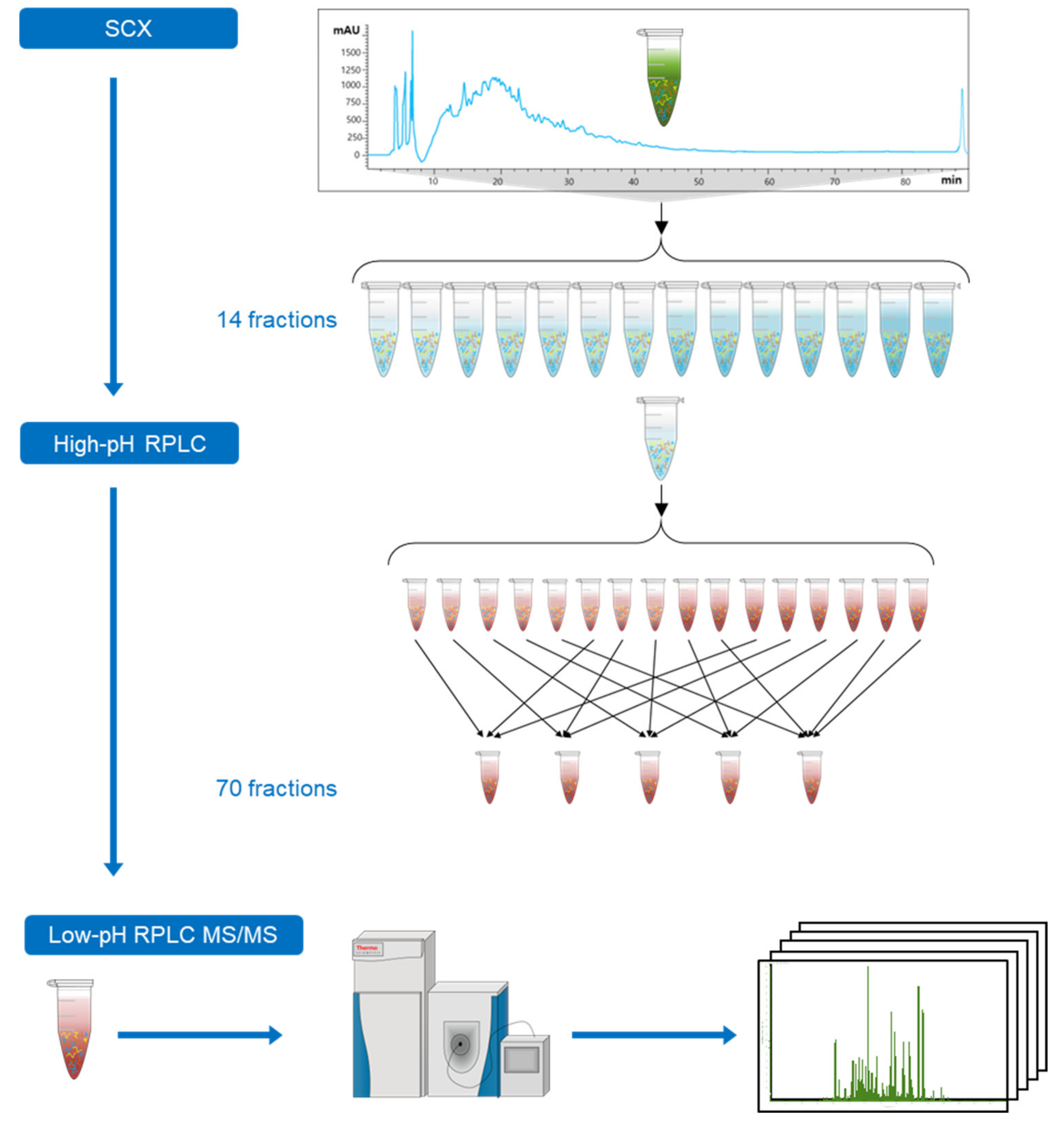
2.1. Design of an Off-line 3D-LC (SCX-HpH RPLC-LpH RPLC) System
2.2. Proteomic Analysis of E. huxleyi Using 3D-LC System
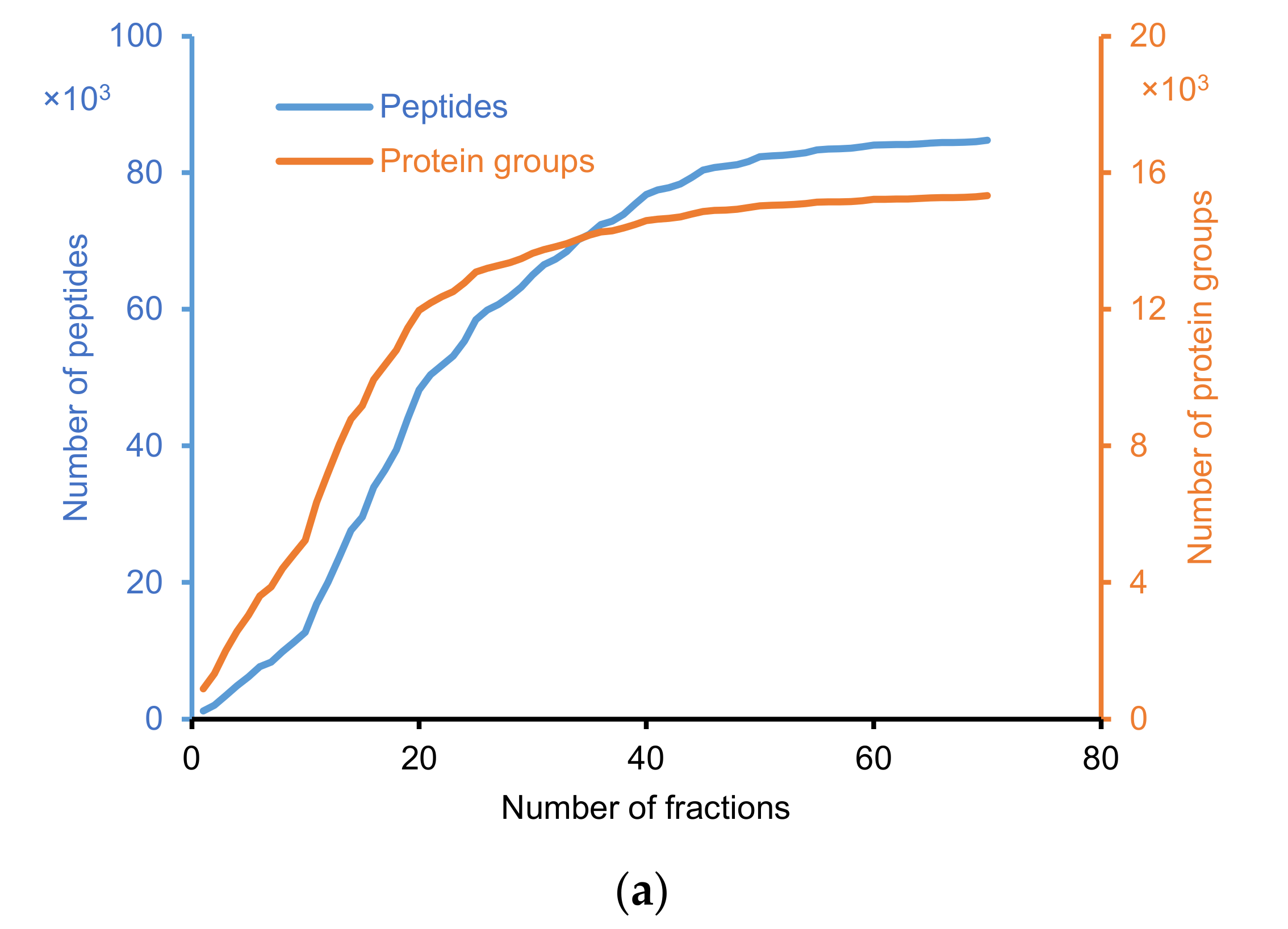
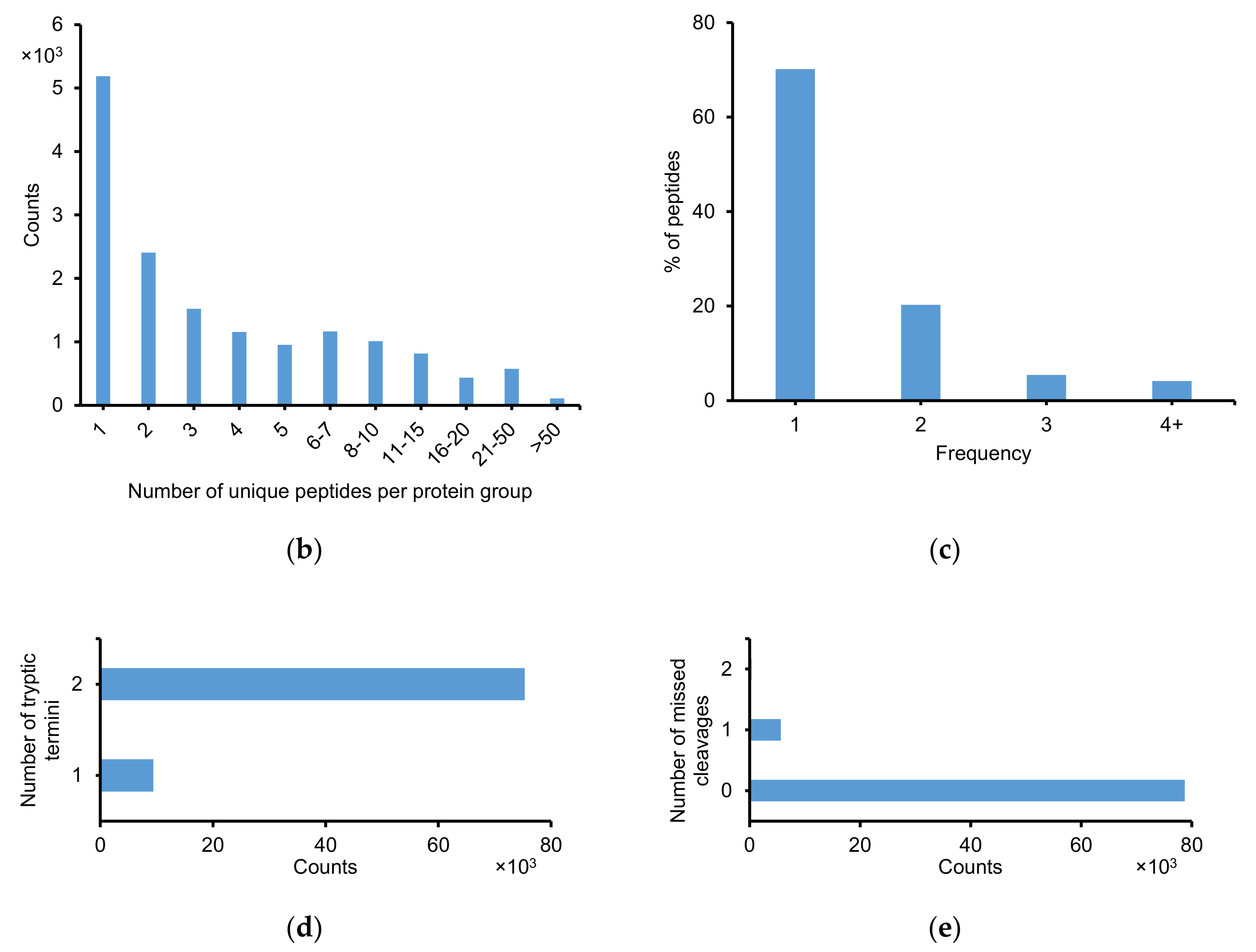
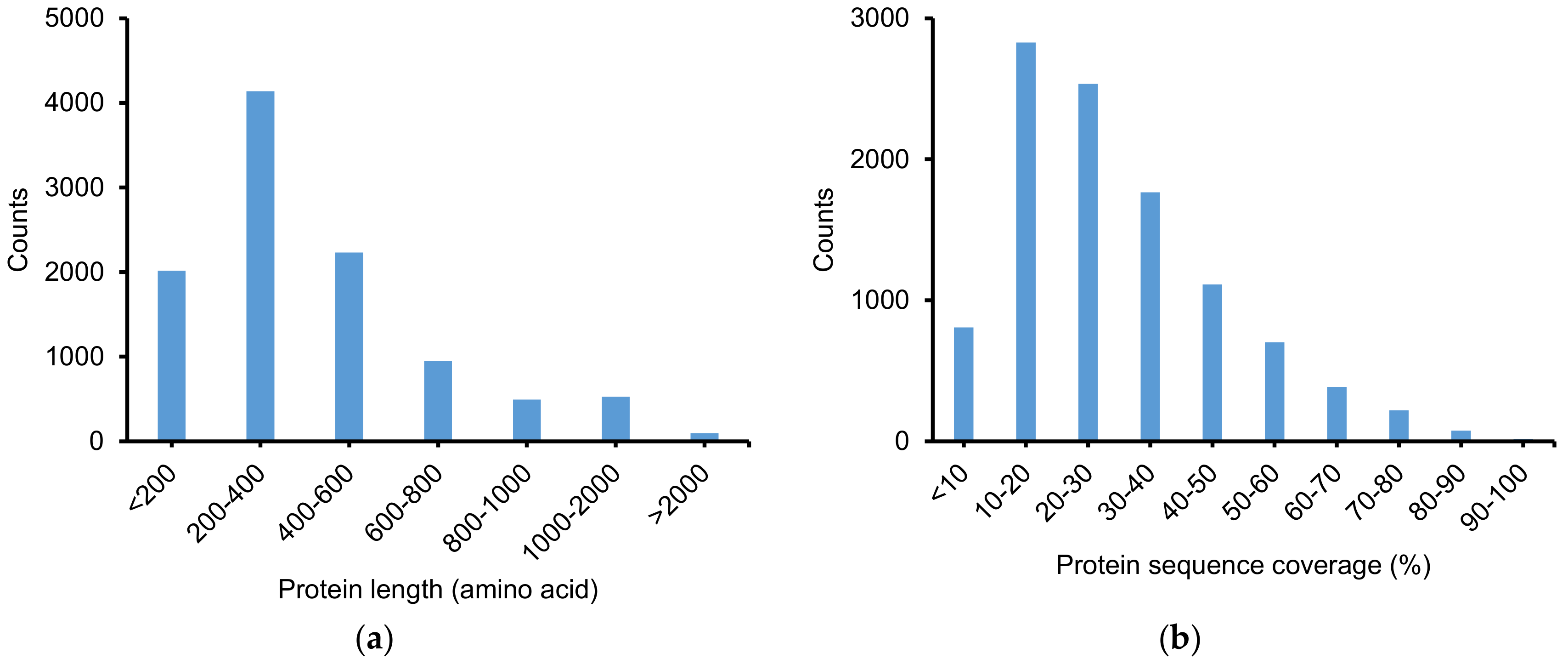
2.2.1. Identification of Peptide and Protein Group
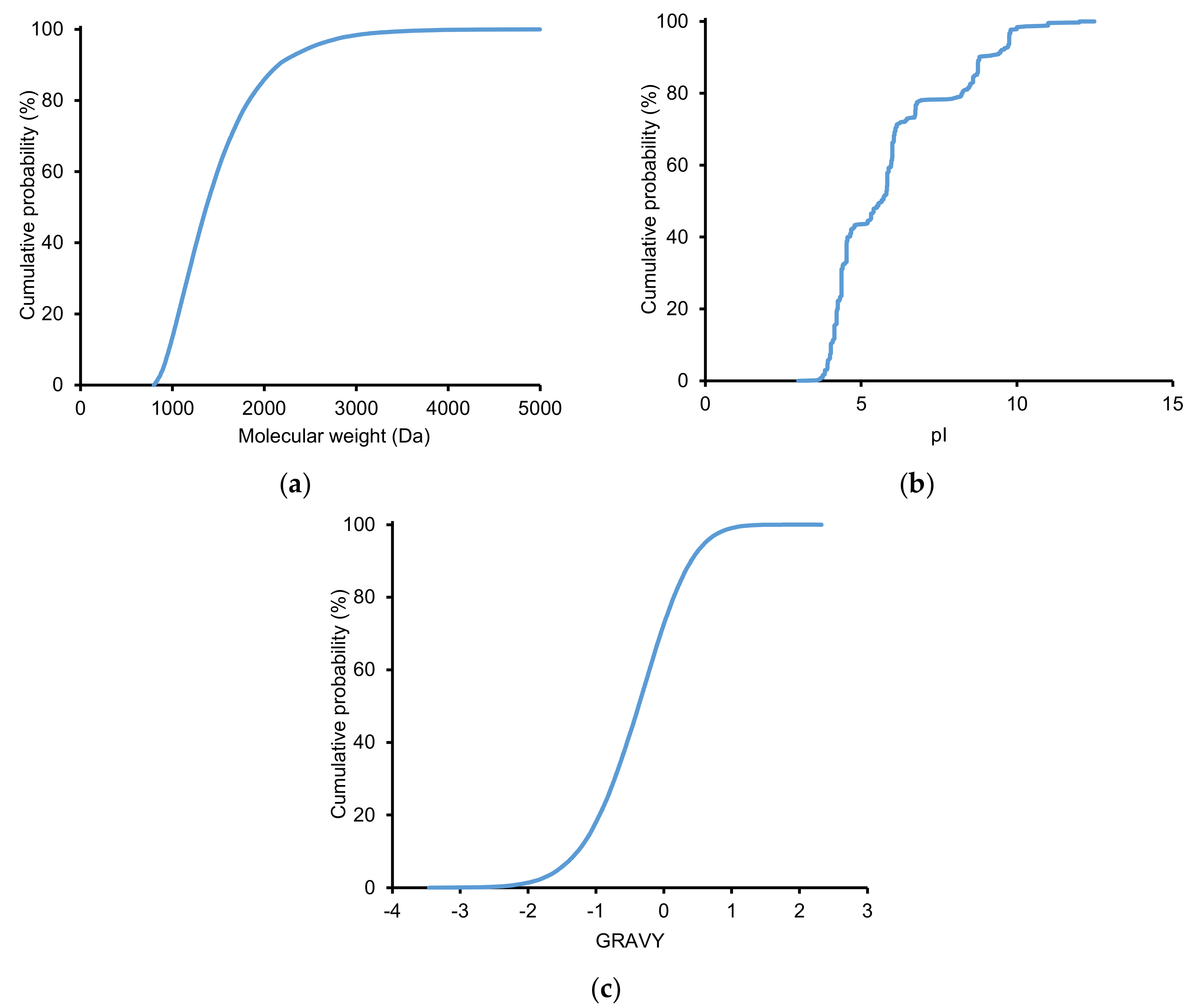
2.2.2. Physicochemical Properties of Identified Peptides
2.2.3. Effects of Strong Cation Exchange (SCX) on Peptide Separation
2.2.4. Protein Identification Using ProteinProphet
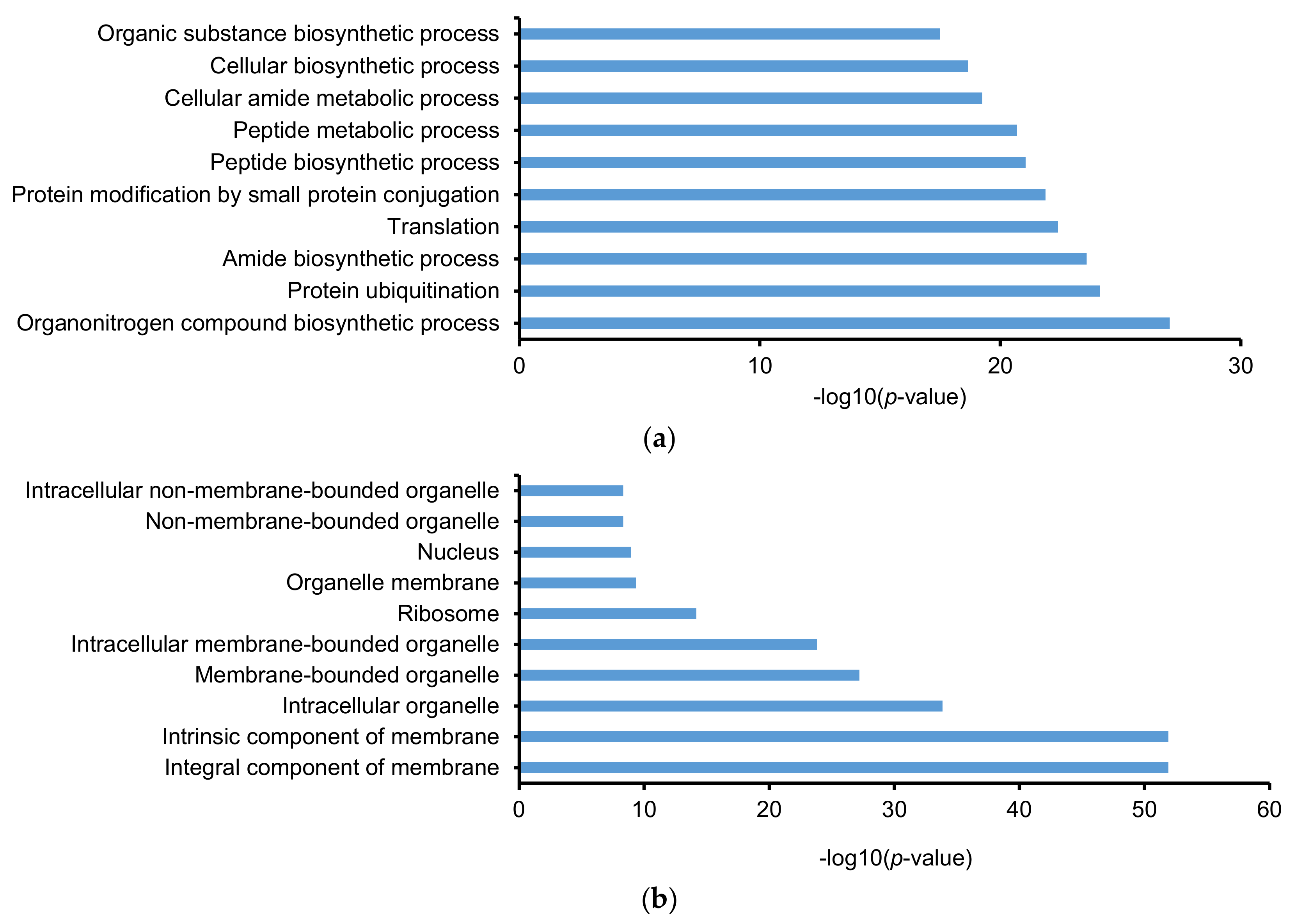
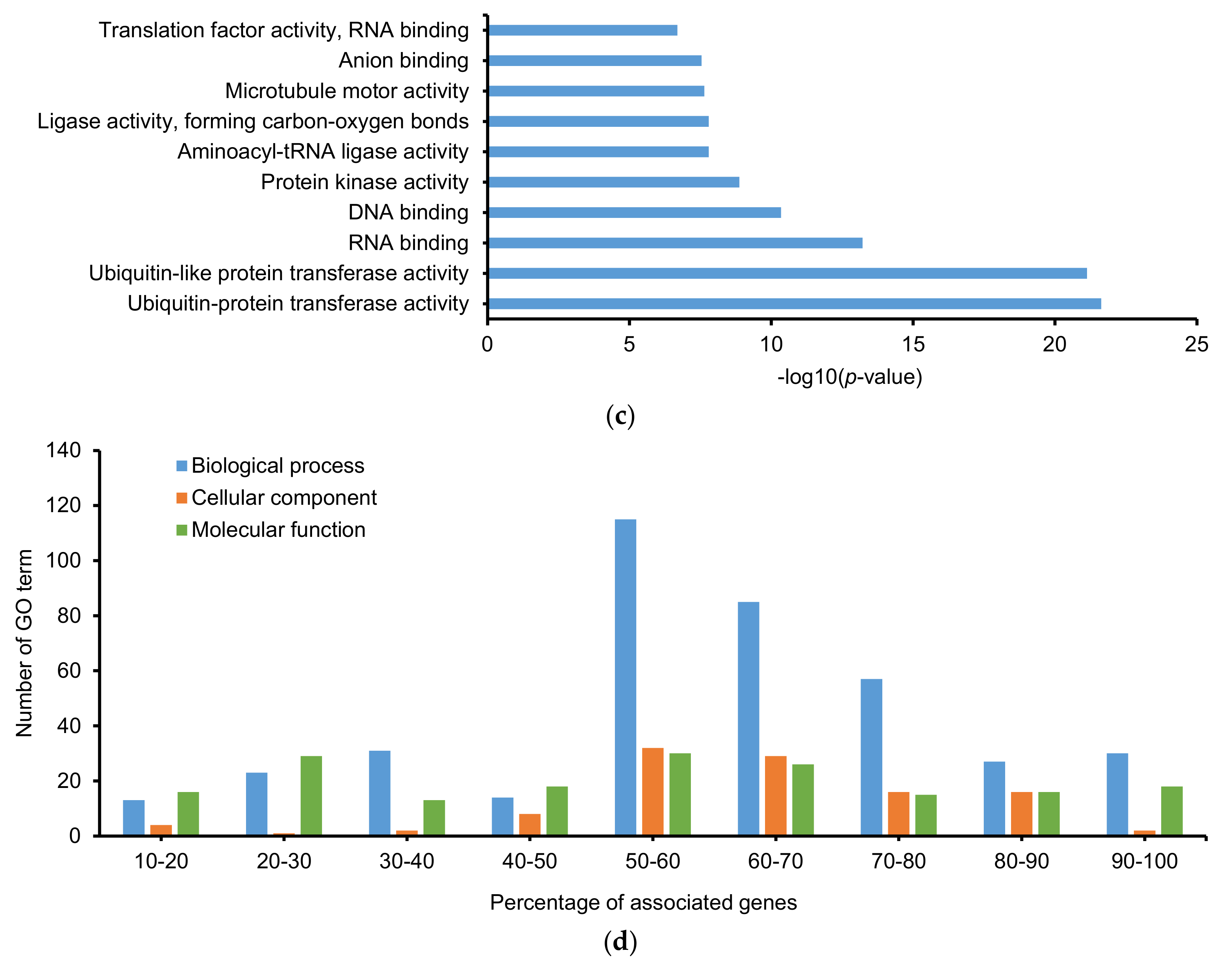
2.3. Gene Ontology (GO) and Kyoto Encyclopedia of Gene and Genomes (KEGG)
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Sample Preparation
4.3.1. Protein Extraction
4.3.2. Automated Filter-Aided Sample Preparation (FASP)
4.3.3. Desalting with Reverse-Phase Solid-Phase Extraction
4.3.4. SCX Fractionation
4.3.5. HpH RPLC Fractionation
4.4. LC–MS/MS Analysis
4.5. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cubillos, J.; Wright, S.; Nash, G.; De Salas, M.; Griffiths, B.; Tilbrook, B.; Poisson, A.; Hallegraeff, G. Calcification morphotypes of the coccolithophorid Emiliania huxleyi in the Southern Ocean: changes in 2001 to 2006 compared to historical data. Marine Ecol. Progress Series 2007, 348, 47–54. [Google Scholar] [CrossRef]
- Iglesias-Rodríguez, M.D.; Brown, C.W.; Doney, S.C.; Kleypas, J.; Kolber, D.; Kolber, Z.; Hayes, P.K.; Falkowski, P.G. Representing key phytoplankton functional groups in ocean carbon cycle models: Coccolithophorids. Global Biogeochem. Cy. 2002, 16. [Google Scholar] [CrossRef]
- Obata, T.; Shiraiwa, Y. A novel eukaryotic selenoprotein in the haptophyte alga Emiliania huxleyi. J. Biol. Chem. 2005, 280, 18462–18468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyrrell, T.; Merico, A. Emiliania huxleyi: Bloom observations and the conditions that induce them. In Coccolithophores: From Molecular Processes to Global Impact; Thierstein, H.R., Young, J.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 75–97. [Google Scholar]
- Hallegraeff, G.M. Ocean climate change, phytoplankton community responses, and harmful algal blooms: A formidable predictive challenge. J. Phycol. 2010, 46, 220–235. [Google Scholar] [CrossRef]
- Medlin, L.K.; Barker, G.L.A.; Campbell, L.; Green, J.C.; Hayes, P.K.; Marie, D.; Wrieden, S.; Vaulot, D. Genetic characterisation of Emiliania huxleyi (Haptophyta). J. Marine Sys. 1996, 9, 13–31. [Google Scholar] [CrossRef] [Green Version]
- Wahlund, T.M.; Hadaegh, A.R.; Clark, R.; Nguyen, B.; Fanelli, M.; Read, B.A. Analysis of Expressed Sequence Tags from Calcifying Cells of Marine Coccolithophorid (Emiliania huxleyi). Marine Biotechnol. 2004, 6, 278–290. [Google Scholar] [CrossRef]
- Dyhrman, S.T.; Haley, S.T.; Birkeland, S.R.; Wurch, L.L.; Cipriano, M.J.; McArthur, A.G. Long Serial Analysis of Gene Expression for Gene Discovery and Transcriptome Profiling in the Widespread Marine Coccolithophore Emiliania huxleyi. Appl. Environ. Microbiol. 2006, 72, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Quinn, P.; Bowers, R.M.; Zhang, X.; Wahlund, T.M.; Fanelli, M.A.; Olszova, D.; Read, B.A. cDNA Microarrays as a Tool for Identification of Biomineralization Proteins in the Coccolithophorid Emiliania huxleyi (Haptophyta). Appl. Environmen. Microbiol. 2006, 72, 5512–5526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richier, S.; Kerros, M.-E.; de Vargas, C.; Haramaty, L.; Falkowski, P.G.; Gattuso, J.-P. Light-Dependent Transcriptional Regulation of Genes of Biogeochemical Interest in the Diploid and Haploid Life Cycle Stages of Emiliania huxleyi. Appl. Environ. Microbiol. 2009, 75, 3366–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, B.A.; Kegel, J.; Klute, M.J.; Kuo, A.; Lefebvre, S.C.; Maumus, F.; Mayer, C.; Miller, J.; Monier, A.; Salamov, A.; et al. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 2013, 499, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.M.; Edwards, R.J.; Skipp, P.J.; O’Connor, C.D.; Iglesias-Rodriguez, M.D. Shotgun Proteomic Analysis of Emiliania huxleyi, a Marine Phytoplankton Species of Major Biogeochemical Importance. Marine Biotechnol. 2011, 13, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.M.; Iglesias-Rodriguez, M.D.; Skipp, P.J.; Edwards, R.J.; Greaves, M.J.; Young, J.R.; Elderfield, H.; O’Connor, C.D. Responses of the Emiliania huxleyi Proteome to Ocean Acidification. PLoS ONE 2013, 8, e61868. [Google Scholar] [CrossRef] [PubMed]
- McKew, B.A.; Lefebvre, S.C.; Achterberg, E.P.; Metodieva, G.; Raines, C.A.; Metodiev, M.V.; Geider, R.J. Plasticity in the proteome of Emiliania huxleyi CCMP 1516 to extremes of light is highly targeted. New Phytol. 2013, 200, 61–73. [Google Scholar] [CrossRef] [PubMed]
- McKew, B.A.; Metodieva, G.; Raines, C.A.; Metodiev, M.V.; Geider, R.J. Acclimation of Emiliania huxleyi (1516) to nutrient limitation involves precise modification of the proteome to scavenge alternative sources of N and P. Environmen. Microbiol. 2015, 17, 4050–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamontova, T.; Afonin, A.M.; Ihling, C.; Soboleva, A.; Lukasheva, E.; Sulima, A.S.; Shtark, O.Y.; Akhtemova, G.A.; Povydysh, M.N.; Sinz, A.; et al. Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi. Molecules 2019, 24, 1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Smith, R.D.; Shen, Y. Advanced proteomic liquid chromatography. J. Chromatogr. A 2012, 1261, 78–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van-An, D.; Jeeyun, A.; Na-Young, H.; Jong-Moon, P.; Jeong-Hun, M.; Tae Wan, K.; Hookeun, L. Proteomic Analysis of the Vitreous Body in Proliferative and Non-Proliferative Diabetic Retinopathy. Curr. Proteomics 2020, 17. [Google Scholar] [CrossRef]
- Ferrari, E.; Wittig, A.; Basilico, F.; Rossi, R.; De Palma, A.; Di Silvestre, D.; Sauerwein, W.A.G.; Mauri, P.L. Urinary Proteomics Profiles Are Useful for Detection of Cancer Biomarkers and Changes Induced by Therapeutic Procedures. Molecules 2019, 24, 794. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.; Liu, Y.; Liu, Y.; Wang, Q.; Ye, H.-L.; Cheng, Y.-G.; Kuang, H.-X.; Jiang, X.-C.; Yang, B.-Y. Proteomics Research on the Protective Effect of Mangiferin on H9C2 Cell Injury Induced by H2O2. Molecules 2019, 24, 1911. [Google Scholar] [CrossRef] [Green Version]
- Zimny-Arndt, U.; Schmid, M.; Ackermann, R.; Jungblut, P.R. Classical Proteomics: Two-Dimensional Electrophoresis/MALDI Mass Spectrometry. In Mass Spectrometry of Proteins and Peptides: Methods and Protocols; Lipton, M.S., Paša-Tolic, L., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 65–91. [Google Scholar]
- O’Farrell, P.H. High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 1975, 250, 4007–4021. [Google Scholar]
- Link, A.J.; Eng, J.; Schieltz, D.M.; Carmack, E.; Mize, G.J.; Morris, D.R.; Garvik, B.M.; Yates, J.R. Direct analysis of protein complexes using mass spectrometry. Nat. Biotechnol. 1999, 17, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Balgley, B.M.; DeVoe, D.L.; Lee, C.S. Capillary Isoelectric Focusing-Based Multidimensional Concentration/Separation Platform for Proteome Analysis. Anal. Chem. 2003, 75, 3145–3152. [Google Scholar] [CrossRef] [PubMed]
- Kašička, V. Capillary electrophoresis of peptides. Electrophoresis 1999, 20, 3084–3105. [Google Scholar] [CrossRef]
- Wu, Q.; Yuan, H.; Zhang, L.; Zhang, Y. Recent advances on multidimensional liquid chromatography–mass spectrometry for proteomics: From qualitative to quantitative analysis—A review. Anal. Chim. Acta 2012, 731, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Stoll, D.R.; Lhotka, H.R.; Harmes, D.C.; Madigan, B.; Hsiao, J.J.; Staples, G.O. High resolution two-dimensional liquid chromatography coupled with mass spectrometry for robust and sensitive characterization of therapeutic antibodies at the peptide level. J. Chromatogr. B 2019, 1134–1135, 121832. [Google Scholar] [CrossRef]
- Capriotti, A.L.; Cavaliere, C.; Cavazzini, A.; Gasparrini, F.; Pierri, G.; Piovesana, S.; Laganà, A. A multidimensional liquid chromatography–tandem mass spectrometry platform to improve protein identification in high-throughput shotgun proteomics. J. Chromatogr. A 2017, 1498, 176–182. [Google Scholar] [CrossRef]
- Fournier, M.L.; Gilmore, J.M.; Martin-Brown, S.A.; Washburn, M.P. Multidimensional Separations-Based Shotgun Proteomics. Chem. Rev. 2007, 107, 3654–3686. [Google Scholar] [CrossRef]
- Duong, V.-A.; Park, J.-M.; Lee, H. Review of Three-Dimensional Liquid Chromatography Platforms for Bottom-Up Proteomics. Int. J. Mol. Sci. 2020, 21, 1524. [Google Scholar] [CrossRef] [Green Version]
- Di Palma, S.; Hennrich, M.L.; Heck, A.J.R.; Mohammed, S. Recent advances in peptide separation by multidimensional liquid chromatography for proteome analysis. J. Proteomics 2012, 75, 3791–3813. [Google Scholar] [CrossRef]
- Giddings, J.C. Two-dimensional separations: concept and promise. Anal. Chem. 1984, 56, 1258A–1270A. [Google Scholar] [CrossRef]
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, S.; Adhikari, S.; Deng, Z.; Wang, L.; Chen, L.; Ke, M.; Yang, P.; Tian, R. Simple and Integrated Spintip-Based Technology Applied for Deep Proteome Profiling. Anal. Chem. 2016, 88, 4864–4871. [Google Scholar] [CrossRef]
- Jungblut, P.R.; Holzhütter, H.G.; Apweiler, R.; Schlüter, H. The speciation of the proteome. Chem. Central J. 2008, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlüter, H.; Apweiler, R.; Holzhütter, H.-G.; Jungblut, P.R. Finding one’s way in proteomics: a protein species nomenclature. Chem. Central J. 2009, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiede, B.; Koehler, C.J.; Strozynski, M.; Treumann, A.; Stein, R.; Zimny-Arndt, U.; Schmid, M.; Jungblut, P.R. High resolution quantitative proteomics of HeLa cells protein species using stable isotope labeling with amino acids in cell culture(SILAC), two-dimensional gel electrophoresis(2DE) and nano-liquid chromatograpohy coupled to an LTQ-OrbitrapMass spectrometer. Mol. Cell. Proteomics 2013, 12, 529–538. [Google Scholar] [PubMed] [Green Version]
- Takemori, A.; Butcher, D.S.; Harman, V.M.; Brownridge, P.J.; Shima, K.; Higo, D.; Ishizaki, J.; Hasegawa, H.; Suzuki, J.; Yamashita, M.; et al. PEPPI-MS: polyacrylamide gel-based prefractionation for analysis of intact proteoforms and protein complexes by mass spectrometry. J. Proteome Res. 2020. [Google Scholar] [CrossRef]
- Zhao, P.; Schulz, T.C.; Sherrer, E.S.; Weatherly, D.B.; Robins, A.J.; Wells, L. The human embryonic stem cell proteome revealed by multidimensional fractionation followed by tandem mass spectrometry. Proteomics 2015, 15, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Motoyama, A.; Xu, T.; Ruse, C.I.; Wohlschlegel, J.A.; Yates, J.R. Anion and Cation Mixed-Bed Ion Exchange for Enhanced Multidimensional Separations of Peptides and Phosphopeptides. Anal. Chem. 2007, 79, 3623–3634. [Google Scholar] [CrossRef] [Green Version]
- Wolters, D.A.; Washburn, M.P.; Yates, J.R. An Automated Multidimensional Protein Identification Technology for Shotgun Proteomics. Anal. Chem. 2001, 73, 5683–5690. [Google Scholar] [CrossRef]
- Dai, J.; Jin, W.-H.; Sheng, Q.-H.; Shieh, C.-H.; Wu, J.-R.; Zeng, R. Protein Phosphorylation and Expression Profiling by Yin-Yang Multidimensional Liquid Chromatography (Yin-Yang MDLC) Mass Spectrometry. J. Proteome Res. 2007, 6, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, R.C.; Spicer, V.; Harder, M.; Antonovici, M.; Ens, W.; Standing, K.G.; Wilkins, J.A.; Krokhin, O.V. Practical Implementation of 2D HPLC Scheme with Accurate Peptide Retention Prediction in Both Dimensions for High-Throughput Bottom-Up Proteomics. Anal. Chem. 2008, 80, 7036–7042. [Google Scholar] [CrossRef]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Orthogonality of Separation in Two-Dimensional Liquid Chromatography. Anal. Chem. 2005, 77, 6426–6434. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, F.; Gritsenko, M.A.; Wang, Y.; Clauss, T.; Liu, T.; Shen, Y.; Monroe, M.E.; Lopez-Ferrer, D.; Reno, T.; et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 2011, 11, 2019–2026. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Shen, Y.; Camp, D.G.; Smith, R.D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 2012, 9, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Sun, J.; Yu, W.; Jones, A.; Oeller, P.; Keller, M.; Woodnutt, G.; Short, J.M. Global Proteome Discovery Using an Online Three-Dimensional LC−MS/MS. J. Proteome Res. 2005, 4, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Law, H.C.H.; Kong, R.P.W.; Szeto, S.S.W.; Zhao, Y.; Zhang, Z.; Wang, Y.; Li, G.; Quan, Q.; Lee, S.M.Y.; Lam, H.C.; et al. A versatile reversed phase-strong cation exchange-reversed phase (RP–SCX–RP) multidimensional liquid chromatography platform for qualitative and quantitative shotgun proteomics. Analyst 2015, 140, 1237–1252. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Wang, F.; Song, C.; Sun, Z.; Cheng, K.; Tan, Y.; Wang, H.; Zou, H. Large-Scale Proteome Quantification of Hepatocellular Carcinoma Tissues by a Three-Dimensional Liquid Chromatography Strategy Integrated with Sample Preparation. J. Proteome Res. 2014, 13, 3645–3654. [Google Scholar] [CrossRef]
- Shen, Z.; Want, E.J.; Chen, W.; Keating, W.; Nussbaumer, W.; Moore, R.; Gentle, T.M.; Siuzdak, G. Sepsis Plasma Protein Profiling with Immunodepletion, Three-Dimensional Liquid Chromatography Tandem Mass Spectrometry, and Spectrum Counting. J. Proteome Res. 2006, 5, 3154–3160. [Google Scholar] [CrossRef]
- Betancourt, L.H.; De Bock, P.-J.; Staes, A.; Timmerman, E.; Perez-Riverol, Y.; Sanchez, A.; Besada, V.; Gonzalez, L.J.; Vandekerckhove, J.; Gevaert, K. SCX charge state selective separation of tryptic peptides combined with 2D-RP-HPLC allows for detailed proteome mapping. J. Proteomics 2013, 91, 164–171. [Google Scholar] [CrossRef]
- Bandhakavi, S.; Stone, M.D.; Onsongo, G.; Van Riper, S.K.; Griffin, T.J. A Dynamic Range Compression and Three-Dimensional Peptide Fractionation Analysis Platform Expands Proteome Coverage and the Diagnostic Potential of Whole Saliva. J. Proteome Res. 2009, 8, 5590–5600. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Lu, Y.; Ficarro, S.B.; Adelmant, G.; Jiang, W.; Luckey, C.J.; Marto, J.A. Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nature Commun. 2013, 4, 2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boichenko, A.P.; Govorukhina, N.; van der Zee, A.G.J.; Bischoff, R. Multidimensional separation of tryptic peptides from human serum proteins using reversed-phase, strong cation exchange, weak anion exchange, and fused-core fluorinated stationary phases. J. Separ. Science 2013, 36, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shen, X.; Sun, L. Strong cation exchange-reversed phase liquid chromatography-capillary zone electrophoresis-tandem mass spectrometry platform with high peak capacity for deep bottom-up proteomics. Anal. Chim. Acta 2018, 1012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Spicer, V.; Ezzati, P.; Neustaeter, H.; Beavis, R.C.; Wilkins, J.A.; Krokhin, O.V. 3D HPLC-MS with Reversed-Phase Separation Functionality in All Three Dimensions for Large-Scale Bottom-Up Proteomics and Peptide Retention Data Collection. Anal. Chem. 2016, 88, 2847–2855. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Gussakovsky, D.; Neustaeter, H.; Spicer, V.; Krokhin, O.V. Sequence-Specific Model for Peptide Retention Time Prediction in Strong Cation Exchange Chromatography. Anal. Chem. 2017, 89, 11795–11802. [Google Scholar] [CrossRef]
- Lorne Burke, T.W.; Mant, C.T.; Black, J.A.; Hodges, R.S. Strong cation-exchange high-performance liquid chromatography of peptides: Effect of non-specific hydrophobic interactions and linearization of peptide retention behaviour. J. Chromatogr. A 1989, 476, 377–389. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, H.; Smith, K.; Wu, S. Two-dimensional separation using high-pH and low-pH reversed phase liquid chromatography for top-down proteomics. Int. J. Mass Spectr. 2018, 427, 43–51. [Google Scholar] [CrossRef]
- Motoyama, A.; Yates, J.R. Multidimensional LC Separations in Shotgun Proteomics. Anal. Chem. 2008, 80, 7187–7193. [Google Scholar] [CrossRef] [Green Version]
- Metwally, H.; McAllister, R.G.; Konermann, L. Exploring the Mechanism of Salt-Induced Signal Suppression in Protein Electrospray Mass Spectrometry Using Experiments and Molecular Dynamics Simulations. Anal. Chem. 2015, 87, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Huang, K.; Cross, F.R.; Cowburn, D.; Chait, B.T. Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. USA 1999, 96, 6591–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, S.-E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable Isotope Labeling by Amino Acids in Cell Culture, SILAC, as a Simple and Accurate Approach to Expression Proteomics. Mol. Cell. Proteom. 2002, 1, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Bach, L.T.; Mackinder, L.C.M.; Schulz, K.G.; Wheeler, G.; Schroeder, D.C.; Brownlee, C.; Riebesell, U. Dissecting the impact of CO2 and pH on the mechanisms of photosynthesis and calcification in the coccolithophore Emiliania huxleyi. New Phytol. 2013, 199, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isenberg, H.D.; Lavine, L.S.; Weissfellner, H. The Suppression of Mineralization in a Coccolithophorid By an Inhibitor of Carbonic Anhydrase. J. Protozool. 1963, 10, 477–479. [Google Scholar] [CrossRef]
- Forgac, M. Structure and Properties of the Clathrin-Coated Vesicle and Yeast Vacuolar V-ATPases. J. Bioenerget. Biomembr. 1999, 31, 57–65. [Google Scholar] [CrossRef]
- Kirchhausen, T. Clathrin. Ann. Rev. Biochem. 2000, 69, 699–727. [Google Scholar] [CrossRef]
- Young, J.R.; Henriksen, K. Biomineralization Within Vesicles: The Calcite of Coccoliths. Rev. Mineral. Geochem. 2003, 54, 189–215. [Google Scholar] [CrossRef]
- Durak, G.M.; Taylor, A.R.; Walker, C.E.; Probert, I.; de Vargas, C.; Audic, S.; Schroeder, D.; Brownlee, C.; Wheeler, G.L. A role for diatom-like silicon transporters in calcifying coccolithophores. Nat. Commun. 2016, 7, 10543. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.J.Y.; Klute, M.J.; Herman, E.K.; Read, B.; Dacks, J.B. Losses, Expansions, and Novel Subunit Discovery of Adaptor Protein Complexes in Haptophyte Algae. Protist 2015, 166, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Paasche, E. A review of the coccolithophorid Emiliania huxleyi (Prymnesiophyceae), with particular reference to growth, coccolith formation, and calcification-photosynthesis interactions. Phycologia 2001, 40, 503–529. [Google Scholar] [CrossRef]
- Martiny, A.C.; Coleman, M.L.; Chisholm, S.W. Phosphate acquisition genes in Prochlorococcus ecotypes: Evidence for genome-wide adaptation. Proc. Natl. Acad. Sci. USA 2006, 103, 12552–12557. [Google Scholar] [CrossRef] [Green Version]
- Hoch, M.P.; Snyder, R.A.; Jeffrey, W.H.; Dillon, K.S.; Coffin, R.B. Expression of glutamine synthetase and glutamate dehydrogenase by marine bacterioplankton: Assay optimizations and efficacy for assessing nitrogen to carbon metabolic balance in situ. Limnol. Oceanogr. Methods 2006, 4, 308–328. [Google Scholar] [CrossRef] [Green Version]
- Sowell, S.M.; Abraham, P.E.; Shah, M.; Verberkmoes, N.C.; Smith, D.P.; Barofsky, D.F.; Giovannoni, S.J. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J. 2011, 5, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Abida, H.; Ruchaud, S.; Rios, L.; Humeau, A.; Probert, I.; De Vargas, C.; Bach, S.; Bowler, C. Bioprospecting marine plankton. Mar. Drugs 2013, 11, 4594–4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.T.; Phyo, M.Y. Marine Cyanobacteria: A Source of Lead Compounds and their Clinically-Relevant Molecular Targets. Molecules 2020, 25, 2197. [Google Scholar] [CrossRef]
- Azam, M.S.; Choi, J.; Lee, M.-S.; Kim, H.-R. Hypopigmenting Effects of Brown Algae-Derived Phytochemicals: A Review on Molecular Mechanisms. Mar. Drugs 2017, 15, 297. [Google Scholar] [CrossRef] [Green Version]
- Favre, L.; Ortalo-Magné, A.; Pichereaux, C.; Gargaros, A.; Burlet-Schiltz, O.; Cotelle, V.; Culioli, G. Metabolome and proteome changes between biofilm and planktonic phenotypes of the marine bacterium Pseudoalteromonas lipolytica TC8. Biofouling 2018, 34, 132–148. [Google Scholar] [CrossRef]
- Favre, L.; Ortalo-Magné, A.; Kerloch, L.; Pichereaux, C.; Misson, B.; Briand, J.-F.; Garnier, C.; Culioli, G. Metabolomic and proteomic changes induced by growth inhibitory concentrations of copper in the biofilm-forming marine bacterium Pseudoalteromonas lipolytica. Metallomics 2019, 11, 1887–1899. [Google Scholar] [CrossRef] [Green Version]
- Maghembe, R.; Damian, D.; Makaranga, A.; Nyandoro, S.S.; Lyantagaye, S.L.; Kusari, S.; Hatti-Kaul, R. Omics for Bioprospecting and Drug Discovery from Bacteria and Microalgae. Antibiotics 2020, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Medhi, K.; Malaviya, P.; Thakur, I.S. Omics approaches for microalgal applications: Prospects and challenges. Biores. Technol. 2019, 291, 121890. [Google Scholar] [CrossRef]
- Guillard, R.R.L. Culture of Phytoplankton for Feeding Marine Invertebrates. In Culture of Marine Invertebrate Animals: Proceedings—1st Conference on Culture of Marine Invertebrate Animals Greenport; Smith, W.L., Chanley, M.H., Eds.; Springer: Boston, MA, USA, 1975; pp. 29–60. [Google Scholar]
- Araie, H.; Sakamoto, K.; Suzuki, I.; Shiraiwa, Y. Characterization of the Selenite Uptake Mechanism in the Coccolithophore Emiliania huxleyi (Haptophyta). Plant Cell Physiol. 2011, 52, 1204–1210. [Google Scholar] [CrossRef] [Green Version]
- Joo, M.; Park, J.-M.; Duong, V.-A.; Kwon, D.; Jeon, J.; Han, M.; Cho, B.-K.; Choi, H.-K.; Lee, C.-G.; Kang, H.-G.; et al. An automated high-throughput sample preparation method using double-filtration for serum metabolite LC-MS analysis. Anal. Methods 2019, 11, 4060–4065. [Google Scholar] [CrossRef]
- Arul, A.-B.; Byambadorj, M.; Han, N.-Y.; Park, J.M.; Lee, H. Development of an Automated, High-throughput Sample Preparation Protocol for Proteomics Analysis. Bull. Korean Chem. Soc. 2015, 36, 1791–1798. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2018, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Mendoza, L.; Shteynberg, D.; Slagel, J.; Sun, Z.; Moritz, R.L. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics 2015, 9, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, A.; Nesvizhskii, A.I.; Kolker, E.; Aebersold, R. Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Anal. Chem. 2002, 74, 5383–5392. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the Emiliania huxleyi cells (strain CCMP371) are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Description | % Associated Genes | Number of Genes | Term p-Value | −log10(p-Value) |
|---|---|---|---|---|---|
| KEGG:03040 | Spliceosome | 69.4 | 120 | 0.0005 | 3.3052 |
| KEGG:00531 | Glycosaminoglycan degradation | 31.0 | 13 | 0.0009 | 3.0608 |
| KEGG:04145 | Phagosome | 38.0 | 40 | 0.0010 | 3.0222 |
| KEGG:03022 | Basal transcription factors | 83.9 | 26 | 0.0017 | 2.7735 |
| KEGG:01040 | Biosynthesis of unsaturated fatty acids | 21.1 | 4 | 0.0020 | 2.6914 |
| KEGG:04141 | Protein processing in endoplasmic reticulum | 67.7 | 105 | 0.0046 | 2.3355 |
| KEGG:00020 | Citrate cycle (TCA cycle) | 78.8 | 26 | 0.0123 | 1.9104 |
| KEGG:00330 | Arginine and proline metabolism | 41.9 | 26 | 0.0201 | 1.6972 |
| KEGG:03050 | Proteasome | 71.9 | 41 | 0.0213 | 1.6717 |
| KEGG:00430 | Taurine and hypotaurine metabolism | 23.1 | 3 | 0.0217 | 1.6635 |
| KEGG:04933 | AGE-RAGE signaling pathway in diabetic complications | 33.3 | 8 | 0.0236 | 1.6280 |
| KEGG:04070 | Phosphatidylinositol signaling system | 45.3 | 39 | 0.0362 | 1.4411 |
| KEGG:03410 | Base excision repair | 43.5 | 27 | 0.0389 | 1.4097 |
| KEGG:03015 | mRNA surveillance pathway | 67.9 | 55 | 0.0409 | 1.3885 |
| KEGG:00520 | Amino sugar and nucleotide sugar metabolism | 67.0 | 59 | 0.0495 | 1.3052 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, G.; Park, J.-M.; Duong, V.-A.; Mok, J.-H.; Jeon, J.; Nam, O.; Lee, J.; Jin, E.; Lee, H. Proteomic Profiling of Emiliania huxleyi Using a Three-Dimensional Separation Method Combined with Tandem Mass Spectrometry. Molecules 2020, 25, 3028. https://doi.org/10.3390/molecules25133028
Yun G, Park J-M, Duong V-A, Mok J-H, Jeon J, Nam O, Lee J, Jin E, Lee H. Proteomic Profiling of Emiliania huxleyi Using a Three-Dimensional Separation Method Combined with Tandem Mass Spectrometry. Molecules. 2020; 25(13):3028. https://doi.org/10.3390/molecules25133028
Chicago/Turabian StyleYun, Goyeun, Jong-Moon Park, Van-An Duong, Jeong-Hun Mok, Jongho Jeon, Onyou Nam, Joonwon Lee, EonSeon Jin, and Hookeun Lee. 2020. "Proteomic Profiling of Emiliania huxleyi Using a Three-Dimensional Separation Method Combined with Tandem Mass Spectrometry" Molecules 25, no. 13: 3028. https://doi.org/10.3390/molecules25133028