
Abstract
A highly stable and magnetized citric acid (CA)-functionalized iron oxide aqueous colloidal solution (Fe3O4@CA) was synthesized by using a simple and rapid method of one-step co-participation via a chemical reaction between Fe3+ and Fe2+ in a NaOH solution at 65 °C, followed by CA addition to functionalize the Fe3O4 surface in 25 min. The NPs were synthesized at lower temperatures and shortened time compared with conventional methods. Surface functionalization is highly suggested because bare Fe3O4 nanoparticles (Fe3O4 NPs) are frequently deficient due to their low stability and hydrophilicity. Hence, 19 nm-sized Fe3O4 NPs coated with CA (Fe3O4@CA) were synthesized, and their microstructure, morphology, and magnetic properties were characterized using X-ray diffraction, transmission electron microscopy, Zeta potential, Fourier transform infrared spectroscopy, and vibrating sample magnetometer. CA successfully modified the Fe3O4 surface to obtain a stabilized (homogeneous and well dispersed) aqueous colloidal solution. The Zeta potential value of the as-prepared Fe3O4@CA increases from − 31 to − 45 mV. These CA-functionalized NPs with high magnetic saturation (54.8 emu/g) show promising biomedical applications.
Similar content being viewed by others

Introduction
Fe3O4 NPs with a grain size of smaller than 20 nm display superparamagnetic behavior at high temperatures but exhibit no coercivity and remanence at room temperature1,2,3,4. These particles are extensively utilized for several biomedical and in vivo applications5,6,7,8,9. Fe3O4 NPs, a well-known ferrofluid, has been expansively analyzed, particularly their colloidal dispersion and many potential biomedical applications. The surface of magnetite particles is modified by different coating agents, including protein10, methoxypoly (ethylene glycol)11, dextran12, chitosan13, and silica coating14, to enhance their performance. Controlling the sizes and dispersion of NPs in preferred solvents is technologically challenging due to difficulties faced in their fabrication and handling for biomedical applications, including their clustering/aggregation, homogeneity, hydrophilicity, and biocompatibility15,16. The high surface energies of NPs are attributed to their large surface to volume ratio. NPs tend to aggregate to minimize total surface energy, which exceeds 0.1 N/m for metal oxide surfaces17.
Proper functionalization of NP surface and solvent selection are critical to attain adequate repelling interactions between the NPs to inhibit agglomeration/accretion and improve the thermodynamic stability of the colloidal solution. The surface of Fe3O4 dispersed in aqueous media via citric acid adsorption can be functionalized by utilizing the coordination of one or two carboxylate functionalities of the citric acid depending on the steric necessity and curvature of the surface18. Carboxylates significantly affect the development of Fe3O4 NPs and their magnetic characteristics. Surface modification of aqueous magnetic NPs by using heavy chain fatty acid or thiol is one of the methods to increase the stability of NP suspension19. Co-precipitation is typically used to synthesize water-stable Fe3O4 NPs and considered as the simplest, most cost-effective technique requiring the lowest temperature20. However, its main drawbacks are the agglomeration, broad size distribution, poor Zeta potential values of NPs. Fe3O4 NPs also lack good colloidal stability and have inadequate repulsive forces to prevent agglomeration. The poor colloidal stability and broad size distribution can be attributed to the reaction time and temperature during co-precipitation. To overcome these problems, the Fe3O4 NPs must be stabilized and their size distribution must be reduced by modifying their surfaces with biocompatible materials, in addition to controlling the synthesis procedures. Nevertheless, most of aqueous stabilized Fe3O4 NPs are achieved either at high temperature21,22,23 or long reaction time24,25,26. For example, Elham et al.27 and Arefi et al.28 synthesized citric acid (CA)-stabilized Fe3O4 NPs through two-step co-precipitation that is laborious and time consuming. In addition, Singh et al.29 synthesized CA-coated Fe3O4 NPs through co-precipitation, and the transmission electron microscopy (TEM) results indicated that the NPs have agglomerated and are non-uniform in shape.
To the extent of our knowledge, the stability of Fe3O4@CA NPs has not been reported. Therefore, this study aims (1) to synthesize a highly stable and magnetized Fe3O4@CA aqueous colloidal solution by employing a one-step, fast, and straightforward route (with shortened time and lower temperature than conventional methods) and systematically controlling and manipulating the flow of the reaction procedure and (2) to develop surface functional groups on magnetic NP derivatization through a one-step process.
Materials and methods
Materials
Ferric chloride (FeCl3·6H2O, 99%), ferrous chloride (FeCl2·4H2O, 99%), and sodium hydroxide (NaOH) were acquired from Sigma–Aldrich, and citric acid (CA) were purchased from Merck.
Preparation Fe3O4@CA
Fe3O4 NPs were synthesized through the co-precipitation of ferrous (Fe2+) and ferric (Fe3+) with sodium hydroxide (NaOH). FeCl2.4H2O (2.5 g) and FeCl3.6H2O (4.0 g) were dissolved in 180 mL of distilled water under nitrogen gas. Following the complete dissolution of the mixture at room temperature, 50 mL of sodium hydroxide was drop-wise added to the reaction mixture, which was mechanically stirred at 650 rpm and kept for 10 min at 65 °C under continuous vigorous stirring. For the prevention of Fe3O4 NP agglomeration, 150 mL of CA was added to the reaction mixture, which was then stirred for 10 min (65 °C). Fe3O4@CA NPs were collected through a permanent magnet and thoroughly rinsed four times with distilled water to eliminate unreactive or inert impurities. Finally, the Fe3O4@CA NPs were redispersed in the distilled water after sonication for 5 min, and the resulting suspension (Fe3O4@CA) responded to an external magnetic field as shown in Fig. 1.

Magnetic attraction of Fe3O4@CA NPs: (a) Fe3O4@CANPs in solution state and (b) magnetic attraction of Fe3O4@CA NPs toward a magnet.
Characterization of Fe3O4@CA
X-ray diffraction (XRD) patterns were obtained using an X-ray diffractometer (PANalytical X’pert PRO MRD PW 3040) with CuKa (λ = 1.54050 Å). The size of Fe3O4 NPs and Fe3O4@Au CSNPs were obtained by transmission electron microscopic (TEM) using a Zeiss Libra 120 at 100 kV. Particle size distribution was measured using ImageJ software. The stability (Zeta potential) of Fe3O4@CA NPs was described using a dynamic light scattering (DLS) instrument (ZETASIZER Nanoseries Model ZEN 3600, Malvern Instruments). The surface functional groups of Fe3O4@CA NPs were determined by Fourier transform infrared spectroscopy (PERKIN ELMER System 2000 FT-IR). Magnetic properties were evaluated using vibrating sample magnetometer (VSM, DMS MODEL 8810).
Results and discussion
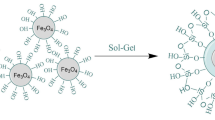
Fe3O4 NP surfaces were functionalized via CA adsorption, which occurs by coordinating one or two of the carboxylate functionalities depending on the need for steric repelling to stabilize the ferrofluids and the curvature or morphology of the surface. Nonetheless, a minimum of one carboxylic acid group is exposed to the solvent, thus accounting for the surface charging. The presence of a carboxylic group surface ligand offers the possibility of developing bonds with proteins, fluorescent dyes, and hormone linkers to facilitate precise targeting in biological systems. The one-step modification of the superparamagnetic Fe3O4 NP surface is presented in Fig. 2, and the as-prepared Fe3O4 NPs were subsequently stabilized with CA to prevent agglomeration.

Steps for CA functionalization of Fe3O4 NP surface in 25 min.
The XRD spectra (Fig. 3) of Fe3O4 NPs confirmed their cubic spinel structure with high crystallinity. These diffraction peaks are narrow and well defined, indicating the high crystallinity of the sample. The positions and intensities of the diffraction peaks for NPs are consistent those of Fe3O4 (Ref. Code 01-075-0033). The synthesis method for Fe3O4 particles via the co-precipitation of Fe2+ and Fe3+ in an aqueous base solution is a relatively established and extensively utilized procedure25,30. The XRD results are consistent with the possible constituents of Fe3O4 particles. The diffraction peaks denote the crystallinity of Fe3O4 NPs as spinel cubic lattice type. Nevertheless, further oxidation of the Fe2O3 phase was not verified by the XRD data due to the similarity between lattice type and constant31. According to a previous study, the CA coating for Fe3O4 NPs does not result in the phase change in the XRD spectra of bare Fe3O429.

XRD spectra of the CA-functionalized Fe3O4 NPs showing the composition and crystal structure of Fe3O4.
The TEM image and size distribution of the as-synthesized Fe3O4 and Fe3O4@CA NPs are presented in (Fig. 4). Figure 4a shows the TEM image of Fe3O4 prior to CA modification. A slightly important change in Fe3O4 agglomeration was induced by CA. From the histogram in Fig. 4c, the average size of the monodispersed Fe3O4@CA NPs is approximately 19 nm. The Fe3O4@CA NPs are spherical in shape with a narrow size distribution after CA modification, particularly at stable synthesis conditions. The TEM images of the CA-functionalized superparamagnetic Fe3O4 NPs show semi-spherical shaped particles and monodispersion. Previous studies used co-precipitation to synthesize CA-coated Fe3O4 and produced agglomerated NPs with average sizes 5128, 5032, 25,33 and 22 nm34, which might be due to the high reaction temperature35.

TEM micrographs of (a) bare Fe3O4, (b) Fe3O4@CA, and (c) the corresponding size distribution of Fe3O4@CA.
Figure 5 presents the stability of Fe3O4 after CA modification to show the role of CA on the stability of Fe3O4 NPs. The Zeta potential magnitude of Fe3O4 NPs was measured immediately after the synthesis of the particles, followed by CA injection on the colloidal Fe3O4 NPs. Dispersion stability can be defined in relation to the Zeta potential value (mV): 0 to ± 5 can cause the rapid agglomeration and precipitation of NP suspension, ± 10 to ± 30 is responsible for the threshold of delicate dispersion, ± 30 to ± 40 denotes the moderate stability of colloidal NPs36,37,38,39, and ± 40 to ± 60 indicates the excellent stability of NP suspension referred to as a high charge on their surface40. The measured results indicate that Zeta potential improves from − 31.3 to − 45.3 mV (Fig. 5a, b), which is higher than the reported values41,42,43 and is attributed to the three carboxylate groups of citrate that dissociate and strongly bind with Fe3O4 NP surface18. In addition, the negative charge of the Zeta potential is due to the electrostatic stabilization provided by the strong adsorption of citrate ions on NP surface. The Zeta potential of CA-coated Fe3O4 has not been reported. The increase in the measured Zeta potential revealed that CA is absorbed onto the Fe3O4 NP surface, thus resulting in a highly negative surface charge. The presence of carboxylate group is confirmed by monitoring the Zeta potential for Fe3O4 NPs. However, this study characterized and analyzed only the results for a moderately polydispersed sample.

Stability measurement of as-synthesized nanoparticles using Zeta potential for (a) bare Fe3O4 and (b) Fe3O4@CA.
The Fourier transform-infrared spectroscopy (FT-IR) spectrum of bare Fe3O4 has been reported28,29. This study aimed to prove the presence of CA on the Fe3O4 surface. FT-IR spectra of CA and Fe3O4@CA NPs are illustrated in Fig. 6a and b. The spectrum peak was assigned to the CA-coated Fe3O4 NPs. The broad band spectrum at 3,384 cm−1 can be referred to as the OH band groups and to the traces of molecular water. The 1722 cm−1 spectrum peak of CA is due to the symmetric C=O stretching from the COOH group. This peak display shifts to a lower wavelength at approximately 1615 cm−1 for the carboxylic group (R-OOH) of the Fe3O4@CA. The peak at 1615 cm−1 determines the binding of CA radical on the surface of Fe3O4 NPs through the chemisorption of carboxylate citrate ions28,29. The peak at 1,384 cm−1 can be ascribed to the asymmetric stretching of C–O from the carboxylic group. The intense peak observed at the IR range at approximately 578 cm−1 in Fe3O4@CA could be assigned to the Fe–O stretching vibrational mode of Fe3O444. Hence, CA binds to the Fe3O4 surface through carboxylate.

FTIR spectra of (a) bare CA and (b) CA conjugated on the surface of Fe3O4 NPs.
The magnetic properties of Fe3O4@CA were determined by VSM analysis at room temperature. The magnetization saturation (emu/g) as a function of the applied magnetic field (Oe) is illustrated in Fig. 7. The magnetization curve shows that the Fe3O4@CA NPs exhibit a superparamagnetic behavior and magnetic saturation (Ms) of approximately 54.8 emu/g, which is higher than that in previous studies25,33 (Table 1) possibly due to the low Fe oxidation state. No hysteresis was observed, and the behavior was completely reversible at 300 K. Neither coercivity nor remanence was observed. Arefi et al.28. reported that the Ms of bare Fe3O4 is reduced after being coated with CA. Alonso et al.23 synthesized Fe3O4 NPs with high crystallinity of approximately 35 nm and high Ms of 65 emu/g by using thermal decomposition. The high Ms value is attributed to the large particle size of Fe3O445. Therefore, the Ms of Fe3O4 NPs decreases with their reduced particle size due to the increase in surface spin disorder46,47. In this case, the size reduction to the nanoscale (below 20 nm) for spherical single-component Fe3O4 greatly influences the magnetic ordering of surface spins, namely, a high degree of disordered surface spins of Fe3O4 NPs. Spherical Fe3O4 NPs could develop surface spin disorder through energy minimization. The disordered surface spins are highly anisotropic, which is in line with the increase in their effective magnetic anisotropy.

Magnetization curve of superparamagnetic (no coercivity or remanence) Fe3O4@CA at room temperature.
For biomedical applications such as in hyperthermia and magnetic resonance imaging (MRI), NPs must have a uniform particle size, exhibit superparamagnetism, and possess high Ms. The as-synthesized Fe3O4@CA has a high magnetic response, which is preferable for biomedical applications28. Our method shows an advantage of having a simple and rapid route to synthesize highly stable (− 45.3 mV), monodispersed, and superparamagnetic Fe3O4@CA (19 nm) compared with conventional techniques.
Conclusion
We developed a simple and rapid synthesis route for highly stable and superparamagnetic Fe3O4@CA through co-precipitation. The proposed method requires simple equipment and cheap materials such as a magnetic stirrer, and the processing time was 25 min at 65 °C. The NPs were achieved at lower temperature, simpler process, and shorter time compared with conventional methods. XRD, TEM, Zeta potential, FT-IR, and VSM were employed to characterize the microstructure and morphology of the synthesized NPs. The presence of carboxylate group is confirmed by FTIR analysis, and the Zeta potential for Fe3O4 particles was monitored. The Zeta potential value of as-prepared Fe3O4@CA increased from − 31.3 to − 45.3 mV. Finally, these NPs are important for several biomedical applications due to their small size, stability, and superparamagnetic behavior.
References
Ángeles-Pascual, A. et al. Structure, magnetic and cytotoxic behaviour of solvothermally grown Fe3O4@ Au core-shell nanoparticles. Mater. Charact. 142, 237–244 (2018).
Khaniabadi, P. M. et al. Magnetic iron oxide nanoparticles as T2 MR imaging contrast agent for detection of breast cancer (MCF-7) cell. Avicenna J Med Biotechnol 9, 181 (2017).
Dheyab, M. A., Aziz, A. A., Jameel, M. S., Khaniabadi, P. M. & Mehrdel, B. Mechanisms of effective gold shell on Fe3O4 core nanoparticles formation using sonochemistry method. Ultrason. Sonochem. 104, 865 (2019).
4Dheyab, M. et al. in Journal of Physics: Conference Series. 012003.
Lai, W.-Y. et al. In vivo investigation into effectiveness of Fe3O4/PLLA nanofibers for bone tissue engineering applications. Polymers 10, 804 (2018).
Wang, K. et al. Fe3O4@ astragalus polysaccharide core-shell nanoparticles for iron deficiency anemia therapy and magnetic resonance imaging in vivo. ACS Appl. Mater. Interfaces. 11, 10452–10461 (2019).
Rajkumar, S. & Prabaharan, M. Noble Metal-Metal Oxide Hybrid Nanoparticles 607–623 (Elsevier, Amsterdam, 2019).
Khaniabadi, P. M., Shahbazi-Gahrouei, D., Majid, A. M. S. A. & Khaniabadi, B. M. Study the Anti-MUC1 antibody-based iron oxide nanoparticles on three-dimension spheroid and breast cancer (MCF-7) cell imaging. Pol. J. Med. Phys. Eng. 25, 69–77 (2019).
Khaniabadi, P. M. et al. In vitro study of SPIONs-C595 as molecular imaging probe for specific breast cancer (MCF-7) cells detection. Ir. Biomed. J. 21, 360 (2017).
10Zorin, V. et al. Journal of Physics: Conference Series. 031009 (IOP Publishing).
PolymersAli, S. et al. Synthesis, characterization, and relaxometry studies of hydrophilic and hydrophobic superparamagnetic Fe3O4 nanoparticles for oil reservoir applications. Colloids Surf. A 543, 133–143 (2018).
Patil, R. M. & Shete, P. B. Biodistribution Interaction Nanostructures of Hybrid (Elesiver, Amsterdam, 2018).
Ge, J., Zhai, M., Zhang, Y., Bian, J. & Wu, J. Biocompatible Fe3O4/chitosan scaffolds with high magnetism. Int. J. Biol. Macromol. 128, 406–413 (2019).
Iqbal, M. Z. et al. A facile fabrication route for binary transition metal oxide-based janus nanoparticles for cancer theranostic applications. Nano Res. 11, 5735–5750 (2018).
Liu, J. et al. Highly water-dispersible biocompatible magnetite particles with low cytotoxicity stabilized by citrate groups. Angew. Chem. Int. Ed. 48, 5875–5879 (2009).
Nigam, S., Barick, K. & Bahadur, D. Development of citrate-stabilized Fe3O4 nanoparticles: conjugation and release of doxorubicin for therapeutic applications. J. Magn. Magn. Mater. 323, 237–243 (2011).
Gavilán, H., Brollo, M., Gutiérrez, L., Veintemillas-Verdaguer, S. & Morales, M. Controlling the Size and Shape of Uniform Magnetic Iron Oxide Nanoparticles for Biomedical Applications (CRC Press, Boca Raton, 2018).
Sahoo, Y. et al. Aqueous ferrofluid of magnetite nanoparticles: fluorescence labeling and magnetophoretic control. J. Phys. Chem. B 109, 3879–3885 (2005).
Shen, L., Stachowiak, A., Fateen, S.-E.K., Laibinis, P. E. & Hatton, T. A. Structure of alkanoic acid stabilized magnetic fluids. A small-angle neutron and light scattering analysis. Langmuir 17, 288–299 (2001).
Dheyab, M. A., Aziz, A. A., Jameel, M. S., Noqta, O. A. & Mehrdel, B. Synthesis and coating methods of biocompatible iron oxide/gold nanoparticle and nanocomposite for biomedical applications. Chin. J. Phys. 36, 305–325 (2019).
Eom, Y., Abbas, M., Noh, H. & Kim, C. Morphology-controlled synthesis of highly crystalline Fe3O4 and CoFe2O4 nanoparticles using a facile thermal decomposition method. RSC Adv. 6, 15861–15867 (2016).
Smith, M., McKeague, M. & DeRosa, M. C. Synthesis, transfer, and characterization of core-shell gold-coated magnetic nanoparticles. MethodsX 6, 333–354 (2019).
Nemati, Z. et al. Enhanced magnetic hyperthermia in iron oxide nano-octopods: size and anisotropy effects. J. Phys. Chem. C 120, 8370–8379 (2016).
Kataria, N. & Garg, V. Green synthesis of Fe3O4 nanoparticles loaded sawdust carbon for cadmium (II) removal from water: regeneration and mechanism. Chemosphere 208, 818–828 (2018).
Kalantari, K. et al. Size-controlled synthesis of Fe3O4 magnetic nanoparticles in the layers of montmorillonite. J. Nanomater. 2014, 181 (2014).
Das, R. et al. Tunable high aspect ratio iron oxide nanorods for enhanced hyperthermia. J. Phys. Chem. C 120, 10086–10093 (2016).
Cheraghipour, E., Javadpour, S. & Mehdizadeh, A. R. Citrate capped superparamagnetic iron oxide nanoparticles used for hyperthermia therapy. J. Biomed. Sci. Eng. 5, 715 (2012).
Arefi, M., Miraki, M. K., Mostafalu, R., Satari, M. & Heydari, A. Citric acid stabilized on the surface of magnetic nanoparticles as an efficient and recyclable catalyst for transamidation of carboxamides, phthalimide, urea and thiourea with amines under neat conditions. J. Iran. Chem. Soc. 16, 393–400 (2019).
Singh, D. et al. Citric acid coated magnetic nanoparticles: synthesis, characterization and application in removal of Cd(II) ions from aqueous solution. J. Water Process Eng. 4, 233–241 (2014).
Zhou, H. et al. Ultrasensitive DNA monitoring by Au–Fe3O4 nanocomplex. Sens. Actuators B 163, 224–232 (2012).
Levy, L., Sahoo, Y., Kim, K.-S., Bergey, E. J. & Prasad, P. N. Nanochemistry: synthesis and characterization of multifunctional nanoclinics for biological applications. Chem. Mater. 14, 3715–3721 (2002).
Patel, U., Chauhan, K. & Gupte, S. Synthesis, characterization and application of lipase-conjugated citric acid-coated magnetic nanoparticles for ester synthesis using waste frying oil. 3 Biotech 8, 211 (2018).
Li, L. et al. Effect of synthesis conditions on the properties of citric-acid coated iron oxide nanoparticles. Microelectron. Eng. 110, 329–334 (2013).
Singha, D. et al. Citric acid coated magnetic nanoparticles: synthesis, characterization and application in removal of Cd(II) ions from aqueous solution. J. Water Process Eng. 4, 233–241 (2014).
Zheng, B. et al. Plant-mediated synthesis of platinum nanoparticles and its bioreductive mechanism. J. Colloid Interface Sci. 396, 138–145 (2013).
Jameel, M. S., Aziz, A. A. & Dheyab, M. A. Comparative analysis of platinum nanoparticles synthesized using sonochemical-assisted and conventional green methods. Nano-Struct. Nano-Obj. 23, 100484. https://doi.org/10.1016/j.nanoso.2020.100484 (2020).
Dheyab, M. A., Owaid, M. N., Rabeea, M. A., Aziz, A. A. & Jameel, M. S. Mycosynthesis of gold nanoparticles by the Portabello mushroom extract, Agaricaceae, and their efficacy for decolorization of Azo dye. Environ. Nanotechnol. Monit. Manage. https://doi.org/10.1016/j.enmm.2020.100312 (2020).
Rabeea, M. A., Owaid, M. N., Aziz, A. A., Jameel, M. S. & Dheyab, M. A. Mycosynthesis of gold nanoparticles using the extract of Flammulina velutipes, Physalacriaceae, and their efficacy for decolorization of methylene blue. J. Environ. Chem. Eng. 103, 841 (2020).
Owaid, M. N., Rabeea, M. A., Aziz, A. A., Jameel, M. S. & Dheyab, M. A. Mushroom-assisted synthesis of triangle gold nanoparticles using the aqueous extract of fresh Lentinula edodes (shiitake), Omphalotaceae. Environ. Nanotechnol. Monit. Manage. 12, 100270 (2019).
Riddick, T. M. Control of Colloid Stability Through zeta Potential (Livingston, Wynnewood, PA, 1968).
Rehana, D., Haleel, A. K. & Rahiman, A. K. Hydroxy, carboxylic and amino acid functionalized superparamagnetic iron oxide nanoparticles: synthesis, characterization and in vitro anti-cancer studies. J. Chem. Sci. 127, 1155–1166 (2015).
Bini, R. A., Marques, R. F. C., Santos, F. J., Chaker, J. A. & Jafelicci, M. Jr. Synthesis and functionalization of magnetite nanoparticles with different amino-functional alkoxysilanes. J. Magn. Magn. Mater. 324, 534–539 (2012).
43Catalano, E. & Di Benedetto, A. Journal of Physics: Conference Series. 012010 (IOP Publishing).
Yamaura, M. et al. Preparation and characterization of (3-aminopropyl) triethoxysilane-coated magnetite nanoparticles. J. Magn. Magn. Mater. 279, 210–217 (2004).
Jayakumar, T., Thomas, P. A. & Geraldine, P. Protective effect of an extract of the oyster mushroom, Pleurotus ostreatus, on antioxidants of major organs of aged rats. Exp. Gerontol. 42, 183–191 (2007).
Bakoglidis, K., Simeonidis, K., Sakellari, D., Stefanou, G. & Angelakeris, M. Size-dependent mechanisms in AC magnetic hyperthermia response of iron-oxide nanoparticles. IEEE Trans. Magn. 48, 1320–1323 (2012).
Phan, M.-H. et al. Exchange bias effects in iron oxide-based nanoparticle systems. Nanomaterials 6, 221 (2016).
Acknowledgements
This work was supported by the Malaysia Ministry of Education FRGS funding [USM Grant Number 203/PFIZIK/6711768], and the authors also acknowledge the School of Physics, Universiti Sains Malaysia for providing the facilities for this research work.
Author information
Authors and Affiliations
Contributions
M.A.D., A.A.A. and M.S.J. conceived the idea and designed the experiments. A.A.A. supervised all the experiments and analyses. M.A.D., M.S.J. and O.A.N. prepared materials, performed characterizations, measurements and analyzed the results. A.A.A., P.M.K. and B.M. commented on manuscript writing. M.A.D. wrote the manuscript and all authors discussed the results and commented on the manuscript of the work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dheyab, M.A., Aziz, A.A., Jameel, M.S. et al. Simple rapid stabilization method through citric acid modification for magnetite nanoparticles. Sci Rep 10, 10793 (2020). https://doi.org/10.1038/s41598-020-67869-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67869-8
This article is cited by
-
Properties of Natural Rubber: Magnetite Decorated with Silver Nanoparticles for Catalytic Degradation of Model Organic Contaminant
Journal of Inorganic and Organometallic Polymers and Materials (2024)
-
Biocatalyst Based on Magnetic Nanoparticles with Cu(II), Mn(II), Zn(II) and Immobilised Catalase
Journal of Cluster Science (2024)
-
A narrative review of the synthesis, characterization, and applications of iron oxide nanoparticles
Discover Nano (2023)
-
Multi-round recycling of green waste for the production of iron nanoparticles: synthesis, characterization, and prospects in remediation
Discover Nano (2023)
-
Influence of the modifiers in polyol method on magnetically induced hyperthermia and biocompatibility of ultrafine magnetite nanoparticles
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
