Possible Three-Dimensional Topological Insulator in Pyrochlore Oxides


National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba Central 2, Umezono 1-1-4, Tsukuba, Ibaraki 305-8568, Japan
*
Author to whom correspondence should be addressed.
Symmetry 2020, 12(7), 1076; https://doi.org/10.3390/sym12071076
Submission received: 25 May 2020
/
Revised: 9 June 2020
/
Accepted: 12 June 2020
/
Published: 1 July 2020
(This article belongs to the Special Issue Vortex, Topology and Singularity in Quantum Systems)
Abstract
:A Kene–Mele-type nearest-neighbor tight-binding model on a pyrochlore lattice is known to be a topological insulator in some parameter region. It is an important task to realize a topological insulator in a real compound, especially in an oxide that is stable in air. In this paper we systematically performed band structure calculations for six pyrochlore oxides A2B2O7 (A = Sn, Pb, Tl; B = Nb, Ta), which are properly described by this model, and found that heavily hole-doped Sn2Nb2O7 is a good candidate. Surprisingly, an effective spin–orbit coupling constant λ changes its sign depending on the composition of the material. Furthermore, we calculated the band structure of three virtual pyrochlore oxides, namely In2Nb2O7, In2Ta2O7 and Sn2Zr2O7. We found that Sn2Zr2O7 has a band gap at the k = 0 (Γ) point, similar to Sn2Nb2O7, though the band structure of Sn2Zr2O7 itself differs from the ideal nearest-neighbor tight-binding model. We propose that the co-doped system (In,Sn)2(Nb,Zr)2O7 may become a candidate of the three-dimensional strong topological insulator.
{kind=link}
{kind=link}
{kind=link}
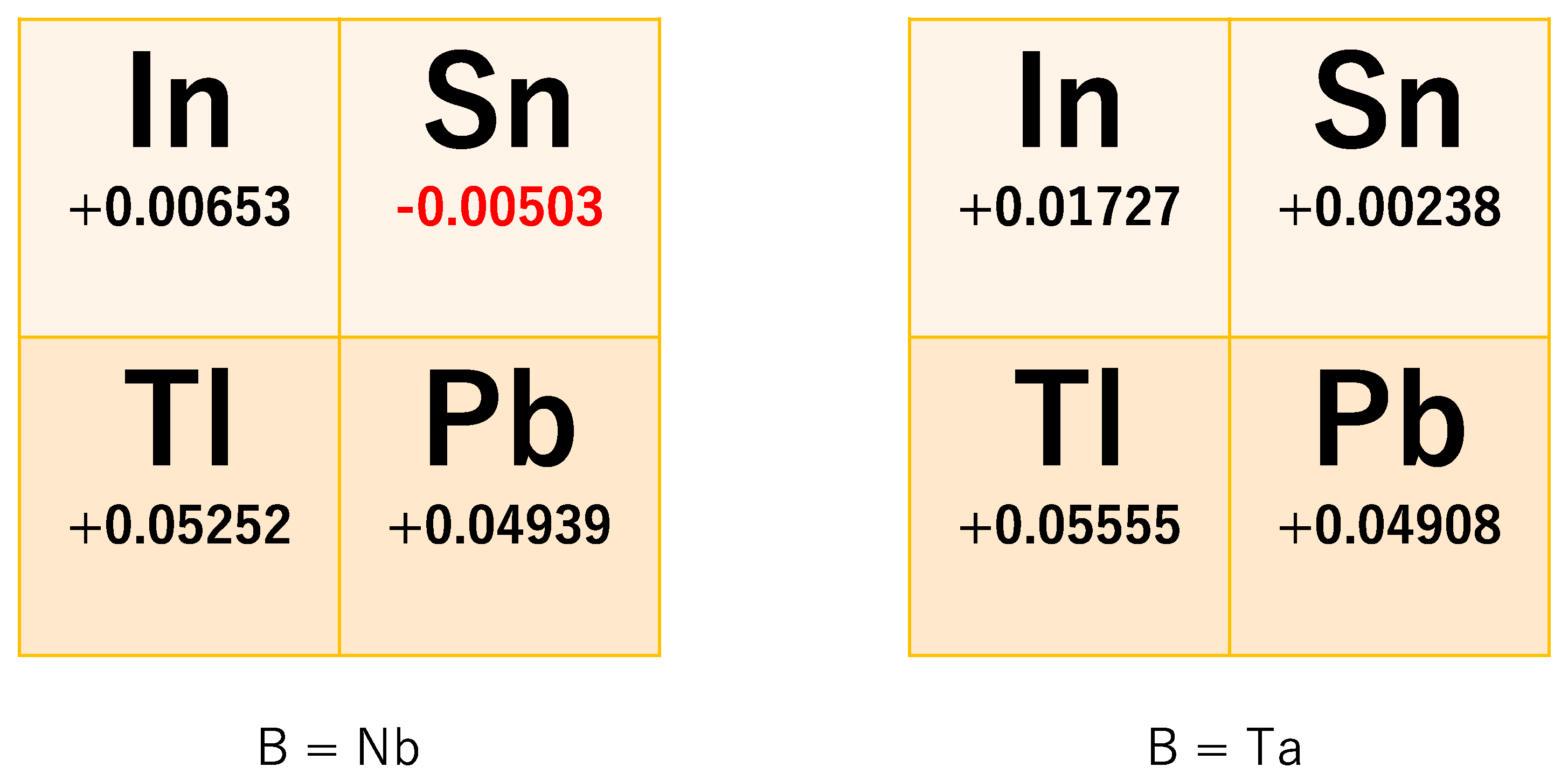{kind=link}
{kind=link}
1. Introduction
Topologically non-trivial states in materials have been paid much attention in the last decades. Among them, topological insulators (TIs) have attracted attention, not only from a basic but also from an application point of view. In TI, while the bulk is insulating, the surface has a metallic state. Since this surface state is robust against perturbations that do not violate the time reversal symmetry, it has been pointed out that it can be applied to a quantum computing device. Considering such applications, the material is desired to be three-dimensional and stable in the air, namely oxide. However, very little is known about oxides that are candidates for TI.
Outstanding works by Kane and Mele [1,2] have revealed that graphene may show a quantum spin Hall (QSH) effect if the spin–orbit coupling (SOC) is sufficiently strong. This effect can be explained as follows: Graphene has Dirac points that are protected by space–group symmetry, and this degeneracy is lifted by the SOC. This SOC opens a gap and makes the system a TI. A conventional (quantum) Hall effect is caused by an external magnetic field, while in this case the SOC plays the role of “magnetic field”, depending on the direction of the electron spin. Although the SOC in real graphene is too small to open a gap, subsequent works predicted that the HgTe–CdTe quantum well is a good candidate showing a QSH effect [3]. This prediction has been subsequently verified experimentally [4].
Later on, the Kane–Mele model [1,2] has been extended to other lattice models. Guo and Franz applied this model to a two-dimensional (2D) Kagome lattice [5] and three-dimensional (3D) pyrochlore lattice [6]. Both of these models are known to have a flat band (FB) that has no energy dispersion when only the nearest neighbor transfer is considered. Adding SOC with the type of Kane–Mele model, these lattice models show various topological properties.
The Guo and Franz (GF) model can be described as follows [6]:
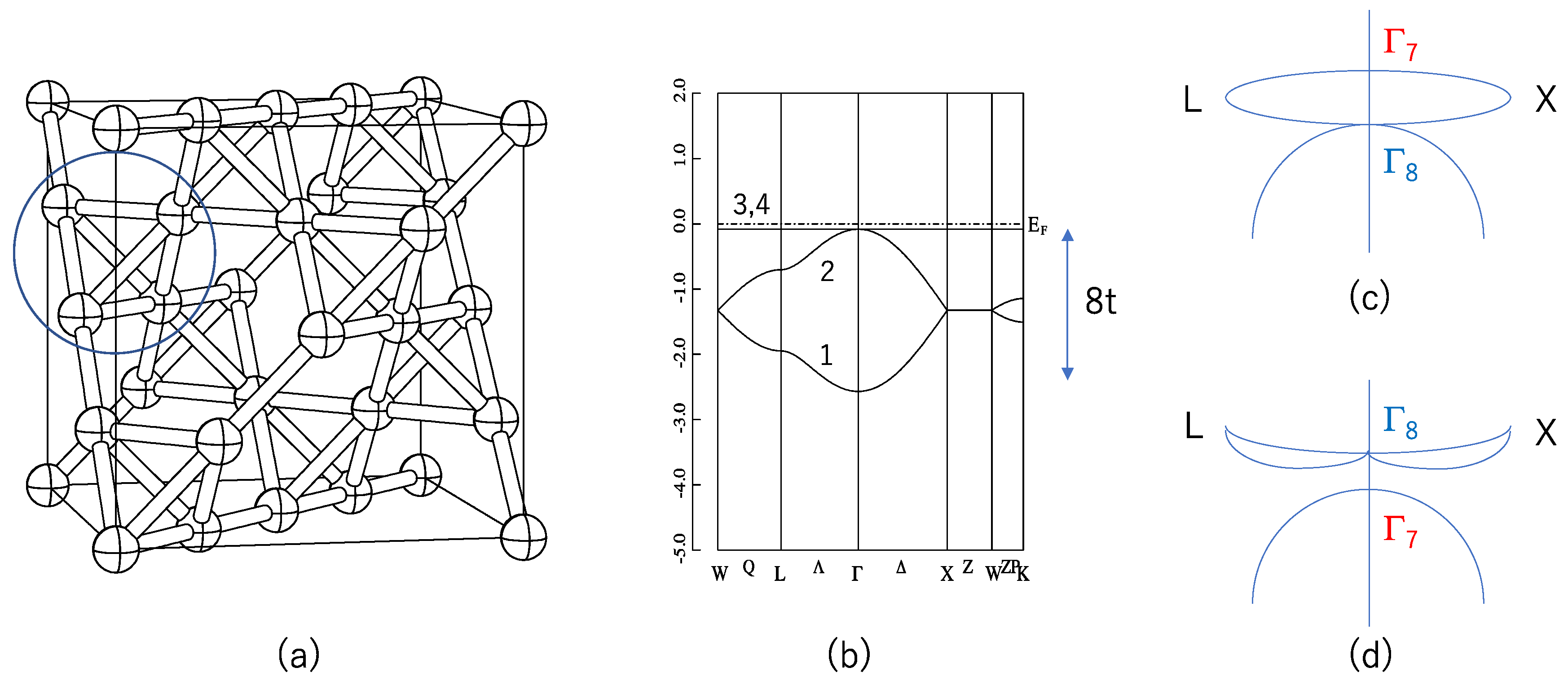
where the first term of Equation (1) denotes the non-interacting tight-binding Hamiltonian, and <ij> denotes the nearest neighbors. This lattice sum is taken on the pyrochlore lattice shown in Figure 1a. This term can also be regarded as the Mielke model without an electron–electron interaction [7,8]. The second term denotes SOC with an effective coupling constant λ. σαβ denotes the spin αβ component of the Pauli matrices. In the pyrochlore lattice there are four sites in the primitive cell, and we obtain four bands. Here, dij1,2 denote the nearest-neighbor vectors traversed between the second neighbor sites i and j. The summation <<ij>> is taken for these second neighbor sites. In case of λ = 0, this Hamiltonian is rigorously diagonalized and the eigenvalues are
where . E3 and E4 do not depend on the wave vector k, so they are called flat bands (FBs). These energy eigenvalues are shown in Figure 1b. Note that these FBs touch the dispersive band at k = 0. In other words, the state at k = 0 is three-fold (six-fold if we consider spin) degenerated. Therefore, in case of half-filling, i.e., two of the four bands are filled, the system is a zero-gap semiconductor. FB itself induces many attractive physical properties [7,8,9,10], but in this paper we focus on the topological aspect of this model.
Next, we include SOC. This SOC lifts the abovementioned six-fold degeneracy into two-fold (Γ7) and four-fold (Γ8) degenerated states. Which of these states have higher energy depends on the sign of λ. A schematic picture is shown in Figure 1c,d. When λ > 0, the energy of Γ7 is higher than that of Γ8, i.e., E(Γ7) > E(Γ8). In this case, four-fold degenerated FBs split into Γ7 and Γ8 at the Γ point, but are still degenerated at the zone boundary points X and L. Therefore, the system remains gapless when λ > 0. On the contrary when λ < 0, the energy of Γ7 is lower than that of Γ8, i.e., E(Γ7) < E(Γ8). In case of half-filling and λ < 0, the system has a band gap Δ = 24 |λ|. Guo and Franz [6] have shown that this is a 3D strong TI having topological indices (1;000), where (ν0;ν1ν2ν3) denote the four-component Z2 topological indices [11,12]. Each νi has a binary value 0 or 1, and if all νi are zero, i.e., (0;000), the system is an ordinary band insulator; if ν0 = 1 it is a strong TI, which means topologically protected surface states exist on all surfaces and they are robust with regard to non-magnetic disorder.
It is very important to apply these results to real materials. In this sense, the GF model defined on the pyrochlore lattice is very attractive since the pyrochlore oxides A2B2O7 include two independent pyrochlore sublattices [13]. However, even though there are hundreds of pyrochlore oxides [13], there are very few compounds whose electronic states near the Fermi level (EF) can be approximated by the GF model. The reason for this is as follows: In the GF model, the transfer integral t is isotropic, while in most of the pyrochlore oxides the relevant orbital is an anisotropic d- or f- orbital.
In previous papers [14,15,16] we have shown that six pyrochlore oxides A2B2O7 (A = Sn, Pb, Tl; B = Nb, Ta) have band structures that are well described in the GF model. In those papers, we mainly focused on the possible ferromagnetism induced by the quasi-FB. We have already shown [14] that Sn2Nb2O7 has E(Γ7) < E(Γ8), i.e., λ < 0. On the other hand, Sn2Ta2O7 has λ > 0. It means that Sn2Nb2O7 is a good candidate for a strong TI when the holes are sufficiently doped (Sn2Nb2O7 itself is away from half-filling), but Sn2Ta2O7 is not. After this work, Zhou et al. proposed a novel topological state for ferromagnetic Sn2Nb2O7 induced by quasi-FB [17], and Zhang et al. proposed that Tl2Nb2O6+x (0 ≤ x ≤ 1) leads to various topological phases accompanied with lattice distortion [18].
The purpose of this paper is to extend the above discussion to other pyrochlore oxides of which the band structure near EF is approximated by the GF model. We calculated the band structure of six pyrochlore oxides A2B2O7 (A = Sn, Pb, Tl; B = Nb, Ta) from first principles. Additionally, we calculated the band structure of three hypothetical compounds, In2Nb2O7, In2Ta2O7 and Sn2Zr2O7, which may be potential candidates for a TI. We found that only Sn2Nb2O7 and Sn2Zr2O7 gives λ < 0, though the band structure of Sn2Zr2O7 cannot fit very well with the GF model.
2. Materials and Computational Methods
We calculated six pyrochlore oxides with the composition A2B2O7 (A = Sn, Pb, Tl; B = Nb, Ta) from first principles. We used the density-functional theory (DFT) and a linearized augmented plane wave with the addition of a local orbital (LAPW+lo) scheme (WIEN2k code [19]). The exchange-correlation potential was constructed within the general gradient approximation [20]. The k-point mesh was set so that the total number of the mesh was about 1000 in the first Brillouin zone. The parameter RKmax, which is a product of the maximum radius of the muffin-tin spheres R and the cut-off wave number of the plane-wave basis Kmax, was set to be 7.0. For simplicity, we assumed that the calculated compounds all have an ideal A2B2O6O’ pyrochlore structure with the space group Fd-3m (#227). Since oxygen atoms occupy two crystallographic sites, we named these sites as O and O’ to distinguish between them. The atomic positions are A(0, 0, 0), B(1/2, 1/2, 1/2), O(u, 1/8, 1/8) and O’(1/8, 1/8, 1/8). This pyrochlore structure contains 88 atoms in the conventional cubic unit cell. In this work we focus on the A atoms, which form the pyrochlore lattice shown in Figure 1a. O’ atoms are at the center of the A4 tetrahedron shown by a circle in Figure 1a. We optimized this parameter u by minimizing the Hellmann–Feynman force , where H denotes the one-electron Hamiltonian for the atomic coordinates R, and is its eigenfunction. The convergence of the atomic positions was judged by this Hellmann–Feynman force working on each atom, which was to be less than 1.0 mRy/a.u. We also optimized the lattice constant a for Tl2Ta2O7. The optimized value a = 10.716 Å well agreed with an experimental value a = 10.56 Å [21] or 10.651 Å [22]. For the other five compounds, we used the experimental lattice constant.
After the relaxation of the atomic position and lattice constant, a spin–orbit interaction (SOI) is included via a second-variational step using a scalar-relativistic eigenfunction as the basis [23,24]. Hereafter, we distinguish between SOI and SOC. The former is essentially an on-site term, explicitly included in the band calculation [19,23,24]. The latter is the term introduced in the Kane–Mele model [1] and GF model [6], which is essentially a non-local term. We can estimate the SOC from the band calculation by fitting the bands to the GF model, or more simply, just see the energy splitting at the Γ point ΔSO = E(Γ7) – E(Γ8) and use the relation ΔSO = 24 λ [6].
In order to understand the electronic structure of this A2B2O7 system, we also performed band calculations on the virtual compounds In2Nb2O7, In2Ta2O7 and Sn2Zr2O7. For these compounds, we relaxed both of the lattice constant and the atomic positions. The obtained lattice constant is a = 10.611, 10.631 and 10.618 Å for In2Nb2O7, In2Ta2O7 and Sn2Zr2O7, respectively. They are all within the normal range of the lattice constants of pyrochlore oxides [13].
3. Results and Discussions
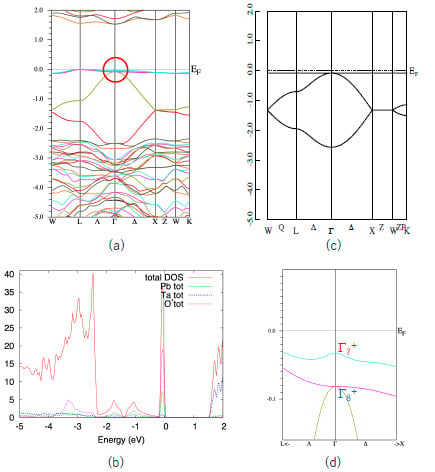
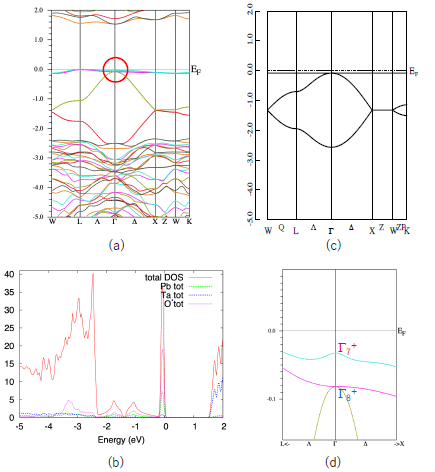
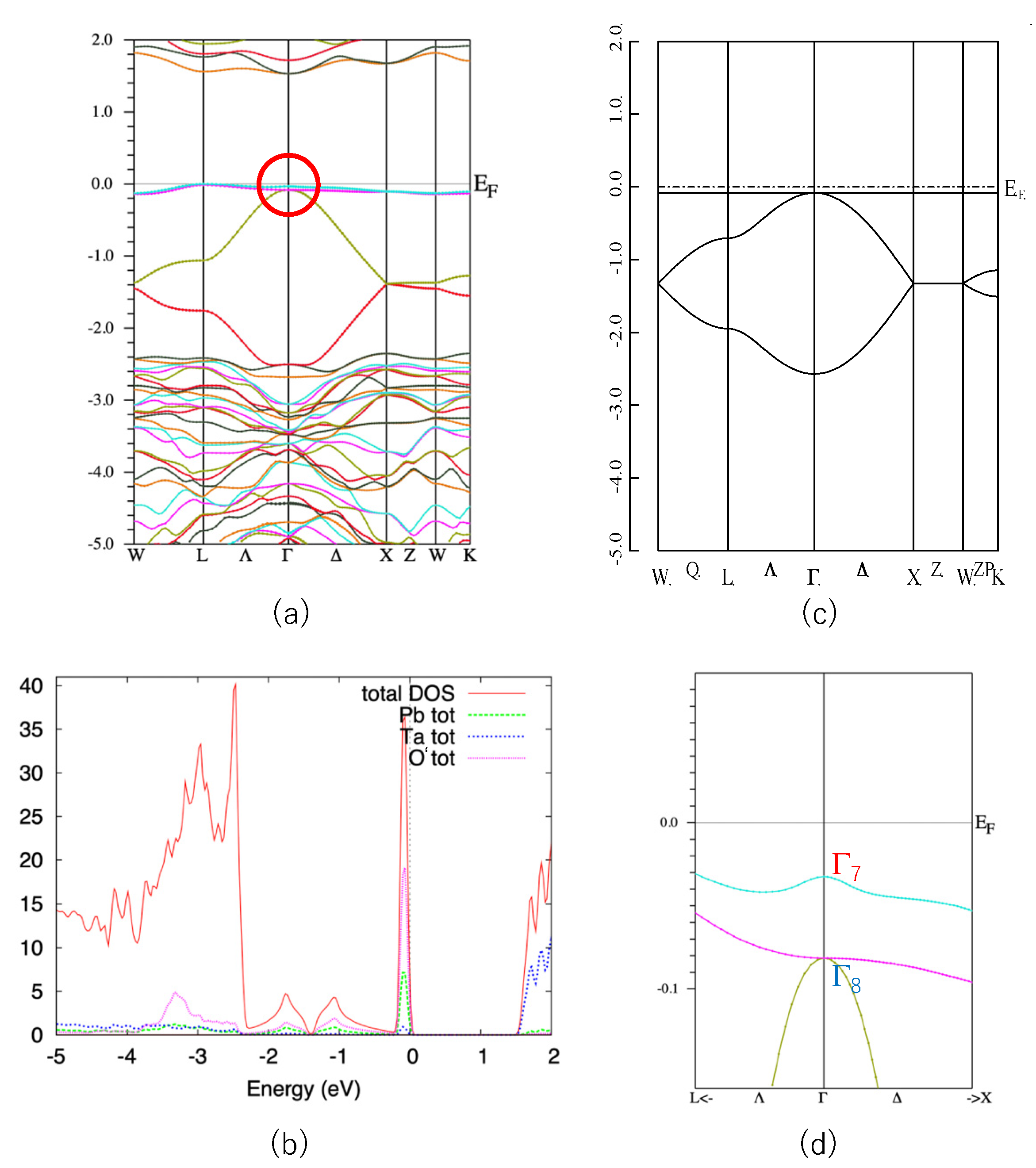
First, we show the band structure of Pb2Ta2O7 in Figure 2a. Similar to other quasi-FB oxides [14,15,16,17,18], this compound also shows a characteristic quasi-FB just below EF. The origin of this quasi-FB is explained as follows. The formal chemical valences of Pb2Ta2O7 are denoted as Pb2+2Ta5+2O2−7. Since Ta5+ and O2− form closed shells, we see that only Pb2+ is chemically active in the first approximation. The electron configuration of Pb2+ is (6s)2, so we can expect that the band gap is opened between the Ta5d band and Pb6s band. This is verified by density-of-states (DOS) analysis shown in Figure 2b. We see that the sharp peak coming from quasi-FB mainly consists of the Pb-s and O’-p components.
As is explained in Section 1, a simple nearest-neighbor tight-binding model on a pyrochlore lattice gives a flat band as in Equation (2). We show the energy dispersion of this model in Figure 2c. The agreement between the actual valence band of Pb2Ta2O7 and the simple GF model is quite good, considering that the GF model includes only one parameter, t (in Figure 2c we set λ = 0). Since the s-orbital is isotropic, the hopping integral t in Equation (1) is also isotropic. Note that in most of the pyrochlore oxides the relevant (frontier) orbitals are d- or f-orbitals, which are anisotropic. We can also show that the more precise 10-orbital model, including four Pb-s and 6 O’-p orbitals, also have a FB [14]. However, we deal with the minimal 4-orbital model because we can utilize the results for the GF model.
Note that the top of the valence band of Pb2Ta2O7 is not a complete FB but a quasi-FB. This small dispersion (about 0.2 eV) is attributed to the hopping terms other than the nearest neighbor one, such as next-nearest-neighbor hopping. However, this small dispersion does not change the topology of the band structure and we can use the result for the GF model.
We can see that the quasi-FB in Pb2Ta2O7 is slightly split due to the SOI. Other bands, e.g., the bands at energy ~1.5 eV, split more than the quasi-FB at the valence band. This is because the conduction band mostly consist of Ta5d orbitals in which the SOI is large. If the valence band purely consists of Pb6s orbitals, the SOI should be zero. Small splitting of the quasi-FB in Pb2Ta2O7 is due to the hybridization of the other orbitals. In Figure 2d we show a blow-up of the band structure of the Pb2Ta2O7 near EF (shown by a circle in Figure 2a).
As shown in Reference [6], the GF model in the half-filled case has a band gap at the Γ point when the SOC is switched on and λ < 0; in this case the system becomes a strong TI with Z2 indices (1;000). On the other hand, it does not have a band gap when λ > 0. We see that E(Γ7) is larger than E(Γ8), i.e., λ > 0 in Pb2Ta2O7. This situation corresponds to the case in Figure 1c. Note that these Z2 indices are purely topological ones and robust for the small perturbations. Therefore, we can expect that these indices are unchanged in the real compounds we calculated, unless there are extra band crossings. Above these reasons, we can investigate the topological properties of the A2B2O7 compounds by calculating the energy difference ΔSO = E(Γ7) − E(Γ8) at the Γ point (k = 0).
Figure 3 shows the energy difference ΔSO = E(Γ7) − E(Γ8) in eight pyrochlore oxides A2B2O7 (A = In, Sn, Tl, Pb; B = Nb, Ta), including two virtual oxides, In2Nb2O7 and In2Ta2O7. Apparently, only Sn2Nb2O7 has a band gap, and the other seven oxides do not have a band gap. This result shows that Sn2Nb2O7 may become a strong TI when the chemical potential is put in this band gap by heavily doping the holes. At present, hole doping in Sn2Nb2O7 has been successful [25], but such a heavy doping is difficult. As for Sn2Nb2O7, combining the topological band structure and the possible ferromagnetism due to quasi-FB, the existence of a novel three-dimensional Weyl point is suggested [17].
As seen in Figure 3, ΔSO (or effective SOC coefficient λ = ΔSO/24) is material-dependent, even in its sign. We can see some trends in this figure:
- When A is the period-6 element of the periodic table, ΔSO is large, and when it is a period-5 element, ΔSO is small. The difference of ΔSO is about 40–50 meV.
- When A is the group-13 element of the periodic table, ΔSO is large, and when it is a period-14 element, ΔSO is small. The difference of ΔSO is about 3–15 meV.
- ΔSO does not so much depend on the B element. The difference of ΔSO is about −0.3–10 meV.
Among these three trends, the first one is most crucial. It suggests that element A should be period-5 like In or Sn for obtaining a small ΔSO. In order to realize a TI in pyrochlore oxide, we should also tune the chemical potential. Half-filling of the 4-orbital GF model means that two of the four bands are occupied, i.e., there are four electrons in the primitive unit cell. Since there are four atomic sites, it turns so that the outermost electron configuration in the real compound is (5s)1 or (6s)1, which means there is one electron in the outermost s-orbital. We call this type of compound the “s1 compound”. This includes Tl2Nb2O7 and so on. A famous mother compound of the high-Tc superconductor BaBiO3 is also an s1 compound [26,27,28,29], but Tl2Nb2O7 has a quasi-FB due to the strong geometric frustration of the pyrochlore lattice. This strong frustration may prevent forming a charge-density wave, which is seen in BaBiO3. On the contrary, a compound containing two electrons in the outermost s-orbital is called the “s2 compound”. As for realizing the TI, the s1 compound is desirable in order to realize the TI, though in general the s1 compound is not so stable.
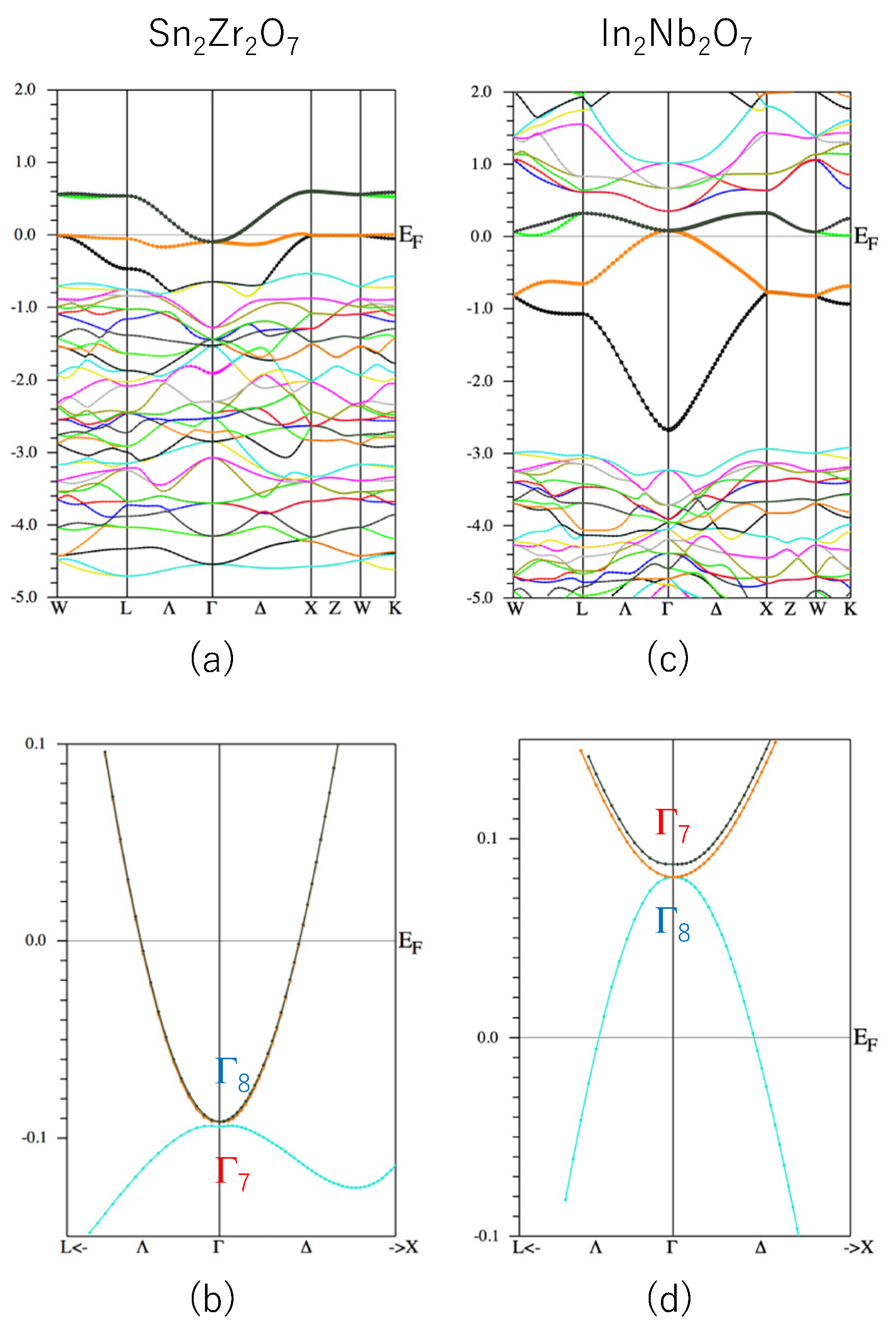
From the above discussion, we consider that a virtual compound Sn2Zr2O7 may be a good candidate for a TI because it is formally described as Sn3+2Zr4+2O2−7, having an s1 ion Sn3+ and the other ions form a closed shell. This compound may also have small λ since it contains an Sn atom on the A site. The calculated band structure of the Sn2Zr2O7 is shown in Figure 4a,b. As is expected, we found Δ = 2.5 meV, which means there is a band gap at the Γ point. However, the valence band Sn2Zr2O7 is apparently not described well by the GF model, as shown in Figure 4a. The reason why Sn2Zr2O7 has such a band structure is not clear, but we note that the band structure of the other pyrochlore oxides with a formal ionic valence A3+2B4+2O2−7 (e.g., Bi2Ti2O7) are not described well by the GF model (or non-interacting Mielke model) [15].
On the other hand, another virtual compound, In2Nb2O7, has a band structure like the GF model (see Figure 4c), but λ > 0 (though not too large, see Figure 3). Sn2Zr2O7 has λ < 0 but the band structure is not like the GF model. Therefore, we propose that a co-doped system, (In,Sn)2(Nb,Zr)2O7, may have both of the desired properties for a TI, namely, a GF-model-like band and λ < 0.
Finally, we comment on the possibility of lattice distortion in the pyrochlore oxides. For example, though in Sn2Nb2O7 there is no experimental signature of lattice distortion from the cubic phase, Pb2Nb2O7 suffers a lattice distortion in low temperature [30]. Lattice distortion can greatly affect the topological properties. Zhang et al. have shown that Tl2Nb2O7 can transform from a semi-metal to a TI by in-plane strain [18]. Since the topological properties are not dependent on the detail of the band structure, it is highly possible that the TI is also induced by in-plane strain in Tl2Ta2O7, In2Nb2O7 and In2Ta2O7, which have similar band structures.
4. Conclusions
We calculated the band structure of six pyrochlore oxides, A2B2O7 (A = Sn, Pb, Tl; B = Nb, Ta). They have quasi-flat valence bands that are characteristic of the Guo–Franz flat-band model. Spin–orbit interaction partially lifts the degeneracy at the Γ point. This results in a strong topological insulator in the s1 compound if the split energy levels satisfy E(Γ7) < E(Γ8). We found only Sn2Nb2O7 satisfies E(Γ7) < E(Γ8) in the above six compounds, though this is an s2 compound. We propose that the virtual compound (In,Sn)2(Nb,Zr)2O7 may have all of the desired properties for a TI, i.e., a Guo–Franz-model-like band, an s1 compound and E(Γ7) < E(Γ8).
Author Contributions
Conceptualization: I.H. and T.Y.; methodology: I.H.; software: I.H.; investigation: I.H.; data curation: I.H.; writing—original draft preparation: I.H.; writing—review and editing: T.Y.; visualization: I.H.; project administration: I.H.; funding acquisition: I.H. and T.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Japan Society for the Promotion of Science, grant number 19K03731.
Acknowledgments
The authors would like to thank Y. Higashi, M. Suzuki, Y. Yanagi, K. Kawashima and H. Aoki for fruitful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kane, C.L.; Mele, E. Quantum Spin Hall Effect in Graphene. Phys. Rev. Lett. 2005, 95, 226801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, C.L.; Mele, E. Z2 Topological Order and the Quantum Spin Hall Effect. Phys. Rev. Lett. 2005, 95, 146802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernevig, B.A.; Hughes, T.L.; Zhang, S.-C. Quantum Spin Hall Effect and Topological Phase Transition in HgTe Quantum Wells. Science 2006, 314, 1757–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, M.; Wiedmann, S.; Brüne, C.; Roth, A.; Buhmann, H.; Molenkamp, L.; Qi, X.-L.; Zhang, S.-C. Quantum Spin Hall Insulator State in HgTe Quantum Wells. Science 2007, 318, 766–770. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.-M.; Franz, M. Topological insulator on the kagome lattice. Phys. Rev. B 2009, 80, 113102. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.-M.; Franz, M. Three-Dimensional Topological Insulators on the Pyrochlore Lattice. Phys. Rev. Lett. 2009, 103, 206805. [Google Scholar] [CrossRef] [Green Version]
- Mielke, A. Ferromagnetic ground states for the Hubbard model on line graphs. J. Phys. A Math. Gen. 1991, 24, L73–L77. [Google Scholar] [CrossRef]
- Mielke, A. Ferromagnetism in the Hubbard model on line graphs and further considerations. J. Phys. A Math. Gen. 1991, 24, 3311–3321. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, F.; Wu, Y.-S. Exotic electronic states in the world of flat bands: From theory to material. Chin. Phys. B 2014, 23, 077308. [Google Scholar] [CrossRef]
- Derzhko, O.; Richter, J.; Maksymenko, M. Strongly correlated flat-band systems: The route from Heisenberg spins to Hubbard electrons. Int. J. Mod. Phys. B 2015, 29, 1530007. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Kane, C.L.; Mele, E. Topological Insulators in Three Dimensions. Phys. Rev. Lett. 2007, 98, 106803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.E.; Balents, L. Topological invariants of time-reversal-invariant band structures. Phys. Rev. B 2007, 75, 121306. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, M.A.; Aravamudan, G.; Subba Rao, G.V. Oxides pyrochlore—A review. Prog. Solid State Chem. 1983, 15, 55–143. [Google Scholar] [CrossRef]
- Hase, I.; Yanagisawa, T.; Aiura, Y.; Kawashima, K. Possibility of flat-band ferromagnetism in hold-doped pyrochlore oxides Sn2Nb2O7 and Sn2Ta2O7. Phys. Rev. Lett. 2018, 120, 196401. [Google Scholar] [CrossRef] [PubMed]
- Hase, I.; Yanagisawa, T.; Kawashima, K. Computational design of flat band system. Nanoscale Res. Lett. 2018, 13, 63. [Google Scholar] [CrossRef]
- Hase, I.; Yanagisawa, T.; Kawashima, K. Flat band in pyrochlore oxides: A first-principles study. Nanomaterials 2019, 9, 876. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Jin, K.-H.; Huang, H.; Wang, Z.; Liu, F. Weyl points created by a three-dimensional flat band. Phys. Rev. B 2019, 99, 201105R. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Luo, K.; Chen, Z.; Zhu, Z.; Yu, R.; Fang, C.; Weng, H. Topological phases in pyrochlore thallium niobate Tl2Nb2O6+x. NPJ Comput. Mater. 2019, 5, 105. [Google Scholar] [CrossRef] [Green Version]
- Blaha, P.; Schwarz, K.; Madsen, G.K.H.; Kvasnicka, D.; Luitz, J. WIEN2k, an Augmented Plane Wave + Local Orbitals Program for Calculating Crystal Properties; Vienna University of Technology: Vienna, Austria, 2001. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3875. [Google Scholar] [CrossRef] [Green Version]
- Ramadass, N.; Palanisamy, N.; Gopalakrishnan, J.; Aravamudan, G.; Sastri, M.V.C. Some ABO3 Oxides with defect pyrochlore structure. Solid State Commun. 1975, 17, 545–575. [Google Scholar] [CrossRef]
- Belyaev, I.N.; Lupeiko, T.G.; Nalbandyan, V.B.; Abanina, E.V. Exchange reactions in systems comprising alkali metal, silver, and thallium sulphates, niobates, and tantalates. Russ. J. Inorg. Chem. 1978, 23, 18–22. [Google Scholar]
- MacDonald, A.H.; Pickett, W.E.; Koelling, D.D. A linearised relativistic augmented-plane-wave method utilising approximate pure spin basis function. J. Phys. C Solid State Chem. 1980, 13, 2675–2683. [Google Scholar] [CrossRef]
- Singh, D. Plane Waves, Pseudopotentials and the LAPW Method; Springer: Berlin/Heidelberg, Germany, 1994. [Google Scholar]
- Kikuchi, N.; Samizo, A.; Ikeda, S.; Aiura, Y.; Mibu, K.; Nishio, K. Carrier Generation in a p-type Oxide Semiconductor Sn2(Nb2-xTax)O7. Phys. Rev. Mater. 2017, 1, 021601R. [Google Scholar] [CrossRef]
- Sleight, A.W. Valency, valence degeneracy, ferroelectricity and superconductivity. Prog. Solid State Chem. 2009, 37, 251–261. [Google Scholar] [CrossRef]
- Taraphder, A.; Krishnamurthy, H.R.; Pandit, R.; Ramakrishnan, T.V. Negative-U extended hubbard model for doped barium bismathates. Phys. Rev. B 1981, 52, 1368. [Google Scholar] [CrossRef] [Green Version]
- Varma, C.M. Missing valence states, diamagnetic insulators, and superconductors. Phys. Rev. Lett. 1988, 61, 2713–2716. [Google Scholar] [CrossRef]
- Hase, I.; Tanagisawa, T. Madelung energy of the valence-skipping compound BaBiO3. Phys. Rev. B 2007, 76, 174103. [Google Scholar] [CrossRef] [Green Version]
- Ubic, R.; Reaney, I.M. Structure and Dielectric Properties of Lead Pyrochlores. J. Am. Ceram. Soc. 2012, 85, 2472–2478. [Google Scholar] [CrossRef]
Figure 1.
(a) Pylochlore lattice in the face-centered cubic Bravais lattice. The large circle shows the 4 atomic sites in the primitive unit cell, forming a regular tetrahedron. These tetrahedra shear corners and form a three-dimensional network; (b) energy dispersion of the Guo and Franz (GF) model with λ = 0 (Equation (2)). Numbers 1–4 denote the band index. Energy offset is added to simulate the band structure of Pb2Ta2O7. Note that the total bandwidth is large (= 8t), while the flat bands (FBs) (E3 and E4) does not have any dispersion; (c) a schematic picture of the energy dispersion of the GF model for λ > 0. At the Γ point the degeneracy is partly lifted, but at the X and L points the degeneracy is protected by symmetry, so the system remains gapless; (d) a schematic picture of the energy dispersion of the GF model for λ < 0. In this case the system opens a gap, and this becomes a strong TI [6].
Figure 1.
(a) Pylochlore lattice in the face-centered cubic Bravais lattice. The large circle shows the 4 atomic sites in the primitive unit cell, forming a regular tetrahedron. These tetrahedra shear corners and form a three-dimensional network; (b) energy dispersion of the Guo and Franz (GF) model with λ = 0 (Equation (2)). Numbers 1–4 denote the band index. Energy offset is added to simulate the band structure of Pb2Ta2O7. Note that the total bandwidth is large (= 8t), while the flat bands (FBs) (E3 and E4) does not have any dispersion; (c) a schematic picture of the energy dispersion of the GF model for λ > 0. At the Γ point the degeneracy is partly lifted, but at the X and L points the degeneracy is protected by symmetry, so the system remains gapless; (d) a schematic picture of the energy dispersion of the GF model for λ < 0. In this case the system opens a gap, and this becomes a strong TI [6].

Figure 2.
(a) Band structure of Pb2Ta2O7. (b) DOS curve of Pb2Ta2O7. (c) Energy dispersion of the GF model with λ = 0. (d) Blow-up figure of the valence band of Pb2Ta2O7 near Γ point, shown in the circle on Figure 1a. The units of the vertical axes are eV for (a), (c) and (d), and eV−1 for (b).
Figure 2.
(a) Band structure of Pb2Ta2O7. (b) DOS curve of Pb2Ta2O7. (c) Energy dispersion of the GF model with λ = 0. (d) Blow-up figure of the valence band of Pb2Ta2O7 near Γ point, shown in the circle on Figure 1a. The units of the vertical axes are eV for (a), (c) and (d), and eV−1 for (b).

Figure 3.
Effective spin–orbit coupling constant λ for eight A2B2O7 pyrochlore oxides (units are eV). The left panels are for B = Nb, and the right panels are for B = Ta. A-site elements are arranged in the order of the periodic table. For example, the upper-left column in the left panel means In2Nb2O7.
Figure 3.
Effective spin–orbit coupling constant λ for eight A2B2O7 pyrochlore oxides (units are eV). The left panels are for B = Nb, and the right panels are for B = Ta. A-site elements are arranged in the order of the periodic table. For example, the upper-left column in the left panel means In2Nb2O7.

Figure 4.
(a) Band structure of Sn2Zr2O7 and (b) its blow-up figure of the valence band near the Γ point. (c) Band structure of In2Nb2O7 and (d) its blow-up figure of the valence band near the Γ point. The units of the vertical axes are eV.
Figure 4.
(a) Band structure of Sn2Zr2O7 and (b) its blow-up figure of the valence band near the Γ point. (c) Band structure of In2Nb2O7 and (d) its blow-up figure of the valence band near the Γ point. The units of the vertical axes are eV.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hase, I.; Yanagisawa, T. Possible Three-Dimensional Topological Insulator in Pyrochlore Oxides. Symmetry 2020, 12, 1076. https://doi.org/10.3390/sym12071076
AMA Style
Hase I, Yanagisawa T. Possible Three-Dimensional Topological Insulator in Pyrochlore Oxides. Symmetry. 2020; 12(7):1076. https://doi.org/10.3390/sym12071076
Chicago/Turabian StyleHase, Izumi, and Takashi Yanagisawa. 2020. "Possible Three-Dimensional Topological Insulator in Pyrochlore Oxides" Symmetry 12, no. 7: 1076. https://doi.org/10.3390/sym12071076
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.