
Abstract
Cervical cancer is the second most common leading cause of women's death due to cancer worldwide, about 528,000 patients’ cases and 266,000 deaths per year, related to human papillomavirus (HPV). Peptide-based vaccines being safe, stable, and easy to produce have demonstrated great potential to develop therapeutic HPV vaccine. In this study, the major histocompatibility complex (MHC) class I, class II T cell epitopes of HPV16-E7 were predicted. Therefore, we designed a plan to find the most effective peptides to prompt appropriate immune responses. For this purpose, retrieving protein sequences, conserved region identification, phylogenic tree construction, T cell epitope prediction, epitope-predicted population coverage calculation, and molecular docking were performed consecutively and most effective immune response prompting peptides were selected. Based on different tools index, six CD8+ T cells and six CD4+ epitopes were chosen. This combination of 12 epitopes created a putative global vaccine with a 95.06% population coverage. These identified peptides can be employed further for peptide analysis and can be used as a peptide or poly-epitope candidates for therapeutic vaccine studies to treat HPV-associated cancers.
Similar content being viewed by others


Introduction
Cervical cancer is the second most common cause of women's death in world (266,000 deaths every year), related to human papillomavirus (HPV) (Li et al. 2010). At least 170 HPV genotypes have been categorized into low-risk and high-risk types. Infection by high-risk HPV types, particularly HPV-16, is the primary etiological factor in the development of cervical cancer. Persistent infection with HPV-16 contributes to progress and maintenance of cervical cancer by more than 50% compared to other factors (Doorbar 2005). The genome of HPV encodes early proteins (E1, E2, E3, E4, E5, E6, and E7) and structural proteins (L1 and L2). Among nonstructural proteins, E7 and E6 proteins interact with negative regulator of the cell cycle, leading the host cell transformation and cellular immortalization (Namvar et al. 2020).
Prophylactic vaccines being available in many countries decrease the high prevalence and the deaths of cervical cancers. Despite the fact that these vaccines have been effective in preventing HPV infections, there are few reports available which explain their therapeutic effect on established infections. The optimal coverage of these vaccines for a large number of women with developed high-grade dysplasia, carcinoma in situ, and cervical cancer is not determined yet (Li et al. 2017). Therefore, despite advances in radiotherapy, chemotherapy and surgery treatments in women, cervical cancer is the leading cause of death yet, so alternative methods such as therapeutic vaccines are establishing(de Oliveira et al. 2015; Liu et al. 2012). Consequently, various types of therapeutic vaccines (cell-based vaccines, RNA/DNA, protein/peptide, and live bacterial/viral vectors) have been explored for treating HPV-associated diseases (Chabeda et al. 2018).
Most of these vaccines couldn’t suppress tumor growth in animal models successfully and usually don’t show remarkable therapeutic efficacy. Vaccines involving SGN-00101 (a fusion protein of heat shock protein and HPV16 E7) (Roman et al. 2007), ZYC-101a (a DNA vaccine against E6 and E7 of HPV 16 and 18) (Coukos et al. 2005), L1VLP-E7 (chimeric VLPs of HPV 16 L1 and E7) (Kawana et al. 2012), TA-HPV (a recombinant vaccinia virus expressing E6 and E7) (Kaufmann et al. 2002), TA-CIN (a fusion protein consisting of E6, E7 and L2) (Van der Burg et al. 2001), and PC10VAC01 (HPV16 E7+ adenylate cyclase) have passed clinical trials as well; (Khallouf et al. 2014).To achieve effective treatment, The peptide-based vaccines that are safe, stable, highly selective, multivalent and easy to produce, and transport have demonstrated great potential in developing therapeutic HPV vaccine (Panahi et al. 2018).
Therapeutic vaccines, unlike traditional preventative vaccines, activate the cell-mediated immune response. In human immune system, T-lymphocytes (CD4+ and CD8+) play crucial role in identifying and eliminate cancerous cells. CD8+ T-lymphocyte, known as killer cell acts in cell-mediated response and protects the cell from many viruses such as HPV by activating major histocompatibility complex (MHC) class I. MHC class I binds to immunogenic peptide and then these complexes transformed the cell surface. The CD8+ T-lymphocyte recognizes the presented complexes through T cell receptors and releases cytotoxic substances to destroy infected cells. Furthermore, the CD4+ T cells perform co-stimulation, proper activation of CD8+ T cells and triggering of immune response, so CD4+ T cells are recognized as T helper (Th) cells. CD4+ T-lymphocytes are stimulated by the peptides and MHC class II complexes located on antigen-presenting cells. Unlike traditional virus vaccines that induce antibody, peptide vaccines activate T cell response, effectively. Peptide vaccine can induce specific T-cell responses using specific peptide antigens that can only be presented by class I and II MHC molecules to T cells. Therefore, the selection of target antigens is a key factor for designing a therapeutic vaccine. Besides, the implementation of informatics tools and online databases can reduce time and experimental costs to develop vaccines (Kaliamurthi et al. 2018; Van Der Burg 2008).
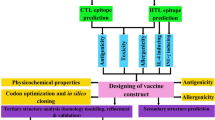
In this investigation, an immunoinformatics method was applied for predicting and assessing T-cell antigenic sites of the E7 protein of HPV types 16 to find out potential peptides for the development of therapeutic vaccines against HPV type 16. Therefore, we designed a plan to find the most effective peptides to prompt an appropriate immune response. Retrieval of protein sequences, conserved region identification, phylogenic tree drawing, T cell epitope prediction, epitope-predicted population coverage calculations, and molecular docking were performed, sequentially. Selected peptides in the present study can be applied for further peptide analysis and for assessing their potential as a peptide or poly-epitope therapeutic vaccine for HPV-associated cancers.
Methods
Retrieving Protein Sequences
HPV16-E7 complete protein sequences of the worldwide population were recovered from the National Centre for Biotechnology Information (NCBI) database, (https://ncbi.nlm.nih.gov) in FASTA format. CustalW2 software (T-coffee server) was used to align the obtained sequences. The sequences were trimmed and analyzed by Bioedit software (version 7.1.3.0).
Conserved Region Identification
After alignments of datasets (https://tcoffee.crg.cat/), to find conserved and mutation regions in aligned sequences Shannon entropy value was evaluated by Bioedit software. The entropy plot was used to identify the variation of amino acid in each position in the protein sequences.
Phylogenic Tree
MEGA software (version.7) was used to construct a phylogenic tree. The phylogenic measurements were performed by maximum likelihood statistical analysis in 100 bootstrap replications and Jones–Taylor–Thornton (JTT) model. The average evolutionary divergence over all sequence pairs was estimated, and the similarity between isolates was also calculated by MEGA software (version 7). To create consensus sequences, the selected sequences were entered into BioEdit software, and analyzed.
T Cell Epitope Prediction
IEDB MHC class I binding tool (https://tools.iedb.org/mhci/) was used for CD8+ epitope prediction with default recommended method and IEDB HLA allele reference set. IEDB MHC class II binding tool (https://tools.iedb.org/mhcii/) with “human, HLA-DR” allele reference set was used for CD4+ epitope prediction (Wang et al. 2008, 2010). Epitopes are 9 or 10(MHC class I) and 16 (MHC class II) amino acids in length.
Calculating Epitope-Predicted Population Coverage
Population protection coverage (PPC) is the percentage of the population that has at least one allele that could display at least one epitope within a group. The selected conserved CD8+ and CD4+ epitopes were entered into the IEDB population coverage tool (https://tools.iedb.org/ tools/population/iedb_input) to calculate epitope-predicted population coverage.
Molecular Docking
To predict the chemical structure of the selected peptides as a ligand, PEP-FOLD3 was used, accessible from: (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/). PEP-FOLD3 carried out with 100 simulations; each simulation showed a different zone of the conformational space. It restore a file of all the models created, the detail of the clusters, and the best conformation of the five best clusters. Prediction of the structure from the input peptide sequence was performed by the sOPEP model suboptimal conformation sampling method. Amino acid sequences of selected HLA were retrieved from IPD-IMGT/HLA Database (https://www.ebi.ac.uk/ipd/imgt/hla/allele.html). To find a three-dimensional (3D) structure of HLA as target receptors, the FASTA format of any HLA was given as an input to the SWISS-MODEL database to search template with high homology or build a model within the duration of 30 min to 1 h (https://swissmodel.expasy.org/interactive). Hex 8.0.0 tool was employed for docking the elite peptide models with determined HLA. The peptide as ligand and the HLA as receptor were selected, and docking control were activated under 100 solution and correlation type in “shape+Electro+DARS” mode. The results were revealed as RSM, E shape, and total.
Results
Shannon’s Entropy Plot
The 1397 protein sequences of HPV-E7 were extracted from the NCBI database. All of the sequences were aligned using ClustalW software (T-coffee server). Aligned sequences were trimmed (includes exchanging unknown spots with determined amino acid and removing short sequences) and analyzed (attained Entropy plot) using BioEdit software. Entropy values were varied from 0.0 (high conservation) to 1.0 (high variable). Amino acid residues that have entropy value of less than 0.1 as a threshold were considered as conserved.
Shannon's entropy plot showed that except for some, other amino acid residues in HPV16-E7 are located below 0.1 and are conserved among sequences (Fig. 1). So variability analysis showed that HPV-E7 protein is a conserved protein totally, this feature facilitated the selection of representative sequences for the next steps.

Shannon’s entropy plot of HPV16-E7. The plot shows that E7 approximately conserved protein. Among peptides, 33th peptide undergoes the most variation across isolate
Phylogenetic Tree for Selection of Vaccinal Sequences
The JTT model and maximum likelihood statistical method were employed for tree-building using MEGA software. The 1387 complete protein sequences of HPV-E7 were inserted to MEGA software, and a phylogenetic tree was constructed. The genetic distances or number of substitutions are displayed at the bottom of the tree, and numbers at the branch nodes are related to 100 bootstrap repetitions (%).The obtained result revealed that the overall mean distance was 0.019. To design a broad-spectrum strain vaccine, we selected fourteen isolates from Iran, USA, Japan, Sweden, Germany, Congo, Brazil, India, Thailand, South Korea, Costa Rica, and Taiwan for further analysis. Among these sequences, the consensus sequence was created with a 70% threshold frequency.
The position of 29th amino acid in these 14 sequences showed the most variability compared to the other amino acids (was the most variable amino acid in the sequence). Asparagine was more frequent than cysteine and glutamine in this position. Asparagine is larger than two other mentioned amino acids. It could link to oligosaccharides with n-glycosylated bonds that lead to more complexity and immunogenicity (larger size of the asparagine compared to two other mentioned amino acids and its ability to participate in n-glycosylation bond with oligosaccharides lead to more complexity and immunogenicity).
CD8+ T-Cell Epitope Prediction and Population Coverage Calculation
In total, CTL epitopes were predicted from E7 based on the HLA allele reference set by IEDB (percentile rank < 2). Among epitopes, the 28 epitopes showed a lower percentile rank and binding affinity for HLA alleles utilizing the IEDB analysis resource Consensus tool (Kim et al. 2012), which mixes ANN aka NetMHC (Andreatta and Nielsen 2016; Lundegaard et al. 2008; Nielsen et al. 2003), SMM (Peters and Sette 2005) and Comblib (Sidney et al. 2008) predictions. The percentile rank, the MHC I restricted alleles and methods that used for different peptide prediction are arranged in Table 1. Epitopes that bound weakly to MHC allele were rejected (percentile rank > 2). According to these results, where multiple epitopes had identical HLA binding profiles, a single representative epitope was selected. For example, DRAHYNIVTF48–57, RAHYNIVTF49–57, and RAHYNIVTFC49–58 have banding profiles covered by DRAHYNIVTFC. Using this approach, epitopes were reduced from 28 to 16 (Table 1).
The calculation of the population coverage (PC) of the 16 selected MHC Class I epitopes was done utilizing the IEDB PPC tool. It supplied allele frequencies from different region in 5 continents which encompass16 different geographical areas. All of the epitopes were analyzed one by one and data was arranged in Table 2. According to prediction and PPC, six MHC class I epitopes were selected: DLLMGTLGIV81–90 with high binding affinity to HLA-A*02:01, HLA-A*02:03, HLA-A*02:06 and highest PC in the world (41.35%), Asia [East (35.99%), Northeast (30.11%), Southeast (30.46%.), Southwest ( 27.03%)], Africa [East (20.47%), North (25.99%), West (20.75%)], Europe 47.37%, DRAHYNIVTFC48–58 with high binding affinity to HLA-A*32:01, HLA-B*15:01, HLA-B*35:01, HLA-B*57:01, HLA-A*23:01, HLA-A*24:02 and great PC in world 27.97. Asia [East (30.71%), Northeast (20.44%), South (22.08%)], Europe (33.69%), Africa [Central (29.85%), East (23.69%), North (36.44%) South (28.64%), West (47.38%)], AEPDRAHYNI45–54 with high binding affinity to HLA-A*24:02, HLA-B*51:01, HLA-B*44:02 and great PC in world (32.97%), Asia [East (51.71%), Northeast (30.57%), Oceania (54.11%), South (24.65%), Southeast (24.22%)], Europe (35.4%), America [North (34.55%), South (29.37%), West indies (31.71%)], YMLDLQPETT11–20 with high binding affinity to HLA-A*02:01, HLA-A*02:06 and great PC in the world (40.6%), Asia [East (35.65%), Northeast (20.34%), Southeast (20.92%), Southwest (27.03%)], East of Africa (20.47%), America [North (44.14%), South (21.85%), West Indies (32.19%)], GIVCPICSQK88–97 with high binding affinity to HLA-A*11:01, HLA-A*68:01, HLA-A*03:01 and great PC in the world (35.75%), Asia [Northeast (47.38%), Oceania (38.45%), South (42.69%), Southeast (37.5%), Southwest (27.27%)], Europe (35.75%), Africa [South (25.62%), West (20.04%)], America [North (29.55%), South (23.47%), West Indies (27.65%)] and DLQPETTDLYCYE14–26 with great affinity to HLA-B*35:01, HLA-B*15:01, HLA-A*26:01, HLA-A*01:01, HLA-B*44:03, HLA-A*68:01, HLA-A*26:01and high PC in the world (43.79%), Asia [East (50.33%), Northeast (26.36%), Oceania (20.77%), South (41.19%), Southwest (35.9%)], Europe (54.75%), Africa [Central (28.95%), East (24.35%), North (36.41%), South (28.26%), West Indies (44.65%)] (Table 3).
The cumulative PC of six selected MHC class I epitopes were also measured. The great PC achieved for individual epitope was 43.79%, meanwhile epitope combination show more PC (95.06%). The highest PC was observed in West Indies (93.51%), West Africa (81.6%), Southeast Asia (89.42), Southwest Asia (83.61%), South Asia (87.77%), Northeast Asia (89.5%), North America (94.46%), North Africa (83.85%), Europe (98.39%), Oceania (91.28%), and East Asia (95.01%) (Table 2).
CD4+ T-Cell Epitope Prediction and Population Coverage Calculation
As mentioned above, T-helper epitopes were predicted from E7 based on the HLA allele reference set by IEDB (percentile rank < 10). Among epitopes, the 29 epitopes showed a lower percentile rank and binding affinity for HLA alleles using the IEDB analysis resource Consensus tool (Wang et al. 2008, 2010). The percentile rank, MHC II-restricted alleles and methods used for different peptide prediction are arranged in Table 4.
The calculation of the population coverage (PC) of the 29 selected MHC Class I epitopes was done utilizing the IEDB PPC tool. It supplied allele frequencies from different region in 5 continents which covered 16 different geographical areas. All of the epitopes were analyzed one by one, and data was arranged in Table 4, and according to the prediction and PPC, six MHC class II epitopes were selected: TLHEYMLDLQPETTDL, LHEYMLDLQPETTDLY, HGDTPTLHEYMLDLQP, EQLNDSSEEEDTDEID, VQSTHVDIRTLEDLLM, and YCYEQLNDSSEEEDTD.
Composition of MHC Class I and MHC Class II Epitopes to Create Potential Vaccines with > 90% Population Protection Coverage
A putative global vaccine with a population coverage of 95.06% was generated by the composition of 12 epitopes (six CD8+ and six CD4+) (Table 5).
Molecular Docking Studies
To know selected peptides binding affinity to HLA molecules, we performed molecular docking using Hex 8.0.0 software using 3D peptide (Table 6) and 3D HLA as.pdb formatted. Based on the energy values estimated, the docking result showed that selected peptides bind to the target HLA strongly and release higher binding energy.
Table 7 depicts the interaction of CD8+ MHC class I epitopes with different HLA alleles in docking investigation. Docking of all the epitopes were performed with structurally determined targets. Overall, 32 docked complexes showed a variety of binding affinities that showed as global energy (− 216.09 to − 612.62 kcal/mol), RSM-1. Also, the interaction of CD4+ MHC class II epitopes with different HLA alleles in docking investigation. All data were summarized in Table 8, which shows a range of binding affinities in terms of global energy (− 201.54 to − 603.96 kcal/mol), RSM-1. Also, the model complexes and amino acid interaction between candidate peptides and their HLA represent in Fig. 2.

Docked selected epitopes at their MHC-I binding site. The LLMGTLGIV, RAHYNIVTF, and IVCPICSQK randomly were selected among high global energy. a Peptide LLMGTLGIV at its receptor HLA-A*02:01. b Peptide RAHYNIVTF at its receptor HLA-B*35:01. c Peptide IVCPICSQK at its receptor HLA-A*03:01. d Peptide IVCPICSQK at its receptor HLA-A*68:01
Discussion
Cervical cancer is globally the second most common leading cause of women's death by cancer. Infection by high-risk human papillomavirus (HPV), particularly those related to HPV-16, is the primary etiological factor in the development of cervical cancer. One promising immunotherapeutic procedure for eliminating HPV-associated tumors is therapeutic HPV vaccines, which are generally designed to boost production and activation CD4+ and CD8+ T cells (de Oliveira et al. 2015).
Numerous therapeutic vaccines couldn’t suppress tumor growth in animal models successfully and usually didn’t show remarkable therapeutic efficacy. Vaccines involving SGN-00101 (a fusion protein of heat shock protein and HPV16 E7) (Roman et al. 2007), ZYC-101a (a DNA vaccine against E6 and E7 of HPV 16 and 18) (Coukos et al. 2005), L1VLP-E7 (chimeric VLPs of HPV 16 L1 and E7) (Kawana et al. 2012), TA-HPV (a recombinant vaccinia virus expressing E6 and E7) (Kaufmann et al. 2002), TA-CIN (a fusion protein consisting of E6, E7 and L2) (Van der Burg et al. 2001), and PC10VAC01 (HPV16 E7+ adenylate cyclase) passed clinical trials (Khallouf et al. 2014). The clinical trials showed that initial efforts at stimulating immune responses through specific T cells via therapeutic vaccinations were not acceptable completely. Consequently, there is still an important requirement to develop new treatment approaches for HPV-associated cancers (Chu et al. 2018). The peptide-based vaccines that are safe, specific, and easy to produce have demonstrated great potential for the development of therapeutic HPV vaccine. Immunoinformatics has been used for predicting antigenic peptide B-cell and T-cell epitopes for peptide vaccine design. Then binding affinity of predicted epitopes -MHC interactions were evaluated by molecular docking studies since eliciting a proper CTL response is highly dependent on the binding affinity of peptides to an antigen-presenting MHC-I protein. Usually, the docking method and results should be validated against the available experimental peptide-MHC complex structures [18].
The present study employed immunoinformatics and docking to predict and evaluate candidate peptides of E7 protein (belonging to HPV types 16) binding affinity.
In silico analysis for epitope prediction was used in many studies against various pathogens previously (Abu-Haraz et al. 2017; Khan et al. 2006; Kumar et al. 2015; Nezafat et al. 2014; Srivastava et al. 2016; Tsang et al. 2017), but the prediction and selection of appropriate peptides to induce strong responses remain ambiguity. Several therapeutic HPV vaccines have been designed to stimulate CD4+ and CD8+ T lymphocytes targeting of two key HPV oncoproteins, E6 or E7. Some studies recommended that use E6 or E7 synthetic long peptide (SLPs) instead, peptides led to the increase of T lymphocyte function in vivo (Bijker et al. 2007; Vambutas et al. 2005).
In the present study, an optimized selection of conserved E7 epitopes utilized to design HPV16 therapeutic vaccine utilizing. A final combination of six conserved CD8+ and six conserved CD4+ epitopes from predicted MHC class I and II epitopes that originally identified in IEDB, had a cumulative world population coverage value 95.06%. Besides, according to Serrano et al. article that published in 2017: “Asia accumulates the vast majority of cervical cancer cases (285,000 cases; 54.0%) and deaths (144,000 deaths; 54.7%), followed by Africa (99,000 new cases and 60,000 deaths) and the Americas (83,000 new cases and 36,000 deaths). Of note, China and India account for 62,000 and 123,000 new cases and 30,000 and 67,000 deaths, respectively “(Serrano et al. 2018). The efforts have also done to design a putative vaccine with approximately 90%, 90%, 70%, 88%, and 80% targeting Asia, Africa, America, China, India, respectively.
The first investigation about the HPV epitope was performed in 1993. The RAHYNIVTF, HPV16-E7 sequence as an MHC-I epitope, can induce CD8+ T cells responses and remove HPV-related tumors in mice (Feltkamp et al. 1993). In addition, The RAHYNIVTF has been previously utilized in various therapeutic vaccines that elicited excellent immune protection against tumor, which expresses HPV-16 E7 protein in C57BL/6 mice. In 2002, Zwaveling et al. achieved considerable success in a preclinical model when mice were vaccinated with an H-2Db-restricted HPV16 E7 CTL epitope (E749-57, RAHYNIVTF) emulsified in IFA (Zwaveling et al. 2002). This sequence is one of the final HPV16-E7 predicted epitopes in our study.
In 1995, the immunogenicity of 9 HLA-A*0201-binding epitopes of HPV16 E6, and E7 was evaluated in vivo in HLA-A*0201 Kb transgenic mice and in vitro in CTL cultures by Ressing et al. Three of them (11–20, YMLDLQPETT; 82–90, LLMGTLGIV; 86–93, TLGIVCPI) were also very potential to CTL induction. The HPV16 E7-containing HLA-A*0201+ cervical carcinoma cell line CaSki was lysed with human CTL clones specific for these three peptides. Not only these E7-derived peptides, are probable to represent naturally processed human CTL epitopes of HPV16 but also these three HPV16-encoded peptides have the highest affinity of binding to the HLA-A*0201 molecule (Ressing et al. 1995). First and second peptides were involved in our study.
In 2004, Nakagawa et al. described E7 7–15 of HPV 16 [TLHEYMLDL] as a new antigenic epitope that has oncogenic activities. E7 7–15 was distinguished between peptides formerly exhibited binding ability to human leukocyte antigen (HLA)-A2.1 molecule, but it was identified probably to be limited by the HLA-B48 molecule (Nakagawa et al. 2004). This peptide also was predicted as a part of the sequences in our study.
In 2018, Krishna et al. evaluated HPV16-specific CD8+ T lymphocytes immunogenicity and recognized 16 strong and 29 moderately immunogenic CTL-epitopes from HPV16 E2, E6, and E7 antigens including GIVCPICSQK restricted by 12 common HLA class I alleles(Krishna et al. 2018).
Other peptides were assumed as novel peptides that need a further experimental survey for confirmation.
Among all of the predicted peptides in our study: RTLEDLLMGTL (Blatnik et al. 2018), HYNIVTFCCK (Krishna et al. 2018), HGDTPTLHEY (Krishna et al. 2018), RLCVQSTHV (Feltkamp et al. 1993), TLHEYMLDL (Kruse et al. 2019), (Tsang et al. 2017) from HPV-16 E7 were reported as CD8+ T cells experimentally.
Using molecular docking studies, the binding affinity of the predicted peptide fragments was surveyed in the peptide-MHC interactions, due to the key role of sufficient binding affinity to an antigen-presenting MHC-I protein in triggering CD8+ responses. Usually, the docking method and modeling required to confirm the available experimental peptide–MHC complex structures. However, due to limitations of current docking methods, they cannot always create docking poses similar to the experimentally verified binding modes (Jabbar et al. 2018). In our study, the HEX 8.0.0 was applied for molecular docking analysis, which has several advantages: (1) No information about the structure and binding site of peptide needed. (2) Using Spherical Polar Fourier (SPF) correlations algorithm rather than the Fast Fourier Transform (FFT) based search. SPF with five rotational and one transnational degrees of freedom is preferable as compared to Fast Fourier FFT, which has 3 rotational and 3 transnational degrees of freedom. The preference of SPF versus FFT is because of reduction of the performance time a few minutes. (3) Hex is based on densely sampling of the search space. Hex make clustering for solutions to show similar orientations. Hex gives score and use the calculation of shape complimentary for this scoring and exclude volume, while having an optimal in vacuo electrostatic contribution. (Agrawal et al. 2019; Macindoe et al. 2010).
Conclusion
It is concluded that immunoinformatics and molecular modeling approaches can predict efficient antigenic peptides for the therapeutic HPV vaccine development. In our study, some HPV16-E7 peptides were identified that could be applied for developing therapeutic HPV vaccine with a population coverage 95.06%. Furthermore, we evaluated epitope binding affinities by docking approach, detailed binding free global energy calculation of the peptide-MHC I (or MHC-II) complexes. Our selected peptides can be applied in further peptide analysis and used as peptide or poly-epitope candidate in therapeutic vaccine studies for HPV-associated cancers.
References
Abu-Haraz AH, Abd-Elrahman KA, Ibrahim MS, Hussien WH, Mohammed MS, Badawi MM, Salih MA (2017) Multi epitope peptide vaccine prediction against Sudan Ebola virus using immuno-informatics approaches. Adv Tech Biol Med. https://doi.org/10.4172/2379-1764.1000203
Agrawal P, Singh H, Srivastava HK, Singh S, Kishore G, Raghava GP (2019) Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinformatics 19:426
Andreatta M, Nielsen M (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32:511–517
Bijker MS, van den Eeden SJ, Franken KL, Melief CJ, Offringa R, van der Burg SH (2007) CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J Immunol 179:5033–5040
Blatnik R et al (2018) A targeted LC-MS strategy for low-abundant HLA class-I-presented peptide detection identifies novel human Papillomavirus T-cell epitopes. Proteomics 18:1700390
Chabeda A, Yanez RJ, Lamprecht R, Meyers AE, Rybicki EP, Hitzeroth II (2018) Therapeutic vaccines for high-risk HPV-associated diseases. Papillomavirus Res 5:46–58
Chu X et al (2018) Combined immunization against TGF-β1 enhances HPV16 E7-specific vaccine-elicited antitumour immunity in mice with grafted TC-1 tumours. Artif Cells Nanomed Biotechnol 46:1199–1209
Coukos G, Conejo-Garcia JR, Roden RB, Wu TC (2005) Immunotherapy for gynaecological malignancies. Expert Opin Biol Ther 5:1193–1210
de Oliveira LMF et al (2015) Design, immune responses and anti-tumor potential of an HPV16 E6E7 multi-epitope vaccine. PLoS ONE 10(9):e0138686
Doorbar J (2005) The papillomavirus life cycle. J Clin Virol 32:7–15
Feltkamp MC et al (1993) Vaccination with cytotoxic T lymphocyte epitope-containing peptide protects against a tumor induced by human papillomavirus type 16-transformed cells. Eur J Immunol 23:2242–2249
Jabbar B et al (2018) Antigenic peptide prediction from E6 and E7 oncoproteins of HPV types 16 and 18 for therapeutic vaccine design using immunoinformatics and MD simulation. Anal Front Immunol 9:3000
Kaliamurthi S, Selvaraj G, Kaushik AC, Gu K-R, Wei D-Q (2018) Designing of CD8+ and CD8+-overlapped CD4+ epitope vaccine by targeting late and early proteins of human papillomavirus. Biologics 12:107
Kaufmann AM et al (2002) Safety and immunogenicity of TA-HPV, a recombinant vaccinia virus expressing modified human papillomavirus (HPV)-16 and HPV-18 E6 and E7 genes, in women with progressive cervical cancer. Clin Cancer Res 8:3676–3685
Kawana K, Adachi K, Kojima S, Kozuma S, Fujii T (2012) Suppl 2: therapeutic human papillomavirus (HPV) vaccines: a novel approach. Open Virol J 6:264
Khallouf H, Grabowska AK, Riemer AB (2014) Therapeutic vaccine strategies against human papillomavirus. Vaccines 2:422–462
Khan AM et al (2006) A systematic bioinformatics approach for selection of epitope-based vaccine targets. Cell Immunol 244:141–147
Kim Y et al (2012) Immune epitope database analysis resource. Nucleic Acids Res 40:W525–W530
Krishna S et al (2018) Human papilloma virus specific immunogenicity and dysfunction of CD8+ T cells in head and neck cancer. Cancer Res 78:6159–6170
Kruse S et al (2019) Therapeutic vaccination using minimal HPV16 epitopes in a novel MHC-humanized murine HPV tumor model. Oncoimmunology 8:e1524694
Kumar A, Yadav IS, Hussain S, Das BC, Bharadwaj M (2015) Identification of immunotherapeutic epitope of E5 protein of human papillomavirus-16: an in silico approach. Biologicals 43:344–348
Li J et al (2017) A novel therapeutic vaccine composed of a rearranged human papillomavirus type 16 E6/E7 fusion protein and Fms-like tyrosine kinase-3 ligand induces CD8+ T cell responses and antitumor effect. Vaccine 35:6459–6467
Li Y-L, Qiu X-H, Shen C, Liu J-N, Zhang J (2010) Vaccination of full-length HPV16 E6 or E7 protein inhibits the growth of HPV16 associated tumors. Oncol Rep 24:1323–1329
Liu T-Y, Hussein WM, Toth I, Skwarczynski M (2012) Advances in peptide-based human papillomavirus therapeutic vaccines. Curr Top Med Chem 12:1581–1592
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M (2008) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 36:W509–W512
Macindoe G, Mavridis L, Venkatraman V, Devignes M-D, Ritchie DW (2010) HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res 38:W445–W449
Nakagawa M, Kim KH, Moscicki A-B (2004) Different methods of identifying new antigenic epitopes of human papillomavirus type 16 E6 and E7 proteins. Clin Diagn Lab Immunol 11:889–896
Namvar A, Panahi HA, Agi E, Bolhassani A (2020) Development of HPV 16, 18, 31, 45 E5 and E7 peptides-based vaccines predicted by immunoinformatics tools. Biotechnol Lett 42(3):403–418
Nezafat N, Ghasemi Y, Javadi G, Khoshnoud MJ, Omidinia E (2014) A novel multi-epitope peptide vaccine against cancer: an in silico approach. J Theor Biol 349:121–134
Nielsen M et al (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 12:1007–1017
Panahi HA, Bolhassani A, Javadi G, Noormohammadi Z (2018) A comprehensive in silico analysis for identification of therapeutic epitopes in HPV16, 18, 31 and 45 oncoproteins. PLoS ONE 13(10):e0205933
Peters B, Sette A (2005) Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC Bioinformatics 6:132
Ressing ME et al (1995) Human CTL epitopes encoded by human papillomavirus type 16 E6 and E7 identified through in vivo and in vitro immunogenicity studies of HLA-A* 0201-binding peptides. J Immunol 154:5934–5943
Roman L et al (2007) A phase II study of Hsp-7 (SGN-00101) in women with high-grade cervical intraepithelial neoplasia. Gynecol Oncol 106:558–566
Serrano B, Brotons M, Bosch FX, Bruni L (2018) Epidemiology and burden of HPV-related disease. Best Pract Res Clin Obstet Gynaecol 47:14–26
Sidney J, Assarsson E, Moore C, Ngo S, Pinilla C, Sette A, Peters B (2008) Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res 4:2
Srivastava PN, Jain R, Dubey SD, Bhatnagar S, Ahmad N (2016) Prediction of epitope-based peptides for vaccine development from coat proteins GP2 and VP24 of Ebola virus using immunoinformatics. Int J Pept Res Therap 22:119–133
Tsang KY et al (2017) Identification and characterization of enhancer agonist human cytotoxic T-cell epitopes of the human papillomavirus type 16 (HPV16) E6/E7. Vaccine 35:2605–2611
Vambutas A et al (2005) Therapeutic vaccination with papillomavirus E6 and E7 long peptides results in the control of both established virus-induced lesions and latently infected sites in a pre-clinical cottontail rabbit papillomavirus model. Vaccine 23:5271–5280
Van der Burg S, Kwappenberg K, O’Neill T, Brandt R, Melief C, Hickling J, Offringa R, (2001) Pre-clinical safety and efficacy of TA-CIN, a recombinant HPV16 L2E6E7 fusion protein vaccine, in homologous and heterologous prime-boost regimens. Vaccine 19:3652–3660
Van Der Burg SH (2008) Therapeutic vaccines in cancer: moving from immunomonitoring to immunoguiding. Expert Rev Vaccines 7:1–5
Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B (2008) A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol 4(4):e1000048
Wang P, Sidney J, Kim Y, Sette A, Lund O, Nielsen M, Peters B (2010) Peptide binding predictions for HLA DR. DP and DQ molecules BMC Bioinformatics 11:568
Zwaveling S et al (2002) Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol 169:350–358
Acknowledgements
This work was supported financially by Cancer Control Research Center, Cancer Control Foundation, Iran University of Medical Sciences, Tehran, Iran, as Project No: CCF-97087.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Bahareh Bahmani, Zahra Amini-bayat and Mohammad Mehdi Ranjbar. The first draft of the manuscript was written by Bahareh Bahmani and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bahmani, B., Amini-bayat, Z., Ranjbar, M.M. et al. HPV16-E7 Protein T Cell Epitope Prediction and Global Therapeutic Peptide Vaccine Design Based on Human Leukocyte Antigen Frequency: An In-Silico Study. Int J Pept Res Ther 27, 365–378 (2021). https://doi.org/10.1007/s10989-020-10089-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-020-10089-5