
Abstract
The [3 + 2] cycloaddition (32CA) reactions of 1-pyrroline-1-oxide with N-vinyl nucleobases leading to bicyclic N,O nucleoside analogues have been studied within the molecular electron density theory (MEDT) at the MPWB1K/6-311G(d,p) computational level. These non-polar zwitterionic type 32CA reactions take place through a one-step mechanism with minimal global electron density transfer (GEDT) at the TSs and the exo/ortho approach mode as the energetically favoured reaction path. The 32CA reactions of N-vinyl nucleobases with thymine and cytosine substituents respectively show the activation enthalpies of 15.2 and 12.5 kcal mol−1 in toluene. The reactions are irreversible due to strong exothermic character of − 35.4–− 26.4 kcal mol−1 in toluene. The bonding evolution theory (BET) study suggests that these 32CA reactions take place through the coupling of pseudoradical centres with earlier C–C bond formation and the formation of new C–C and C–O covalent bonds has not been started in the TSs. Non-covalent interactions (NCI) are predicted at the TSs from the visualization of NCI gradient isosurfaces.
Similar content being viewed by others
Introduction
Nucleoside analogues [1] constitute an important class of compounds in medicinal chemistry due to their unique therapeutic potential to mimic physiological nucleosides. The sphere of antiviral research experienced a major breakthrough with the evolution of nucleoside analogues [1, 2] that have been identified to exhibit broad spectrum activity for the treatment of chronic hepatitis B in 2011 [3] and for coronaviruses in 2019 [4]. Recently, Thomson and Lamont have also identified the use of modified nucleoside analogues as antibacterial agents [5]. The mechanism of drug resistance by nucleoside analogues is also well documented to establish them as important antimetabolites in the treatment of malignancies and tumours [6]. Owing to the important involvement of naturally occurring nucleosides in DNA and RNA synthesis, modified nucleoside analogues have been designed by chemists to act on the DNA/RNA chain terminators to achieve interesting biological results [7]. One of these modification strategies is to replace the carbohydrate moiety of the natural nucleoside with an isoxazolidine nucleus [8,9,10]. As model strategy of synthesis, the [3 + 2] cycloaddition (32CA) reactions of nitrones and N-vinyl nucleobases provide a straightforward route to these modified nucleosides [11]. As a unique initiative to modified nucleosides, Procopio and coworkers [11, 12] synthesized conformationally locked bicyclic N,O nucleoside analogues of antiviral drugs from 32CA reactions of the cyclic nitrone, 1-pyrroline-1-oxide 1 to N-vinyl nucleobases 2 and 3 (see Scheme 1).

32CA reactions of 1-pyrroline-1-oxide (1) with N-vinyl nucleobases (2, 3)
This reaction proceeded with exclusive regio and stereoselectivity [12] and induced a restricted conformational mobility in the nucleoside analogues due to the presence of a five-membered ring fused with the isoxazolidine system. The reactivity, selectivity and sequential bonding rearrangements are worth investigating theoretically due to the unique structural framework.
The evolution of computational chemistry [13] in the twentieth and twenty-first century has gradually attracted theoretical chemists to analyse the reactivity and selectivity of molecules. 32CA reactions have been targeted in several computational studies due to their varied range of mechanistic implications. In 2014, Jasiński et al. [14] proposed polar mechanism for 32CA reactions of nitrones to substituted nitroethenes, while the competition between one-step and two-step mechanisms in polar 32CA reactions of C,N-disubstituted nitrone to nitroethenes has been explained in 2018 [15]. Stepwise zwitterionic mechanisms for 32CA reactions [16, 17] have also been reported. Additionally, zwitterionic or biradical adducts with “extended conformation” may exist in reaction environment independently of [3 + 2] cycloadducts, which was reported by Jasiński [18] for 32CA reactions of nitroacetylene with allenyl type three-atom components. Recently in 2020 [19], one-step non-polar mechanism has been reported for 32CA reaction between (Z)-C,N-diphenyl nitrone and 1,2-bismethylene-3,3,4,4,5,5-hexamethylcyclopentane and this reaction proceeds without the intervention of a biradical intermediate. Thus, the mechanism of 32CA reactions shares an important place in the top shelf of theoretical organic chemistry.
A new theoretical outlook on organic reactions, called the molecular electron density theory [20, 21] (MEDT), was proposed in 2016 by Domingo to analyse the changes in electron density and hence the molecular reactivity of organic reactions. The MEDT perspective has been successfully applied to analyse several organic systems [21], majority of them dedicated to Diels-Alder (DA) and [3 + 2] cycloaddition (32CA) reactions. We have recently carried out MEDT [22,23,24,25,26] studies to analyse 32CA reactions of acyclic nitrones, nitrile oxides and azides leading to isoxazolidines [22, 23], spiroisoxazolines [24, 25] and 1,5-disubstituted 1,2,3-triazoles [26].
Herein, a theoretical analysis for the synthesis of bicyclic N,O nucleoside analogues experimentally performed by Procopio and coworkers [12] (Scheme 1) is presented within the molecular electron density theory [20, 21] (MEDT) framework. N-vinyl nucleobases with thymine and cytosine substituents are selected as the computational models. This study presents the first MEDT report to analyse the synthesis of bicyclic N,O-modified nucleosides and has been shaped into the following five sections:
-
(1)
Initially, the topological analysis of the electron localisation function [27, 28] (ELF) of the reactants 1, 2 and 3 (Scheme 1) is performed. ELF, constructed by Becke and Edgecombe in 1990 [27], establishes a quantitative connection between the electronic structure and molecular reactivity. Subsequent illustrations of the ELF attractors by Silvi and Savin in 1994 [28] have allowed characterizing the core, bonding and non-bonding regions in chemical structures. Domingo [20, 21] applied the ELF analysis to classify the three-atom components (TACs) participating in 32CA reactions, which has consequently defined their reactivity profile [29]. TACs having two pseudoradical centres (monosynaptic basin integrating less than 1e) are called pseudodiradical type [21], while TACs having one pseudoradical centre are called pseudo(mono)radical type [21]. TACs with a carbenoid centre (monosynaptic basin integrating 2e) are classified as carbenoid type [21] and finally, the TACs which do not have pseudoradical or carbenoid centres are classified as zwitterionic TACs. The activation energy of 32CA reactions increases in the order pdr-type < pmr-type ≈ cb-type < zw-type [29].
-
(2)
Analysis of the conceptual density functional theory [30, 31] (CDFT) indices at the ground state of the reactants is done to initially predict the reactivity and polar character of the 32CA reactions.
-
(3)
Energy profile of the stationary states along all possible regio- and stereochemical channels of the 32CA reactions is studied. The global electron density transfer [32] (GEDT) at the TSs is calculated to finally comment on the polar character of the 32CA reactions.
-
(4)
ELF [27, 28] topological analysis along with the Thom’s catastrophe theory [33] is used to obtain the sequential bonding changes along the reaction path from bonding evolution theory [34] (BET) analysis.
-
(5)
Topological analysis of the quantum theory of atoms in molecules [35, 36] (QTAIM) at the TSs is done to predict the nature of bonding and finally, the NCI [37] gradient isosurfaces at the TSs are plotted and analysed.
Computational methods
All stationary states were optimized using Berny analytical gradient optimization method [38] with the MPWB1K functional [39] in conjunction with the 6-311G(d,p) basis set [40]. This computational level has been successfully applied in MEDT studies [21, 24, 25, 29] of 32CA reactions. The absence of imaginary frequencies for local minimum and one imaginary frequency at the TSs was ensured through frequency calculations at the same level on the optimized geometries. Intrinsic reaction coordinate [41,42,43] (IRC) calculations were performed to verify the minimum energy reaction pathway (MERP) in mass-weighted Cartesian coordinates between the TSs, reactants and products.
The CDFT [30, 31] indices are namely the electronic chemical potential [30, 44] μ, chemical hardness [30, 45] η, electrophilicity index [30, 46] ω and relative nucleophilicity index [47] N by Eqs. (1)–(4)
where EHOMO and ELUMO are the HOMO and LUMO energies and EHOMO(tetracyanoethylene) is the HOMO energy of tetracyanoethylene as the reference.
The global electron density transfer [32] (GEDT) was calculated from the natural population analysis (NPA [48, 49]) by Eq. (5)
where qA is the net charge and the sum is taken over all the atoms of the nucleophile.
The electrophilic Pk+ and nucleophilic Pk− Parr functions [50] are calculated using Eqs. (6) and (7):
where ρsra (r) and ρsrc (r) are the Mulliken atomic spin densities (MASD) of radical anion and radical cation respectively.
The conductor-like polarizable continuum model (PCM) in the framework of the self-consistent reaction field [51,52,53] (SCRF) was used for solvent calculations in toluene. The enthalpies, Gibbs free energies and entropies were calculated in gas phase and toluene at the experimental temperature [12] 383 K (110 °C) and 1 atm.
ELF [27, 28] topological analysis and QTAIM [35, 36] parameter calculations were performed using the Multiwfn [54] software. ELF basin analysis was done with high-quality grid with spacing 0.06 Bohr. The UCSF Chimera software [55] was used to visualize the ELF localization domains and attractors. All computations were performed using the Gaussian 03 suite of programs [56].
Results and discussion
ELF topological analysis of 1-pyrroline-1-oxide 1, N-vinyl nucleobases 2 and 3
Topological analysis of the ELF [27, 28] establishes a quantitative correlation between the electronic structure and the reactivity of three-atom components [20, 21, 29] (TACs) participating in 32CA reactions. Consequently, the topological analysis of the ELF of 1-pyrroline-1-oxide 1 was performed to predict its reactivity in 32CA reactions. In addition, the ELF of N-vinyl nucleobases 2 and 3 was also analysed. The ELF localization domains are given in Fig. 1.

MPWB1K/6-311G(d,p) ELF localisation domains represented at an isosurface value of ELF = 0.85 of 1-pyrroline-1-oxide 1, N-vinyl nucleobases 2 and 3. Protonated basins are shown in blue, disynaptic basins are shown in green, monosynaptic basins are shown in red and core basins are shown in magenta colours
ELF topology of 1-pyrroline-1-oxide 1 shows the presence of two monosynaptic basins, V′(O1) and V(O1), integrating a total population of 5.88 e, a V(N2,C3) disynaptic basin integrating 3.80 e and a V(N2,O1) disynaptic basin integrating 1.55 e. The V(O1) and V′(O1) monosynaptic basins can be associated with the non-bonding electron density on the O1 oxygen. V(N2,C3) and V(N2,O1) disynaptic basins can be associated respectively with the underpopulated N–C double bond and N–O single bond. Thus, 1-pyrroline-1-oxide 1 can be classified as zwitterionic TAC owing to the absence of pseudoradical or carbenoid centres. ELF of the N-vinyl nucleobases 2 and 3 shows the presence of two disynaptic basins, V(C4,C5) and V′(C4,C5), integrating a total of 3.53 and 3.52 e, respectively, associated with the underpopulated C4–C5 double bond.
After establishing the bonding pattern of the reagents, the atomic charge distribution of 1-pyrroline-1-oxide 1 and N-vinyl nucleobases 2 and 3 was analysed through NPA [48, 49] (Fig. 2). In 1-pyrroline-1-oxide 1 (calculated dipole moment 4.28 D), the O1 oxygen is negatively charged by − 0.542 e, the N2 nitrogen is positively charged by 0.121 e and the C3 carbon shows a negligible charge of − 0.008 e. The computed dipole moment and charge distribution suggests charge separation in the nitrone; however, it differs from the expected charges from Lewis’s bonding model. The term “zwitterionic” used by Domingo [21] for this classification refers to the specific bonding pattern of the resonance Lewis structure proposed by Huisgen for the “1,3-dipoles” participating in 32CA reactions [57]. This term is not synonymous to the dipolar electronic structure of the nitrones.

MPWB1K/6-311G(d) calculated natural atomic charges, in average number of electrons e, of 1-pyrroline-1-oxide 1, N-vinyl nucleobases 2 and 3. Negative charges are coloured in red, and positive charges in blue
In the N-vinyl nucleobases 2 and 3, the unsubstituted carbon C5 shows higher negative charge value compared with C4 bearing positive charge 0.019 e in 2 and negligible negative charge 0.007 in 3, which indicates the changes in electronic rearrangement of the alkyne system due to thymine and cytosine substitution.
Analysis of the CDFT indices of the reactants
The concept of “Conceptual DFT” dates back to the pioneering work of Parr [58], and has been subsequently reviewed by Geerlings, Proft and Langenaeker in 2003 [31], and recently, by Domingo et al. in 2016 [30]. CDFT [30, 31] has been used as a powerful tool to understand the reactivity in numerous studies devoted to Diels Alder and 32CA reactions. Within the MEDT, the most relevant CDFT indices are analysed to address the chemical behaviour of the reactants. The standard electrophilicity and nucleophilicity scales [30] are defined at B3LYP/6-31G(d) level and hence have been used in the present CDFT study. The electronic chemical potential [44], μ, chemical hardness [45], ɳ, electrophilicity [46], ω, and nucleophilicity [47], N, at the ground state of 1-pyrroline-1-oxide 1, N-vinyl nucleobases 2 and 3, are listed in Table 1.
The electronic chemical potential [30, 44] μ of 1-pyrroline-1-oxide 1, μ = − 2.90 eV, is slightly lower than that of the vinyl nucleobases 2, μ = − 3.85 eV, and 3, μ = − 3.46 eV, indicating that the corresponding 32CA reactions will have non-polar character.
The electrophilicity [46] ω and nucleophilicity [47] N indices of 1-pyrroline-1-oxide 1 are 0.77 and 3.48 eV respectively, being classified a marginal electrophile and strong nucleophile within the corresponding scales [30].
The electrophilicity ω and nucleophilicity N indices of N-vinyl nucleobase 2 are 1.50 and 2.80 eV respectively, being classified a strong electrophile and moderate nucleophile, while vinyl nucleobase 3 is classified a moderate electrophile and strong nucleophile.
In 2004, Domingo [59] established that the asynchronicity in bond formation is controlled by electrophilic ethylene derivative participating in the 32CA reaction irrespective of the polar character of the reaction. As a result, the formation of the first new single bond begins from the most electrophilic centre of the ethylene derivative. Analysis of atomic spin densities (ASD) of the radical ions determines the electron density changes at the reactive sites and the local CDFT index, namely the electrophilic Pk+ and nucleophilic Pk− Parr functions [50] is analysed to obtain a quantitative comprehension. Consequently, for 32CA reactions of 1 with N-vinyl nucleobases 2 and 3, the electrophilic Pk+ Parr functions [50] of 2 and 3 were analysed (Fig. 3). The nucleophilic Pk− Parr function of 1 was also computed. The Mulliken atomic spin densities (MASDs) are given in Fig. 3.

Three-dimensional representation of the Mulliken atomic spin densities (MASDs) (isovalue = 0.0004) of radical anions 2− and 3−, and radical cation 1+ together with the electrophilic Pk+ Parr functions of 2 and 3 and the nucleophilic Pk− Parr functions of 1. Purple regions correspond to positive values of the MASD, while the brown regions correspond to negative regions of the MASD
For the C4–C5 bond, the electrophilic Pk+ Parr functions of C4 and C5 centres in 2, 0.07 and 0.17, respectively and in 3, − 0.03 and 0.17 respectively, indicate that the first single bond starts from the C5 carbon. This agrees well with the ELF topological analysis showing formation of pseudoradical centre at C5 earlier than C4 (the “ELF topological analysis at the TSs” section) in the bonding evolution theory study (the “Bonding evolution theory (BET) study of the 32CA reaction of 1-pyrroline-1-oxide 1 with the N-vinyl nucleobase 2” section) Finally, the O1 oxygen of 1 with Pk− = 0.72 presents the most nucleophilic activation, while the C3 is lesser nucleophilically activated in 1 with Pk− = 0.38.
Analysis of the energy profile associated with the 32CA reactions of 1-pyrroline-1-oxide 1 with the N-vinyl nucleobases 2 and 3
For the 32CA reactions of 1-pyrroline-1-oxide 1 to N-vinyl nucleobases 2 and 3, the two regiochemical paths, labelled ortho and meta, and the two diastereofacial isomeric reaction paths, endo and exo, along these two regiochemical paths, have been considered. The ortho regioisomeric reaction paths are associated with the formation of the C3–C5 and C4–O1 bonds, while the meta paths are associated with the formation of the C3–C4 and C5–O1 bonds (Scheme 2 and Scheme 3). The endo diastereofacial reaction path is associated with the approach of the N-vinyl nucleobases 2 and 3 in such a way that the thymine or cytosine substituent lies on the same side of the pyrroline ring of the nitrone 1, while in the exo diastereofacial reaction path, the thymine or cytosine substituent of the N-vinyl nucleobase lies on the opposite side of the pyrroline ring of the nitrone 1.

Studied reaction paths for 32CA reactions of 1-pyrroline-1-oxide (1) with N-vinyl nucleobases 2

Studied reaction paths for 32CA reactions of 1-pyrroline-1-oxide (1) with N-vinyl nucleobases 3
The search for stationary points along the reaction paths allowed locating and characterizing the reagents, 1, 2 and 3, one TS, TS1 (exo/ortho, 1 + 2), TS2 (endo/ortho, 1 + 2), TS3 (exo/meta, 1 + 2), TS4 (endo/meta, 1 + 2), TS5 (exo/ortho, 1 + 3), TS6 (endo/ortho, 1 + 3), TS7 (exo/meta, 1 + 3), and TS8 (endo/meta, 1 + 3), along each reaction path and the corresponding cycloadducts 4–11, the bicyclic nucleoside analogues. Consequently, these reactions proceed through one-step mechanism. The studied reaction paths for 32CA reaction of 1 with 2 and 3 are respectively shown in Scheme 2 and Scheme 3. The relative energies, enthalpies, entropies and free energies in gas phase and toluene at 383 K are given in Table 2, while the total energies in gas phase and toluene are given in S1 and S2 in the supplementary material.
The activation enthalpies range from 11.0 (TS5) to 16.0 (TS4) in gas phase and from 12.5 (TS5) to 19.6 (TS4) kcal mol−1 in toluene, with the 32CA reactions being strongly exothermic from 38.6 (8) to 29.4 (7) kcal mol−1 in gas phase and from 35.4 (8) to 26.4 (7) kcal mol−1 in toluene. Some appealing conclusions can be drawn from the relative energies. (i) The most favourable reaction path is associated with the exo/ortho approach mode in both reactions, yielding the experimentally obtained isoxazolidines 4 and 8, the bicyclic N,O-nucleoside analogues, via TS1 and TS5. (ii) The activation enthalpy for 32CA reaction of 1 with 3 with the cytosine substituent is lowered than that for 32CA reaction of 1 with 2 with the thymine substituent by 1.4 kcal mol−1 in gas phase and 2.7 kcal mol−1 in toluene at 383 K. (iii) The 32CA reactions show negative relative entropies of TS1–TS8 between − 43.2 and − 52.0 cal mol−1 K−1 in gas phase and between − 39.9 and − 50.9 cal mol−1 K−1 in toluene. The calculated negative relative entropies of cycloadducts 4–11 are − 47.6 to − 55.1 cal mol−1 K−1 in gas phase and − 45.1 to− 54.3 cal mol−1 K−1 in toluene.
(iv) These 32CA reactions are ortho regioselective and exo stereoselective in refluxing toluene, in complete agreement with the experiments [12]. The activation enthalpy, ∆H of exo/ortho TS1 associated with the 32CA reaction of 1 with 2, is lowered than that of TS2, TS3 and TS4 by 2.4, 2.8 and 4.4 kcal mol−1 respectively in toluene at 383 K, while the exo/ortho TS5 associated with the 32CA reaction of 1 with 3 is lowered than that of TS6, TS7 and TS8 by 2.5, 3.3 and 3.8 kcal mol−1 respectively in toluene at 383 K. The activation free energy, ∆G of exo/ortho TS1 associated with the 32CA reaction of 1 with 2, is lowered than that of TS2, TS3 and TS4 by 4.2, 4.3 and 5.9 kcal mol−1 respectively in toluene at 383 K. The activation free energy of exo/ortho TS5 associated with the 32CA reaction of 1 with 3 is lowered than that of TS6, TS7 and TS8 by 2.0, 2.2 and 5.4 kcal mol−1 respectively in toluene at 383 K. This suggests that for 32CA reaction of 1 with 2, the endo/ortho, exo/meta and endo/ortho are forbidden from kinetic point of view. For 32CA reaction of 1 with 3, the endo/ortho and exo/meta channels are less favoured but not forbidden, and the endo/meta channel is forbidden from kinetic point of view.
(v) The formation of isoxazolidines is strongly exothermic, which makes the reactions irreversible. (vi) The reaction enthalpy of isoxazolidine 4 is lowered than that of 5, 6 and 7 by 3.0, 1.3 and 3.1 kcal mol−1 respectively in toluene at 383 K, while the reaction enthalpy of isoxazolidine 8 is lowered than that of 9, 10 and 11 by 4.9, 5.1 and 6.8 kcal mol−1 respectively in toluene at 383 K. (vii) The inclusion of thermal corrections to the electronic energies increases the activation enthalpies by 0.8–2.1 kcal mol−1 in gas phase and by 0.9–1.9 kcal mol−1 in toluene, while the reaction enthalpies are decreased by 3.1–4.0 kcal mol−1 in gas phase and 3.3–4.1 kcal mol−1 in toluene. (viii) The inclusion of entropies to enthalpies strongly increases the activation free energies by 16.5–19.9 kcal mol−1 in gas phase and by 15.3–19.5 kcal mol−1 in toluene and strongly decreases the reaction enthalpies by 18.2–21.1 kcal mol−1 in gas phase and by 17.3–20.8 kcal mol−1 in toluene. This is due to the unfavourable entropies associated with these 32CA reactions. The activation Gibbs free energy of the isoxazolidines 4 and 8 respectively becomes 30.5 and 30.4 kcal mol−1 in toluene at 383 K. These activation free energies are respectively lowered than that of the other feasible reaction paths by 4.2–5.9 kcal mol−1 and 2.2–5.4 kcal mol−1 in toluene and hence account for the ortho regioselectivity and exo stereoselectivity experimentally observed [12]. The MPWB1K/6-311G(d,p) optimized geometry of TSs is given in Fig. 4 and the geometrical parameters are listed in Table 3.

MPWB1K/6-311G(d,p) optimized gas phase geometries of the TSs involved in the 32CA reactions of 1-pyrroline-1-oxide 1 with N-vinyl nucleobases 2 and 3
In toluene, the distances between C3 and C5 and O1 and C4 interacting centres at the eight TSs are 2.126 and 2.094 Å at TS1, 2.082 and 2.088 Å at TS2, 2.172 and 1.980 Å at TS3, 2.103 and 1.982 Å at TS4, 2.077 and 2.105 Å at TS5, 2.048 and 2.124 Å at TS6, 2.155 and 1.994 Å at TS7 and 2.090 and 1.989 Å at TS8 respectively. These geometrical parameters indicate that while the ortho TSs TS1, TS2, TS5 and TS6 show low ∆d (│dC3–C4−dO1–C5│) values of 0.032, 0.006, 0.028 and 0.076 Å, the meta TSs TS3, TS4, TS7 and TS8 show ∆d (│dC3–C4−dO1–C5│) values of 0.192, 0.121, 0.161 and 0.101 Å. The bond length in gas phase-optimized structures shows minimal differences than that in toluene. These geometrical parameters also suggest that at all eight TSs, the formation of the C–O and C–C single bonds has not yet been started, which is in agreement with the ELF topological studies at the TSs (see the “ELF topological analysis at the TSs” section).
The bond formation process can be quantitatively assessed by the calculation of l index and the asymmetry index ∆l proposed by Jasiński [17], given by Eqs. (8) and (9)
where rTSX−Y is the distance between the reaction centres X and Y in the transition state and rPX−Y is the same distance in the corresponding product.
The asymmetry index for the present reactions is given by,
The calculated l index and the asymmetry index ∆l at the TSs associated with the 32CA reaction of 1 with 2 and 3 are listed in Table 3. The ortho TSs show high asymmetry index ∆l as compared with the meta TSs. The forming C3–C5 bond is more advanced than the forming C4–O1 bond at the ortho TSs TS1, TS2, TS5 and TS6, which is in agreement with the ELF topological analysis along the reaction path showing earlier formation of C3–C5 bond (see the “Bonding evolution theory (BET) study of the 32CA reaction of 1-pyrroline-1-oxide 1 with the N-vinyl nucleobase 2” section). The calculated values in toluene show similar trend as the gas phase, with minimal changes in the calculated l and ∆l values.
Finally, in order to evaluate the polar nature of these 32CA reactions, the GEDT [32] at the TSs was analysed. The gas phase GEDT values at the TSs are 0.02 e at TS1, 0.01 e at TS2, 0.03 e at TS3, 0.03 e at TS4, 0.02 e at TS5, 0.03 e at TS6, 0.03 e at TS7 and 0.03 e at TS8 (see Table 2). In toluene, the GEDT values at the TSs show values between 0.01 and 0.04 e (see Table 2). These values being less than 0.1 e suggest a non-polar character for the 32CA reactions.
Bonding evolution theory (BET) study of the 32CA reaction of 1-pyrroline-1-oxide 1 with the N-vinyl nucleobase 2
The conjunction of ELF topological analysis [27, 28] and Thom’s catastrophe theory [33], termed as the bonding evolution theory (BET [34]), has proven to be a very useful methodological tool to establish the nature of the electronic rearrangement associated along the reaction path. Herein, the BET of the 32CA reactions of 1-pyrroline-1-oxide 1 with N-vinyl nucleobase 2 is studied as the model example. The sequential bonding changes resulting from the BET study are presented in Fig. 5.

Simple representation of the sequential bonding changes of 32CA reaction of 1-pyrroline-1-oxide 1 and N-vinyl nucleobase 2 predicted by BET study
The 32CA reaction of 1 with 2 takes place along seven different phases (see Table 4). Phase I starts at S0-I, dC4–O1 = 2.58 Å and dC3–C5 = 2.78 Å, which corresponds with the first structure of the IRC. ELF of S0-I is similar to that of the separated reagents (see the “ELF topological analysis of 1-pyrroline-1-oxide 1, N-vinyl nucleobases 2 and 3” section).
Phase II starts at S1-I, dC4–O1 = 2.18 Å and dC3–C5 = 2.26 Å with energy cost of 10.5 kcal mol−1. This phase is characterized by the creation of a new V(N2) monosynaptic basin, integrating 0.83 e, associated with the formation of a lone pair at the N2 nitrogen. The electron density of this lone pair mainly comes from the depopulation of the N2–C3 bonding region along Phase I, which experiences depopulation from 3.76 e at S0-I to 3.14 e at S1-I.
Phase III starts at S2-I, dC4–O1 = 2.15 Å and dC3–C5 = 2.23 Å with energy cost of 11.0 kcal mol−1. The two V(C4,C5) and V′(C4,C5) disynaptic basins present at S1-I have merged into a new V(C4,C5) disynaptic basin, integrating 3.33 e, which indicates beginning of the rupture of C4–C5 double bond.
Phase IV starts at S3-I, dC4–O1 = 2.09 Å and dC3–C5 = 2.14 Å with energy cost of 11.6 kcal mol−1, which is characterized by the creation of a new V(C3) and V(C5) monosynaptic basins, integrating 0.34 e and 0.36 e respectively, associated with the formation of a pseudoradical centre at the C3 and C5 carbon. The electron density for formation of pseudoradical centre at C3 comes from the N2–C3 bonding region which experiences depopulation from 3.04 e at S2-I to 2.54 e at S3-I. Together with this change, the V(C4,C5) disynaptic basin experiences depopulation from 3.33 e at S2-I to 2.99 e at S3-I, leading to formation of pseudoradical centre at C5. In this phase, TS1 is found.
Phase V starts at S4-I, dC4–O1 = 1.96 Å and dC3–C5 = 1.96 Å. At the beginning of this phase, the first more relevant change along the IRC takes place. At this structure, while the V(C3) and V(C5) monosynaptic basins present at S3-I are missing, a new V(C3,C5) disynaptic basin, integrating 1.26 e, is created. These topological changes indicate that the formation of the first C3–C5 single bond has begun at a C–C distance of 1.96 Å.
Phase VI starts at S5-I, dC4–O1 = 1.92 Å and dC3–C5 = 1.92 Å, which is characterized by the creation of a new V(C4) monosynaptic basin, integrating 0.18 e, associated with the formation of a pseudoradical centre at the C4 carbon. Together with this change, the V(C4,C5) disynaptic basin experiences a depopulation of 0.23 e along Phase V.
Finally, the last Phase VII starts at S6-I, dC4–O1 = 1.74 Å and dC3–C5 = 1.73 Å, and ends at the cycloadduct 4, dC4–O1 = 1.41 Å and dC3–C5 = 1.52 Å. At S6-I, the second more relevant change along the IRC takes place. At this structure, while the V(C4) monosynaptic basin is missing, a new V(C4,O1) disynaptic basin, integrating 0.69 e, is created. These relevant topological changes indicate that the formation of the second C4–O1 single bond has begun at a C–O distance of 1.73 Å, through the C– to –O coupling of the electron density of the C4 pseudoradical carbon (integrating 0.18 e) and part of the non-bonding electron density of the O1 oxygen [29]. Along this last phase, the molecular electron density is relaxed to reach the structure cycloadduct 4, in which the populations of the V(C4,O1) and V(C3,C5) disynaptic basins reach a population of 1.35 and 1.90 e, respectively.
ELF topological analysis at the TSs
Finally, the ELF topology of the eight gas phase TSs was analysed and compared. The populations of the most significant ELF valence basin at TS1, TS2, TS3, TS4, TS5, TS6, TS7 and TS8 are given in Table 5, while the pictorial representation of ELF localisation domains of TS1–TS4 is given in Fig. 6.

Pictorial representation of MPWB1K/6-311G(d) ELF localisation domains of TS1–TS4 represented at an isosurface value of ELF = 0.85. Protonated basins are shown in blue, disynaptic basins are shown in green and monosynaptic basins are shown in red. The attractors are shown in orange colour
ELF of TS1–TS8 shows the presence of one V(N2) monosynaptic basin, integrating 1.14–1.39 e, whose electron density comes from the depopulation of the N2–C3 bonding region. Note that V(N2,C3) disynaptic basin experiences a depopulation of the electron density from 3.80 e in 1 to 2.54 e, 2.44 e, 2.51 e, 2.52 e, 2.55 e, 2.59 e, 2.55 e and 2.54 e in TS1–TS8, respectively.
At the alkyne framework, the pair of disynaptic basins, V(C4,C5) and V′(C4,C5), associated with C4–C5 bonding region in N-vinyl nucleobases 2 and 3 have experienced depopulation and are merged into one V(C4,C5) disynaptic basin in the TSs integrating a total population of 2.92–3.02 e. The ELF of the TSs also shows the presence of one V(C5) and one V(C3) monosynaptic basin integrating at 0.31–0.46 e and 0.36–0.44 e respectively associated with the formation of pseudoradical centre at C3 and C5. It should be noted that the pseudoradical centre is formed earlier at C5, which begins the formation of the new C–C single bond, in complete agreement with the analysis of the Parr functions (the “Analysis of the CDFT indices of the reactants” section).
Finally, the absence of neither V(C3,C5) nor V(C4,O1) disynaptic basin at these TSs indicates that the formation of new C3–C5 and O1–C4 single bonds has not yet begun, which is consistent with the corresponding C4–O1 and C3–C5 distance above 2.0 Å (see Table 3).
QTAIM topological analysis at TSs
A quantum theory of atoms in molecules [35, 36] (QTAIM) topological analysis of the electron density ρ at the critical points (CPs) corresponding to the molecular region associated with the formation of new C3–C5 and O1–C4 single bonds at the eight TSs was carried out. The calculated QTAIM parameters are given in Table 6.
Laplacian of electron density ∇2ρ(rc) at CPC–C and CPC–O shows positive values at the eight TSs. The low electron density ρ(rc) values together with the positive Laplacian ∇2ρ(rc) at these CPs indicate the absence of any covalent bonding interaction between the reacting nuclei at these TSs, and consequently that the formation of the new C3–C5 and O1–C4 single bonds has not yet started at these TSs, in complete agreement with the ELF topological analysis (the “ELF topological analysis at the TSs” section).
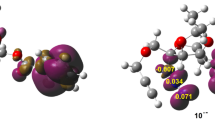
In 2011, García and coworkers [37] proposed the NCI plot program to map and analyse non-covalent interactions in molecular systems. NCI plot uses the reduced density gradient (RDG) which is used to isolate non-covalent interactions in real space. Large negative values of sign (λ2)ρ indicate attractive interactions, while large positive values of sign (λ2)ρ are associated with repulsive interactions. NCI plots of the preferred TSs, TS1 and TS5 of the two studied 32CA reactions are shown in Fig. 7.

NCI gradient isosurfaces of TS1 and TS5. Surfaces are coloured in the (− 0.02, 0.02) au range of sign (λ2)ρ (isosurfaces = 0. au)
TS1 shows non-covalent attractive overlap (blue portions) as well as repulsive overlap (red portions) between O1 and C4 and also between C3 and C5. For TS5, while the non-covalent attractive overlap (blue portions) and repulsive overlap (red portions) are shown between O1 and C4, only the repulsive overlap (red portions) is shown between C3 and C5.
Conclusion
The 32CA reactions of 1-pyrroline-1-oxide 1 with N-vinyl nucleobases 2 and 3 have been studied within MEDT at the MPWB1K/6-311G(d,p) computational level.
Topological analysis of the ELF of 1-pyrroline-1-oxide 1 allows its classification as zwitterionic TAC participating in zw-type 32CA reactions that is consistent with the calculated high activation enthalpies of 12.5–19.6 kcal mol−1 in toluene.
These 32CA reactions take place through a one-step mechanism and the lowest activation enthalpy corresponds to the exo/ortho approach mode. The 32CA reaction of N-vinyl nucleobase with the cytosine substituent is lowered than that of the thymine substituent by 2.7 kcal mol−1 in toluene at 383 K. These 32CA reactions show minimal GEDT at the TSs, which suggests their non-polar character.
The BET analysis shows formation of pseudoradical centre at the unsubstituted C5 carbon of the N-vinyl nucleobase initially and agrees well with the Parr function predictions, consequently leading to the earlier C3–C5 bond formation through coupling of pseudoradical centres. Topological analysis of ELF and AIM indicates early TSs in which the formation of new C–C or C–O covalent bonds has not started.
The present MEDT study allows concluding that the 32CA reactions of 1-pyrroline-1-oxide 1 with N-vinyl nucleobases 2 and 3 leading to bicyclic N,O nucleoside analogues are non-polar zwitterionic type 32CA reactions involving early TSs, and bond formation takes place through one-step mechanism through coupling of the pseudoradical centres.
References
Seley-Radtke KL, Yates MK (2018) The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: early structural modifications to the nucleoside scaffold. Antivir Res 154:66–86. https://doi.org/10.1016/j.antiviral.2018.04.004
Jordheim LP, Durantel D, Zoulim F, Dumontet C (2013) Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov 12:447–464. https://doi.org/10.1038/nrd4010
Fung J, Lai CL, Seto WK, Yuen MF (2011) Nucleoside/nucleotide analogues in the treatment of chronic hepatitis B. J Antimicrob Chemother 66:2715–2725. https://doi.org/10.1093/jac/dkr388
Pruijssers AJ, Denison MR (2019) Nucleoside analogues for the treatment of coronavirus infections. Curr Opin Virol 35:57–62. https://doi.org/10.1016/j.coviro.2019.04.002
Thomson JM, Lamont IL (2019) Nucleoside analogues as antibacterial agents. Front Microbiol 10:952. https://doi.org/10.3389/fmicb.2019.00952
Galmarini CM, Mackey JR, Dumontet C (2001) Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia 15:875–890. https://doi.org/10.1038/sj.leu.2402114
Pan S, Eang G, Schinazi RF, Zhao K (1998) Synthesis of novel isoxazolinyl substituted imidazo[1,2-a]pyridine C-nucleoside analogs. Tetrahedron Lett 39:8191–8194. https://doi.org/10.1016/S0040-4039(98)01872-3
Colacino E, Converso A, De Nino A, Leggio A, Liguori A, Maiuolo L, Napoli A, Procopio A, Siciliano C, Sindona G (1999) Synthesis of isoxazolidino analogues of 2′,3′-dideoxynucleosides. Nucleosides nucleotides nucleic acids 18:581-583. https://doi.org/10.1080/15257779908041501
Chiacchio U, Corsaro A, Iannazzo D, Piperno A, Rescifina A, Romeo R, Romeo G (2001) Diastereoselective synthesis of N,O-psiconucleosides via 1,3-dipolar cycloadditions. Tetrahedron Lett 42:1777–1780. https://doi.org/10.1016/S0040-4039(00)02325-X
Gi HJ, Xiang Y, Schinazi RF, Zhao K (1997) Synthesis of dihydroisoxazole nucleoside and nucleotide analogs. J Org Chem 62:88–92. https://doi.org/10.1021/jo961779r
Nguyen TB, Martel A, Gaulon C, Dahl R, Dujardin G (2010) 1,3-Dipolar cycloadditions of nitrones to heterosubstituted alkenes. Part 1: oxa and aza-substituted alkenes. Org Prep Proced Int 42:387–431. https://doi.org/10.1080/00304948.2010.513886
Procopio A, Alcaro S, Nino AD, Maiuolo L, Ortuso F, Sindona G (2005) New conformationally locked bicyclic N,O-nucleoside analogues of antiviral drugs. Biorg Med Chem Lett 15:545–550. https://doi.org/10.1016/j.bmcl.2004.11.048
Krylov A, Windus TL, Barnes T, Marin-Rimoldi E, Nash JA, Pritchard B, Smith DGA, Altarawy D, Saxe P, Clementi C, Crawford TD, Harrison RJ, Jha S, Pande VS, Head-Gordon T (2018) Perspective: computational chemistry software and its advancement as illustrated through three grand challenge cases for molecular science. J Chem Phys 149:180901. https://doi.org/10.1063/1.5052551
Jasiński R, Ziółkowska M, Demchuk OM, Maziark A (2014) Regio- and stereoselectivity of polar[2+3] cycloaddition reactions between(Z)-C-(3,4,5-trimethoxyphenyl)-N-methylnitrone and selected (E)-2-substituted nitroethenes. Centr Eur J Chem 12:586–593. https://doi.org/10.2478/s11532-014-0518-2
Jasiński R (2018) Competition between one-step and two-step mechanism in polar [3 + 2] cycloadditions of (Z)-C-(3,4,5-trimethoxyphenyl)-N-methyl-nitrone with (Z)-2-EWG-1-bromo-1-nitroethenes. Comput Theo Chem 1125:77–85. https://doi.org/10.1016/j.comptc.2018.01.009
Jasiński R (2015) In the searching for zwitterionic intermediates on reaction paths of [3 + 2] cycloaddition reactions between 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide and polymerizable olefins. RSC Adv 5:101045–101048. https://doi.org/10.1039/C5RA20747A
Jasiński R (2015) A stepwise, zwitterionic mechanism for the 1,3-dipolar cycloaddition between (Z)-C-4-methoxyphenyl-N-phenylnitrone and gem-chloronitroethene catalysed by 1-butyl-3-methylimidazolium ionic liquid cations. Tetrahedron Lett 56:532–535. https://doi.org/10.1016/j.tetlet.2014.12.007
Jasiński R (2015) Nitroacetylene as dipolarophile in [2 + 3] cycloaddition reactions with allenyl-type three-atom components: DFT computational study. Monatsh Chem 146:591–599. https://doi.org/10.1007/s00706-014-1389-0
Jasiński R (2020) A new insight on the molecular mechanism of the reaction between(Z)-C,N-diphenylnitrone and 1,2-bismethylene-3,3,4,4,5,5-hexamethylcyclopentane. J Mol Graph Model 94: 107461. https://doi.org/10.1016/j.jmgm.2019.107461
Domingo LR (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21:1319. https://doi.org/10.3390/molecules21101319
Ríos-Gutiérrez M, Domingo LR (2019) Unravelling the mysteries of the [3+2] cycloaddition reactions. Eur Jour Org Chem 2019:267–282. https://doi.org/10.1002/ejoc.201800916
Domingo LR, Acharjee N (2018) [3+2] Cycloaddition reaction of c-phenyl-n-methyl nitrone to acyclic-olefin-bearing electron-donating substituent: a molecular electron density theory study. ChemistrySelect 3:8373–8380. https://doi.org/10.1002/slct.201801528
Acharjee N, Banerji A (2020) A molecular electron density theory study to understand the interplay of theory and experiment in nitrone-enone cycloaddition. J Chem Sci 132:65. https://doi.org/10.1007/s12039-020-01766-5
Domingo LR, Ríos-Gutiérrez M, Acharjee N (2019) A molecular electron density theory study of the chemoselectivity, regioselectivity, and diastereofacial selectivity in the synthesis of an anticancer spiroisoxazoline derived from α-santonin. Molecules 24:832. https://doi.org/10.3390/molecules24050832
Acharjee N (2020) Theoretical analysis of the regio- and stereoselective synthesis of spiroisoxazolines. J Mol Model 26:117. https://doi.org/10.1007/s00894-020-04372-x
Domingo L R, Acharjee N (2020) A molecular electron density theory study of the Grignard reagent-mediated regioselective direct synthesis of 1,5-disubstituted-1,2,3-triazoles. J Phys Org Chem:e4062. https://doi.org/10.1002/poc.4062
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5403. https://doi.org/10.1063/1.458517
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–686. https://doi.org/10.1038/371683a0
Domingo LR, Ríos-Gutiérrez M, Pérez P (2018) A molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of c,n-dialkyl nitrones with ethylene derivatives. J Org Chem 83: 2182–2197. https://doi.org/10.1021/acs.joc.7b03093
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748. https://doi.org/10.3390/molecules21060748
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev103:1793–1874. https://doi.org/10.1021/cr990029p
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428. https://doi.org/10.1039/C4RA04280H
Thom R (1972) StabilitéStructurelle et Morphogénèse. Intereditions, Paris
Krokidis X, Noury S, Silvi B (1997) Characterization of elementary chemical processes by catastrophe theory. J Phys Chem A 101:7277–7282. https://doi.org/10.1021/jp9711508
Bader RFW (1994) Atoms in molecules: a quantum theory. Clarendon Press, USA
Bader RFW, Essén H (1984) The characterization of atomic interactions. J Chem Phys 80:1943–1960. https://doi.org/10.1063/1.446956
García JC, Johnson ER, Keinan S, Chaudret R, Piquemal JP, Beratan DN, Yang W (2011) NCIPLOT: a program for plotting noncovalent interaction regions. J Chem Theory Comput 7:625–632. https://doi.org/10.1021/ct100641a
Schlegel HB (1982) Optimization of equilibrium geometries and transition structures. J Comput Chem 3:214–218. https://doi.org/10.1002/jcc.540030212
Zhao Y, Truhlar DG (2004) Hybrid meta density functional theory methods for thermochemistry, thermochemical kinetics, and noncovalent interactions: the MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J Phys Chem A 108:6908–6918. https://doi.org/10.1021/jp048147q
Hehre WJ, Radom L, Schleyer PVR, Pople JA (1996) Ab initio molecular orbital theory. Wiley, New York, USA
Fukui K (1970) Formulation of the reaction coordinate. J Phys Chem 74:4161–4163. https://doi.org/10.1021/j100717a029
González C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527. https://doi.org/10.1021/j100377a021
González C, Schlegel HB (1991) Improved algorithms for reaction path following: higher-order implicit algorithms. Chem Phys 95:5853–5860. https://doi.org/10.1063/1.461606
Parr RG, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, Oxford 4:70–86
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423. https://doi.org/10.1016/S0040-4020(02)00410-6
Domingo LR, Pérez P (2011) The nucleophilicity N index in organic chemistry. Org. Biomol Chem 9:7168–7175. https://doi.org/10.1039/C1OB05856H
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746. https://doi.org/10.1063/1.449486
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926. https://doi.org/10.1021/cr00088a005
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494. https://doi.org/10.1039/C2RA22886F
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094. https://doi.org/10.1021/cr00031a013
Cances E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041. https://doi.org/10.1063/1.474659
Barone V, Cossi M, Tomasi J (1998) Geometry optimization of molecular structures in solution by the polarizable continuum model. J Comput Chem 19:404–417. https://doi.org/10.1002/(SICI)1096-987X(199803)19:4<404::AID-JCC3>3.0.CO;2-W
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592. https://doi.org/10.1002/jcc.22885
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. https://doi.org/10.1002/jcc.20084
Gaussian 03, Revision D.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, C. Peng Y, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA, Gaussian, Inc., Wallingford CT, 2004
Huisgen R (1961) 1,3-dipolar cycloadditions. Proc Chem Soc:357–396. https://doi.org/10.1039/PS9610000357
Parr RG, Yang W (1995) Density-functional theory of the electronic structure of molecules. Annu Rev Phys Chem 46:701–728. https://doi.org/10.1146/annurev.pc.46.100195.003413
Aurell MJ, Domingo LR, Pérez P, Contreras R (2004) A theoretical study on the regioselectivity of 1,3-dipolar cycloadditions using DFT-based reactivity indexes. Tetrahedron 60:11503–11509. https://doi.org/10.1016/j.tet.2004.09.057
Acknowledgements
The author acknowledges the help and support of Professor Luis R Domingo, Professor, University of Valencia, Spain, for the important clarifications related to the concept of molecular electron density theory. The author is also thankful to Professor Manas Banerjee, Retired Professor, The University of Burdwan, India for the kind cooperation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that he has no conflict of interest
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
Supplementary Material Tables with the MPWB1K/6-311G(d,p) electronic energies, enthalpies and Gibbs free energies of the stationary points and Cartesian coordinates of the optimized TSs involved in the 32CA reactions of 1-pyrroline-1-oxide 1 with N-vinyl nucleobases 2 and 3, in gas phase and toluene. (DOCX 44 kb)
Rights and permissions
About this article

Cite this article
Acharjee, N. Unravelling the regio- and stereoselective synthesis of bicyclic N,O-nucleoside analogues within the molecular electron density theory perspective. Struct Chem 31, 2147–2160 (2020). https://doi.org/10.1007/s11224-020-01569-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-020-01569-x